

ABSTRACT
Zaire ebolavirus (EBOV), Bundibugyo ebolavirus (BDBV), and Reston ebolavirus (RESTV) belong to the same genus but exhibit different virulence properties. VP24 protein, a structural protein present in all family members, blocks interferon (IFN) signaling and likely contributes to virulence. Inhibition of IFN signaling by EBOV VP24 (eVP24) involves its interaction with the NPI-1 subfamily of karyopherin alpha (KPNA) nuclear transporters. Here, we evaluated eVP24, BDBV VP24 (bVP24), and RESTV VP24 (rVP24) interactions with three NPI-1 subfamily KPNAs (KPNA1, KPNA5, and KPNA6). Using purified proteins, we demonstrated that each VP24 binds to each of the three NPI-1 KPNAs. bVP24, however, exhibited approximately 10-fold-lower KPNA binding affinity than either eVP24 or rVP24. Cell-based assays also indicate that bVP24 exhibits decreased KPNA interaction, decreased suppression of IFN induced gene expression, and a decreased half-life in transfected cells compared to eVP24 or rVP24. Amino acid sequence alignments between bVP24 and eVP24 also identified residues within and surrounding the previously defined eVP24-KPNA5 binding interface that decrease eVP24-KPNA affinity or bVP24-KPNA affinity. VP24 mutations that lead to reduced KPNA binding affinity also decrease IFN inhibition and shorten VP24 half-lives. These data identify novel functional differences in VP24-KPNA interaction and reveal a novel impact of the VP24-KPNA interaction on VP24 stability.
IMPORTANCE The interaction of Ebola virus (EBOV) VP24 protein with host karyopherin alpha (KPNA) proteins blocks type I interferon (IFN) signaling, which is a central component of the host innate immune response to viral infection. Here, we quantitatively compared the interactions of VP24 proteins from EBOV and two members of the Ebolavirus genus, Bundibugyo virus (BDBV) and Reston virus (RESTV). The data reveal lower binding affinity of the BDBV VP24 (bVP24) for KPNAs and demonstrate that the interaction with KPNA modulates inhibition of IFN signaling and VP24 stability. The effect of KPNA interaction on VP24 stability is a novel functional consequence of this virus-host interaction, and the differences identified between viral species may contribute to differences in pathogenesis.
KEYWORDS: STAT1, ebola virus, filovirus, interferons, karyopherin, protein stability
INTRODUCTION
The filovirus family is comprised of three genera, Ebolavirus, Marburgvirus, and Cuevavirus. Within the Ebolavirus genus are five species: Zaire ebolavirus (also known as Ebola virus [EBOV]), Sudan ebolavirus, Bundibugyo ebolavirus, Tai Forrest ebolavirus, and Reston ebolavirus (1). EBOV has been associated with multiple outbreaks of lethal human disease, including the 2014 West African epidemic that resulted in approximately 28,000 infections and 11,000 deaths (2). Other members of the genus have displayed various degrees of virulence. Notably, outbreaks due to Bundibugyo virus (BDBV) resulted in case fatality rates of approximately 30%, a figure lower than that seen in EBOV outbreaks (3). Moreover, BDBV was also less lethal than EBOV in nonhuman primate studies (4). Despite exposure of individuals to infected animals and evidence of seroconversion in humans, Reston virus (RESTV) stands out for never having caused documented human disease, suggesting that RESTV is of low virulence in humans (5).
The molecular determinants of virulence and replication efficiency of members within the Ebolavirus genus remain incompletely defined. However, the capacity of these viruses to suppress interferon (IFN) responses is thought to contribute to their pathogenesis (6). One viral protein that functions as a suppressor of IFN responses is VP24 (6). VP24 is a viral structural protein that also influences viral particle infectivity, virus host range, and viral RNA synthesis (7–16). EBOV VP24 (eVP24), RESTV VP24 (rVP24), and the VP24 of Lloviu virus, an as-yet-uncultured virus classified in the genus Cuevavirus, all inhibit IFN-induced gene expression, whereas VP24s from members of the Marburgvirus genus do not (17–22). Previous studies demonstrated that eVP24 competes with tyrosine-phosphorylated STAT1 (PY-STAT1) for interaction with the NPI-1 subfamily of karyopherin alphas (KPNA): KPNA1, KPNA5, and KPNA6 (18, 19, 21). This results in a block in PY-STAT1 nuclear import by the KPNAs, leading to inhibition of IFN-induced gene expression and impaired antiviral activity. Structural studies defined the eVP24-binding interface with KPNA5 and allowed testing of the competitive inhibition model (21). These studies also defined a high-affinity target site for VP24s on KPNAs that overlaps the binding site for the nonclassical nuclear localization signal (ncNLS) used by PY-STAT1.
Although eVP24 inhibition of IFN signaling has been studied in detail, it has been unclear whether the VP24s from different ebolavirus species differ in how they interact with KPNAs and whether any differences have functional consequences. Here, we compared the VP24s from EBOV, BDBV, and RESTV, viruses with different virulence profiles in humans. We demonstrate different affinities of the VP24s for the NPI-1 subfamily KPNAs, with BDBV VP24 (bVP24) in particular exhibiting lower affinities than either eVP24 or rVP24. This correlates with a relatively reduced level of inhibition of IFN signaling by bVP24. In addition, bVP24 accumulates to lower levels in transfected cells than does eVP24 or rVP24, corresponding to a shorter bVP24 half-life. bVP24 can be stabilized, however, by overexpression of NPI-1 subfamily KNPAs. Assessments using KPNA and eVP24 mutants with different binding affinities support the idea of a direct influence of KPNA interaction on VP24 stability. These findings provide a novel explanation of the functional consequence of the interaction between VP24 and KPNA.
RESULTS
bVP24 binds to the same KPNAs as do eVP24 and rVP24.
To examine the efficiencies with which eVP24, rVP24, and bVP24 bind to the NPI-1 subfamily of KPNAs, coimmunoprecipitation (coIP) experiments with Flag-tagged KPNAs and hemagglutinin (HA)-tagged VP24s were performed. As a positive control, an HA-eVP24-Flag-KPNA5 coIP was included. As a negative control, HA-tagged Marburg virus VP24 (mVP24) was used because it does not interact with KPNAs (22). eVP24, rVP24, and bVP24 were each found to interact with KPNA1 (Fig. 1A), KPNA5 (Fig. 1B), and KPNA6 (Fig. 1C). None of the VP24s detectably interacted with the non-NPI-1 KPNA2 (Fig. 1D). Notable in these experiments was the lower efficiency of bVP24 pulldown, particularly with KPNA1 (Fig. 1A) and KPNA5 (Fig. 1B). bVP24 also accumulated to lower levels in the whole-cell extracts than eVP24 or rVP24. These differences were most pronounced in the absence of KPNA overexpression. Further, when KPNA1, KPNA5, or KPNA6 was coexpressed, the signal of bVP24 in the whole-cell extracts, relative to that of eVP24 or rVP24, was increased.
FIG 1.

eVP24, rVP24, and bVP24 coprecipitate with each member of the NPI-1 subfamily of KPNAs. (A) VP24 coprecipitations with KPNA1. Empty expression plasmid (E) and HA-tagged eVP24, rVP24, bVP24, or mVP24 were transfected alone or in combination with KPNA5 or KPNA1, as indicated. Shown are Western blots of whole-cell extracts (WCE) or anti-Flag immunoprecipitates (IP:Flag). KPNAs were detected by blotting with anti-Flag antibody, and VP24 proteins were detected by blotting with anti-HA antibody. WCEs were blotted with anti-β-tubulin (TubB) as a loading control. The interaction of KPNA5 with eVP24 and the absence of interaction of KPNA5 with mVP24 served as positive and negative controls, respectively. (B) VP24 coprecipitations with KPNA5. (C) VP24 coprecipitations with KPNA6. (D) Absence of VP24 interactions with KPNA2.
bVP24 binds to KPNA1, KPNA5, and KPNA6 with reduced affinities in comparison to eVP24 and rVP24.
The dissociation constants (KD) for eVP24, rVP24, and bVP24 interaction with KPNAs were quantified with biolayer interferometry (BLI) (see Fig. S1 in the supplemental material). eVP24 bound KPNA1, KPNA5, and KPNA6 with dissociation constants of 13.3 ± 3.9 nM, 9.77 ± 1.2 nM, and 5.33 ± 1.5 nM, respectively (Table 1). The affinities of bVP24 for the three KPNAs were 5-fold to 10-fold weaker than those of eVP24, binding KPNA1 with a KD of 69.7 ± 11 nM, KPNA5 with a KD of 43.3 ± 7.1 nM, and KPNA6 with a KD of 49.0 ± 4.2 nM (Table 1). The affinities of rVP24 for KPNA1, KPNA5, and KPNA6, at 5.23 ± 2.6 nM, 7.07 ± 2.5 nM, and 3.17 ± 1.3 nM, respectively, were similar to those of eVP24 (Table 1).
TABLE 1.
Affinities of VP24 proteins for members of the NPI-1 subfamily of KPNAs
| Protein |
KD (nM)a |
||
|---|---|---|---|
| KPNA1 | KPNA5 | KPNA6 | |
| eVP24 | 13.3 ± 3.9 | 9.77 ± 1.2 | 5.33 ± 1.5 |
| bVP24 | 69.7 ± 11 | 43.3 ± 7.1 | 49.0 ± 4.2 |
| rVP24 | 5.23 ± 2.6 | 7.07 ± 2.5 | 3.17 ± 1.3 |
Data represent the means and standard deviations of results from three experiments.
bVP24 shows reduced inhibition of IFN-induced gene expression relative to eVP24 or rVP24.
Next, we assessed the three VP24s for their potency of IFN signaling inhibition. Following transfection of cells with empty vector or HA-tagged VP24 and an ISG54 promoter reporter plasmid, cells were treated with IFN-β and reporter activity was assessed. While bVP24 efficiently inhibited ISG54 reporter activity at higher concentrations, the level of this inhibition was reduced more quickly than that seen with eVP24 or rVP24 at lower concentrations of the protein (Fig. 2A). rVP24 functional activity was similar to that of eVP24, consistent with the KPNA binding data (Fig. 2A) and data from a previous study (19). These findings are consistent with a model where the lower bVP24 binding affinity for KPNAs results in less-efficient IFN inhibition. However, it should also be noted that bVP24 once again appeared to accumulate to lower levels than did eVP24 and rVP24 (Fig. 2B).
FIG 2.
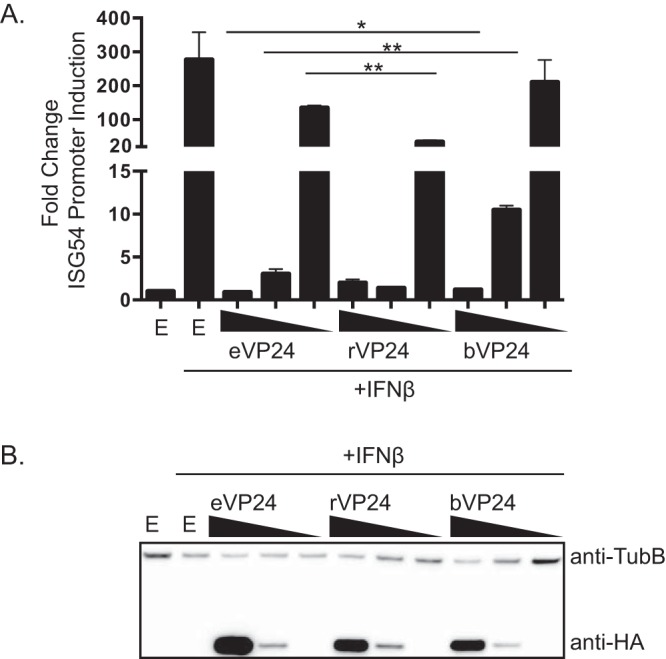
Impact of VP24s on IFN-induced gene expression. (A) The effect of expressing HA-tagged eVP24, rVP24, or bVP24 on IFN-β-induced expression of an ISG54 promoter reporter gene was assessed. Cells were transfected with empty vector (E) or 100 ng, 10 ng, or 1 ng of VP24 expression plasmid (decreasing concentrations are indicated by the wedges), an ISG54 promoter-firefly luciferase reporter plasmid, and a constitutively expressed Renilla luciferase plasmid. IFN-β was added after an overnight incubation, and luciferase values were measured 24 h later. Columns are representative of the means and error bars represent the standard deviations of results from two biological replicates each, including three technical replicates of each condition. Statistical significance was assessed by Student's t test. For eVP24 at 100 ng versus rVP24 at 100 ng, the single asterisk (*) represents P = 0.029; for eVP24 at 10 ng versus bVP24 at 10 ng, double asterisks (**) represent P = 0.0054; for eVP24 at 100 ng versus bVP24 at 100 ng, the single asterisk (*) represents P = 0.0276. (B) Cell lysates from the experiment whose results are shown in panel A were assayed by Western blotting for HA-VP24 expression (anti-HA) and β-tubulin expression (anti-TubB).
bVP24 displays a shorter half-life than eVP24.
Next, VP24 half-life was quantified in the absence or presence of interacting KPNAs. Transfected cells were treated with an inhibitor of protein synthesis, cycloheximide (CHX), and protein lysates were examined for VP24 expression at several time points. Protein was followed over an 8-h time period (Fig. 3), and Western blot signals were quantified and plotted (Fig. 4). In the absence of KPNA overexpression, eVP24 levels modestly decreased by 8 h post-CHX addition. This loss of signal was mostly reversed when KPNA1, KPNA5, or KPNA6 was coexpressed (Fig. 3A and 4A). In contrast, expression of noninteracting KPNA2 did not result in increased eVP24 half-life (Fig. 3B and 4A). bVP24 exhibited a shorter half-life in comparison to eVP24, and its stability also increased upon coexpression of the NPI-1 subfamily KPNAs (Fig. 3C and 4B) but not KPNA2 (Fig. 3D and 4B). Interestingly, rVP24 levels in the absence of KPNA overexpression were high at the beginning of the time course but did drop over time. rVP24 remained relatively stable in the presence of KPNA1, KPNA2, or KPNA6. However, total levels were lower and stability was decreased in the presence of KPNA5 in both experiments (Fig. 3E and F and 4C). These data suggest that interactions with KPNAs can modulate VP24 stability.
FIG 3.

VP24 protein stability is modulated upon coexpression of interacting KPNAs. (A) Abundance of eVP24 in the presence of KPNA1, KPNA5, or KPNA6. 293T cells were transfected with the indicated plasmids and were treated at 24 h posttransfection with cycloheximide (CHX). Starting at time zero, protein lysates were harvested and analyzed by Western blotting (WB) to detect Flag-tagged KPNA, HA-tagged VP24, or β-tubulin (TubB), as indicated. (B) Abundance of eVP24 in the presence of KPNA2. (C) Abundance of bVP24 in the presence of KPNA1, KPNA5, or KPNA6. (D) Abundance of bVP24 in the presence of KPNA2. (E) Abundance of rVP24 in the presence of KPNA1, KPNA5, or KPNA6. (F) Abundance of rVP24 in the presence of KPNA2.
FIG 4.
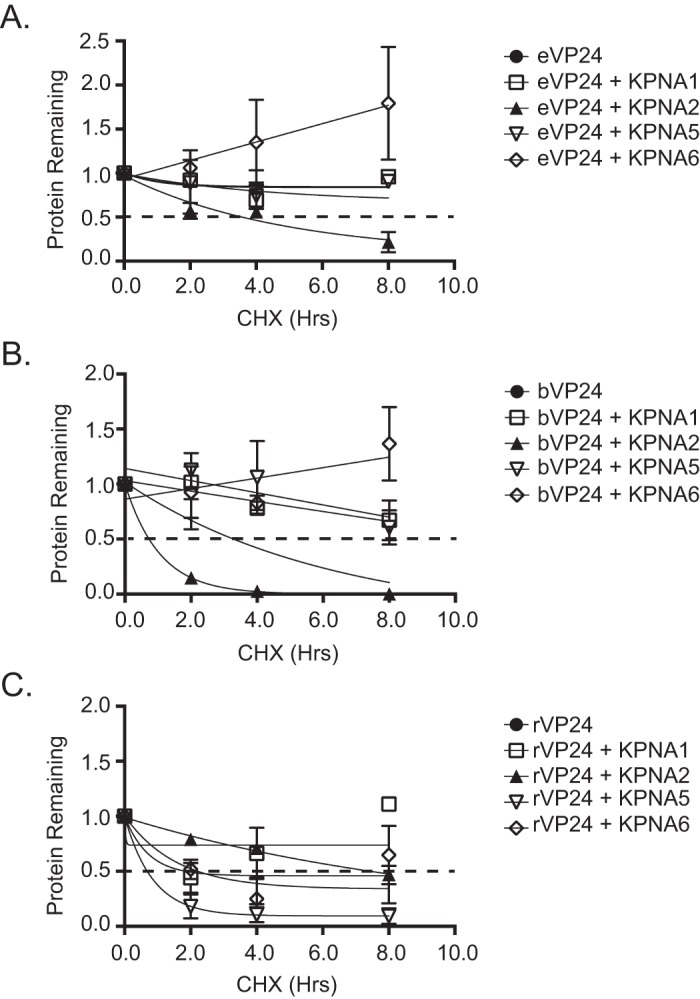
Densitometric analysis demonstrates that KPNA expression modulates eVP24 and bVP24 stability. VP24 abundance over time was measured relative to time zero, and half-live values were calculated by measuring the band intensity using Bio-Rad Image Lab software. VP24 band intensity over time was normalized to β-tubulin band intensity. The dotted black line indicates the 50% point. Data points represent two biological replicates. Protein half-life was calculated by the best-fit curve to the data points. Half-life decay curves for bVP24 (A), eVP24 (B), and rVP24 (C) are shown.
To address the hypothesis that the lower KPNA binding affinity might account for the instability of VP24 in the absence of overexpressed KPNA and for its enhanced stability upon KPNA expression, we tested the half-live values of previously described eVP24 mutants with decreased KPNA binding affinities (21). eVP24 cluster 1A and 1C mutants were previously shown by coimmunoprecipitation to be substantially impaired for interaction with KPNA5 (21), while eVP24 cluster 1D mutant weakly interacted with KPNA5 (Fig. 5A). As shown by BLI, weak binding between the three eVP24 cluster mutants and KPNA1, KPNA5, and KPNA6 occurred at the highest concentration (2 μM) of eVP24 tested; however, a steady-state dissociation constant could not be determined, indicating dissociation constants for the mutant eVP24s much larger than those for the wild-type (WT) eVP24 (Fig. S2). The eVP24 mutants had a shorter half-life than WT eVP24, with the eVP24 cluster 1A mutant exhibiting the most rapid loss of signal (Fig. 5B). Coexpressed with KPNA5, the half-life of eVP24 cluster 1A mutant was substantially extended, whereas expression of KPNA2, which does not interact, failed to enhance eVP24 cluster 1A mutant stability (Fig. 5C). A separate experiment, performed to quantify the levels of the different VP24s, substantiated these conclusions (Fig. 5D). These data suggest that binding to KPNA protects eVP24 from degradation and that weaker binding of eVP24 to KPNA can be compensated for with increased expression of KPNA.
FIG 5.
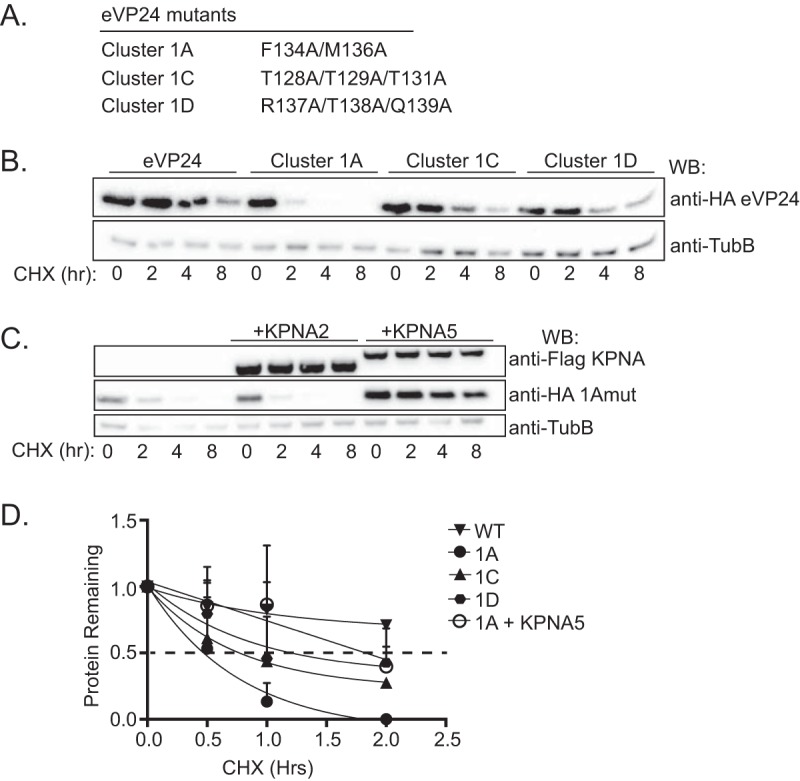
eVP24 residues that are critical for KPNA binding also influence eVP24 protein stability. (A) Previously described eVP24 constructs possessing mutations that impair binding to KPNA. (B) Abundance of WT HA-eVP24 (eVP24) or the indicated HA-eVP24 mutants in transfected cells treated with cycloheximide (CHX). Western blotting to detect Flag-tagged KPNA, HA-tagged VP24, or β-tubulin (TubB) was performed at the indicated times post-CHX addition. (C) Abundance of HA-eVP24 1A mutant in the presence or absence of Flag-tagged KPNA2 or Flag-tagged KPNA5. (D) Densitometric analysis of WT HA-tagged eVP24 or the indicated HA-tagged eVP24 mutants over an 8-h time frame. Data points represent two biological replicates, and the dotted black line indicates the point at which 50% of the protein remains.
To further correlate KPNA binding with VP24 half-life, the stabilities of eVP24 and bVP24 were compared in the absence or presence of WT or mutant KPNA5s that were previously described as being diminished for eVP24 interaction. The KPNA mutants tested included F484A, Y477G, or the KPNA5 ARM10 mutant containing four amino acid changes (Fig. 6A) (21). As described above, eVP24 was once again more stable than bVP24 (Fig. 6B). The levels of expression of the KPNA5 mutants were similar over the 8-h CHX treatment (Fig. 6C). In contrast to the outcome seen when WT KPNA5 was expressed, the interaction-defective KPNAs did not enhance bVP24 stability (Fig. 6D). For eVP24, which is already relatively stable, expression of WT KPNA5 once again resulted in modestly enhanced stability whereas, particularly for the ARM10 KPNA5 mutant, eVP24 stability was clearly lower (Fig. 6E).
FIG 6.
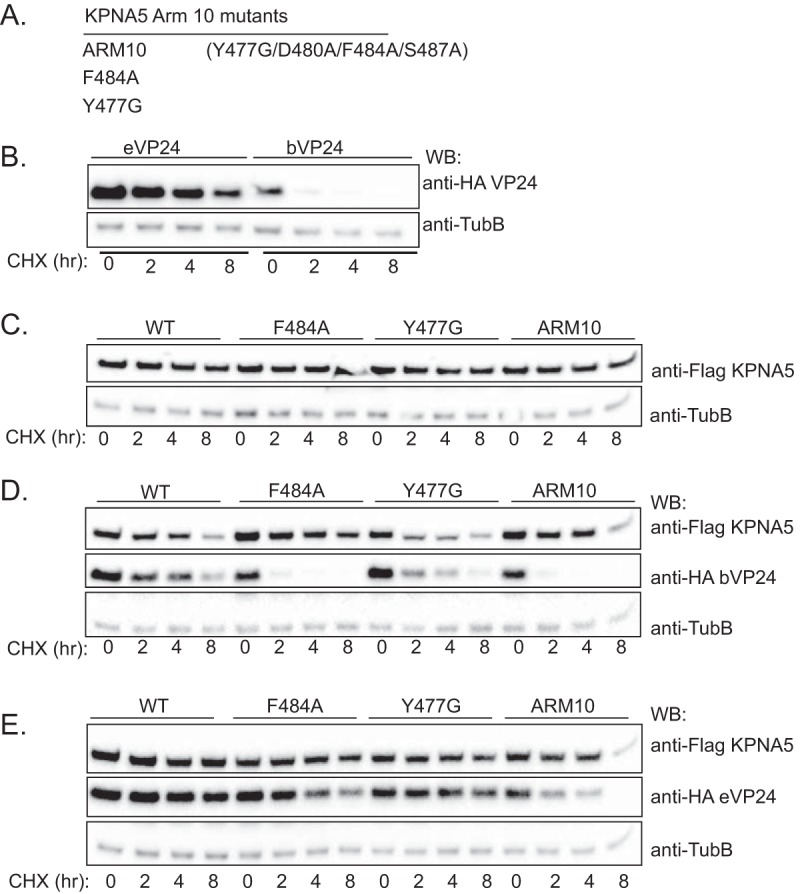
Stabilization of VP24 requires interaction with KPNA. (A) Previously described KPNA5 constructs possessing mutations in ARM domain 10 that impair interaction with eVP24. (B) Protein stability of HA-eVP24 or HA-bVP24 transfected alone. Western blotting to detect Flag-tagged KPNA, HA-tagged VP24, or β-tubulin (TubB) was performed at the indicated times post-CHX addition. (C) Abundance of the indicated Flag-KPNA5 mutants after CHX addition when transfected alone. (D) Stabilities of HA-bVP24 coexpressed with either WT Flag-KPNA5 or the indicated mutants. (E) Stabilities of HA-eVP24 coexpressed with either WT Flag-KPNA5 or the indicated mutants. For panels C to E, labeling is as described for panel B.
To address the impact of endogenous KPNA expression on VP24 stability, we measured VP24 half-life in cells that were transfected with small interfering RNAs (siRNAs) targeting the NPI-1 KPNA subfamily. siRNAs specific for KPNA1, KPNA5, and KPNA6 (si3xRNA) were simultaneously transfected into cells followed by the sequential transfection of VP24 24 h later. As a negative control, cells were transfected with a nonspecific siRNA (siScramble). Only reduction of KPNA5 and KPNA6 was achieved in the cells transfected with siRNAs targeting all three NPI-1 subfamily KPNAs (Fig. 7A). Despite the expression of endogenous KPNA1 in these cells, the eVP24 and bVP24 half-live values were reduced in comparison to those seen with the cells transfected with siScramble (Fig. 7B and C). rVP24 protein stability was also reduced modestly (Fig. 7D). Together, these data suggest that endogenous KPNA levels modulate VP24 stability.
FIG 7.
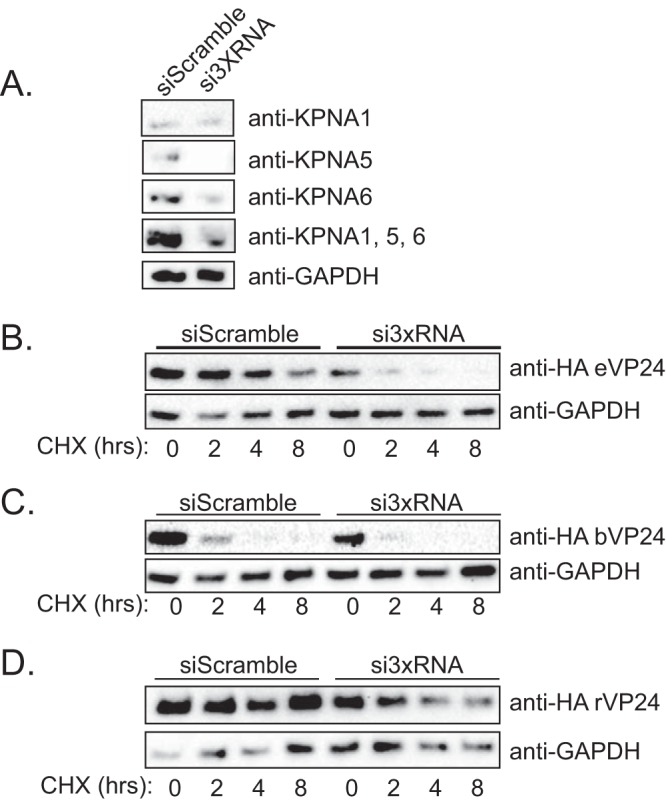
VP24 half-life is altered in cells transfected with siRNA targeting the NPI-1 KPNA subfamily. (A) Following transfection of a scrambled siRNA or pooled siRNAs to knock down KPNA1, KPNA5, and KPNA6 (si3XRNA), Western blotting was performed to detect KPNA1, KPNA5, or KPNA6 or all three simultaneously (KPNA1, KPNA5, and KPNA6). β-Tubulin Western blots served as a loading control. (B to D) Protein half-life decay curves for bVP24 (B), eVP24 (C), and rVP24 (D). VP24 abundance was measured over time in cells transfected with either a scrambled control siRNA or siRNAs targeting the three members of the NPI-1 subfamily of KPNAs (si3xRNA). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Molecular basis for differential KPNA binding by eVP24 and bVP24.
We next sought to clarify the basis for the differential levels of KPNA binding by bVP24 and eVP24. Although bVP24 and eVP24 are highly conserved (86% identity at the amino acid level), analysis of the eVP24/KPNA5 structure indicated that several nonconserved residues are located at the interface (Fig. 8A to C). To identify nonconserved residues located in hot spots for association between eVP24 and KPNA5, the cocrystal structure of eVP24 and KPNA5 C terminus (PDB 4U2X) was analyzed by HyPare (http://bip.weizmann.ac.il/HyPare), an algorithm that calculates the impact of mutations on the affinity between two interacting proteins (23). This process identified eVP24 residues N135, R140, and V141 (bVP24 residues Q135, H140, and A141) (Fig. 8D).
FIG 8.

Comparison of the eVP24 and bVP24 KPNA5 binding interfaces. (A) Structure of the eVP24 (blue and purple) and KPNA5C (orange) (PDB 4U2X) complex with KPNA5C aligned with KPNA1 (light orange) (PDB 3TJ3). Residues conserved between eVP24 and bVP24 are indicated in blue, while nonconserved residues are highlighted in purple. N and C indicate the N terminus and C terminus, respectively. (B) Closeup view (corresponding to the dotted box in panel A) of the interface between eVP24 and KPNA5. Residues P83, N135, R140, and V141 are indicated in purple with side chains. (C) Sequence comparison of eVP24, bVP24, and rVP24 proteins. Alignment of VP24 protein sequences was performed using the Cobalt constraint-based multiple protein alignment tool. Zaire ebolavirus VP24 (eVP24) (NCBI accession number AF086833.2), Reston ebolavirus VP24 (rVP24) (NCBI accession number AF522874.1), and Bundibugyo ebolavirus VP24 (bVP24) (NCBI accession number NC_014373.1) were used in the alignment. Conserved residues are indicated in red, and those not conserved are in blue. The eVP24-KPNA5 binding interfaces identified by Xu et al. (21), comprising cluster 1, cluster 2, and cluster 3, are highlighted in yellow. (D) HyPare analysis of the interaction between eVP24 and KPNA5. Changes in relative levels of binding affinity between eVP24 and KPNA5 are indicated for the predicted outcome of mutating eVP24 residues 16 to 231 to basic residues (lysine [Lys] or arginine [Arg]) (purple line), acidic residues (aspartate [Asp] or glutamate [Glu]) (green line), or alanine (Ala) residues (gray line). The dashed lines indicate the location of residues N135, R140, and V141. Residue P83 was not present in the HyPare analysis. A representation of an alignment of eVP24 and bVP24 residues 1 to 251 is shown at the bottom of the graph, with red indicating conserved residues and blue indicating nonconserved residues.
To examine the contribution of these residues to the differential levels of affinity of eVP24 and bVP24 for KPNAs, we mutated eVP24 residues at positions 135, 140, and 141 to the analogous residues found in bVP24. We also performed the reciprocal mutagenesis, making bVP24 more eVP24-like. An additional nonconserved residue at position 83 was also included in this analysis. We therefore generated mutants eVP24 4x (eVP24 P83S/N135Q/R140H/V141A) and bVP24 4x (bVP24 S83P/Q135N/H140R/A141V) (Table 2). The affinities of these two mutant proteins for the three KPNAs, as determined by BLI, were similar to those of the WT VP24 proteins (Fig. S3). eVP24 4x bound KPNA1, KPNA5, and KPNA6 with dissociation constants of 22.0 ± 5.2 nM, 14.0 ± 2.1 nM, and 7.63 ± 1.2 nM, respectively, while bVP24 4x bound KPNA1 with a KD of 103 ± 14 nM, KPNA5 with a KD of 57.0 ± 10 nM, and KPNA6 with a KD of 85.0 ± 14 nM, representing a less than 1-fold decrease in affinity in all cases (Tables 1 and 2). This suggests that, although three of these residues were identified by HyPare analysis to be important for the interaction between eVP24 and KPNA5, they do not determine the differing affinities of eVP24 and bVP24 for the KPNAs. To verify that these residues were important for the interaction of VP24 with KPNA, we mutated positions 83, 135, and 140 to alanine and position 141 to glycine, generating eVP24 4xA (eVP24 P83A/N135A/R140A/V141G) and bVP24 4xA (bVP24 S83A/Q135A/H140A/A141G). The affinities of these mutants for KPNA1, KPNA5, and KPNA6 were tested using BLI (Fig. S3). A significant decrease in affinity for all three KPNA proteins in comparison to the WT level was detected, with eVP24 4xA binding the three KPNAs with 8-fold to 16-fold-lower affinity than WT eVP24 and bVP24 4xA binding the KPNAs with 7-fold-lower affinity than WT bVP24. This indicates that one or more of these residues are important for the interaction of VP24 and KPNA (Table 2). Together, these data suggest that while the interaction between VP24 proteins and KPNA is evolutionarily conserved across members of the Ebolavirus genus, there is a level of plasticity and redundancy allowed in the amino acids present in these positions.
TABLE 2.
Affinities of mutant eVP24 and bVP24 proteins for members of the NPI-1 subfamily of KPNAs
| Protein |
KD (nM)a |
||
|---|---|---|---|
| KPNA1 | KPNA5 | KPNA6 | |
| eVP24 4x | 22.0 ± 5.2 | 14.0 ± 2.1 | 7.63 ± 1.3 |
| eVP24 4xA | 150 ± 21 | 96.7 ± 7.6 | 92.0 ± 13 |
| bVP24 4x | 103 ± 14 | 57.0 ± 10 | 85.0 ± 14 |
| bVP24 4xA | 560 ± 25 | 376 ± 36 | 420 ± 19 |
| bVP24 Q135N | 60.3 ± 2.0 | 41.1 ± 4.0 | 42.0 ± 3.3 |
| bVP24 Q135A | 98.3 ± 3.4 | 64.0 ± 6.5 | 99.0 ± 4.8 |
Data represent the means and standard deviations of results from three experiments.
Next, we tested the effect of mutating residues at positions 83, 135, 140, and 141 on eVP24 and bVP24 IFN inhibition. The mutants included eVP24 4x, eVP24 4xA, eVP24 N135Q, bVP24 H140R/A141V, bVP24 Q135N, and bVP24 R184Q (Fig. 9A and B). The bVP24 R184Q mutant was included in these studies because this residue is located in the eVP24 cluster 2 KPNA binding interface and is not conserved between eVP24 and bVP24 (Fig. 8C). Cells were transfected with an ISG54 reporter plasmid and empty vector or HA-tagged VP24 proteins as indicated and treated with IFN-β, after which reporter activity was assessed. While eVP24 4x inhibited ISG54 reporter activity to a level similar to that seen with WT eVP24 at the highest concentration, this inhibition decreased more rapidly as VP24 levels were reduced (Fig. 9C). Mutation of the four positions in eVP24 4xA resulted in a loss of inhibition compared to WT eVP24, even at the highest concentration (Fig. 9C). bVP24 mutant H140R/A141V behaved like WT bVP24 (Fig. 9D), consistent with the minimal impact of these residues on binding affinities when combined in the bVP24 4xA mutant. Likewise, bVP24 R184Q inhibited ISG54 activity similarly to WT bVP24 (Fig. 9D). The single point mutation Q135N resulted in a modest gain of inhibitory activity for bVP24, despite the lack of increased binding affinity for the KPNAs or of an obvious change in expression levels (Fig. 9D and Table 2). Conversely, the reciprocal N135Q mutation did not decrease eVP24 inhibitory activity (Fig. 9C). This suggests that in vitro binding data may not fully account for the IFN inhibitory activities of VP24s.
FIG 9.

Nonconserved interface residues dictate the potency of IFN antagonism by eVP24 and bVP24. (A and B) eVP24 (A) and bVP24 (B) mutants used for the experiments represented in this figure. (C) Suppression of IFN-induced ISG54 promoter activity upon expression of bVP24 or eVP24 or the indicated eVP24 mutants with residues changed to correspond to bVP24 residue identities. (D) Suppression of IFN-induced ISG54 promoter activity upon expression of eVP24 or bVP24 or the indicated bVP24 mutants with residues changed to correspond to eVP24. Transfections and reporter gene assays were performed as described for Fig. 2. Columns are representative of the means and error bars represent the standard deviations of results from two biological replicates each with three technical replicates performed for each transfection condition. Statistical significance was assessed by Student's t test, comparing the chimeric VP24 change in reporter activity to that obtained with the corresponding wild-type VP24 plasmid concentration (**, P < 0.1; ***, P < 0.01).
DISCUSSION
In this study, we identified previously unrecognized differences in how VP24 proteins from three ebolavirus species interact with KPNAs. The differences influence IFN inhibitory activity and VP24 stability, affecting the weaker binding bVP24 most dramatically. eVP24 was originally established as a viral immune antagonist that blocks IFN signaling by inhibiting the nuclear import of PY-STAT1 (18, 19). The discovery of this anti-IFN signaling function correlated with eVP24 binding to the KPNA1 nuclear import factor. KPNA1 had been described as the mediator of PY-STAT1 nuclear import, with this import being mediated via a nonclassical NLS. PY-STAT1 nuclear import is critical to allow PY-STAT1 to stimulate the transcription of interferon-stimulated genes (ISGs) and create an antiviral state (24, 25). In the presence of excess eVP24, the interaction between KPNA1 and PY-STAT1 was found to be disrupted, thereby preventing ISG expression (18, 19). Subsequent studies demonstrated that eVP24 and PY-STAT1 can interact with all three members of the NPI-1 subfamily of KPNAs: KPNA1, KPNA5, and KPNA6 (19, 26, 27).
KPNAs possess an N-terminal importin-beta binding domain, 10 armadillo (ARM) repeats, and a C-terminal CAS binding site. ARM2 to ARM4 and ARM6 to ARM8 are responsible for binding to classical mono- and bipartite basic NLSs, whereas dimerized PY-STAT1 interacts more C-terminally within a region encompassing ARM8 to ARM9 (25). Mapping studies of KPNA1 indicated that eVP24 binds at a site that partially overlaps the PY-STAT1 ncNLS binding site (19). Consistent with this observation, an X-ray cocrystal structure demonstrated the interaction of eVP24 with a large binding interface encompassing ARM8 to ARM10 in the C terminus of KPNA5 (21). Mutagenesis studies, based on the eVP24-KPNA5 structural data, demonstrated that loss of this interaction results in loss of inhibition of IFN-induced PY-STAT1 nuclear import and loss of inhibition of IFN-induced ISG expression. These data, along with data from coIP and in vitro binding studies, all point to a competition binding model of eVP24 antagonism of IFN signaling (21). Given this model, it was of interest to determine whether affinities of a given VP24 vary between the three different NPI-1 KPNAs. It was also of interest to determine whether VP24s from different ebolavirus species exhibit various degrees of interaction with KPNAs, as this might result in altered degrees of IFN antagonism.
Previous studies established that VP24s from a mouse-adapted EBOV and from REBOV also interacted with members of the NPI-1 subfamily of KPNAs from human and mouse (19). The mouse-adapted eVP24 amino acid sequence differs by only one amino acid from the parental version of eVP24, whereas rVP24 shares 82% amino acid identity with eVP24 (data not shown). The conservation of KPNA binding activity, despite sequence divergence, suggests a biologically important role for the VP24-KPNA interaction. The coIP assays used to demonstrate these interactions were not quantitative, however, prompting us here to compare KPNA binding affinities using purified, recombinant versions of eVP24, rVP24, and bVP24. bVP24 was chosen because its sequence is also divergent from that of eVP24, exhibiting 86% amino acid identity to eVP24 (data not shown), and because BDBV exhibits attenuation in human outbreaks and in nonhuman primate models relative to EBOV (3, 4). Our results demonstrate that the affinities for KPNA1, KPNA5, and KPNA6 are very similar for a given VP24 but that, while eVP24 and rVP24 exhibit similar KPNA binding affinities, bVP24 binding represents lower affinity. Comparable affinities of the VP24 proteins across the NPI-1 KPNAs would allow the virus to block IFN signaling at equal levels regardless of the ratio of NPI-1 family members present in the cell. However, the impaired binding of bVP24 for each KPNA suggested possible functional differences.
Evaluation of the sequences of eVP24 and bVP24 (Fig. 8) identified several differences at the primary sequence level that may impact interactions between KPNAs and VP24 proteins. Our studies show that mutating eVP24 residues to those found in bVP24 results in limited perturbations to binding affinity or IFN antagonism. Importantly, mutation of these same residues to alanine in either eVP24 or bVP24 results in an approximately 10-fold decrease in affinity. Our data are consistent with the following two conclusions. First, the interface between eVP24 and KPNA is highly redundant and is therefore able to accept multiple mutations without significant loss of binding affinity. Second, although the binding interface is redundant and may accommodate multiple interactions, we observed side chain preferences, as the alanine mutations in either eVP24 or bVP24 appear to lose about 10-fold to 15-fold affinity for KPNA.
The different KPNA binding affinities are of functional significance. rVP24 shows modestly higher binding affinity than does eVP24 for each of the KPNAs (Table 1). This correlates with very similar capacities of eVP24 and rVP24 for suppression of IFN-induced gene expression in titration studies. In contrast, the significantly lower bVP24 binding affinity with KPNA correlates with slightly impaired suppression of IFN responses at lower concentrations, although binding is sufficient to inhibit IFN-induced gene expression at higher concentrations. VP24 proteins also play other roles over the course of the viral replication cycle. VP24, in cooperation with NP and VP35, promotes the formation of nucleocapsid structures and is necessary for newly formed virus particles to be infectious; VP24 also modulates viral RNA synthesis and serves as a host range determinant, although the impact of VP24 on viral RNA synthesis in the context of EBOV infection has been questioned (8, 10, 12–15). Therefore, functional effects of the KPNA interaction on VP24 that extend beyond suppression of IFN responses are of interest. Here, we demonstrate that KPNA has a stabilizing effect on VP24 that may have implications for multiple VP24 functions. The impact of KPNA on stability is most apparent with lower-binding-affinity VP24s. Expressed alone, bVP24 displays a short protein half-life relative to eVP24 and rVP24, as demonstrated by pulse-chase experiments. VP24 half-life is inversely correlated to KPNA binding affinity, as eVP24 proteins with mutations in the KPNA binding interface were also demonstrated to have higher protein turnover than eVP24 or rVP24. Interestingly, when coexpressed with any of the mutant or wild-type eVP24 or bVP24 proteins, only the members of the NPI-1 subfamily of KPNAs had a stabilizing effect on VP24. Conversely, rVP24 appeared to decrease in protein concentration when coexpressed with KPNA5, suggesting that this stabilizing effect could be specific to certain members of the Ebolavirus genus and that KPNA5 may have a unique impact on rVP24. Future studies will need to be performed to determine how KPNA might influence other VP24 functions in the context of virus replication.
An important issue raised by these observations is whether the differences detected impact virulence. While the determinants of filovirus pathogenesis are incompletely defined, robust viral replication and modulation of the innate immune responses are important (14, 28, 29). For example, VP35 mutations that abrogate its inhibition of IFN production result in attenuation of the virus in vivo and in vitro (28, 30–32). In addition, adaptation of EBOV and Marburg virus (MARV) to animal models involves mutations within their respective IFN signaling antagonists, VP24 and VP40, to enhance virulence (15, 16, 19, 29, 33). High levels of viral replication would also require robust packaging of the viral genomes to make infectious particles. The substantial similarities between eVP24 and rVP24 in terms of NPI-1 binding affinities and capacity to suppress IFN signaling would argue against the VP24 IFN-antagonist function playing a significant role in virulence differences. The differences between bVP24 and either eVP24 or rVP24 are more intriguing. The lower IFN-antagonist function detected in our transfection studies, which were seen when smaller amounts of bVP24 plasmid were transfected, might result in increased sensitivity to IFNs early in infection, before substantial bVP24 levels have accumulated. If bVP24 is also less stable in infected cells, then this might impact multiple viral functions. It will therefore be important to examine the role of bVP24 during the course of BDBV infection in vivo.
Although filoviruses are primarily of concern as causes of severe human disease, their evolution as zoonotic pathogens has presumably occurred in nonhuman species. As bats have been implicated as the host reservoir for filoviruses, the interaction between KPNA and VP24 most likely evolved in these mammals (34, 35). While the Egyptian fruit bat (Rousettus aegyptiacus) is believed to act as a reservoir host for Marburg virus, the identities of reservoir hosts for different Ebolavirus species have not been definitively identified (36). It is likely that the reservoir hosts differ for EBOV, RESTV, and BDBV, which would explain differential interactions with human KPNAs. Taking KPNA5 as an example, the KPNAs are well conserved between humans and bats. However, there are differences, with KPNAs from different bats exhibiting different degrees of amino acid identity. Bats can be divided into two large groups, the microbats and megabats. Human KPNA5 shares 95.9% amino acid identity with KPNA5 from a microbat (Myotis lucifigus) and 98.1% with KPNA5 from a megabat (Pteropus vampyrus) (data not shown). It will be of interest, as reservoir hosts are better defined, to compare VP24-KPNA interactions across different reservoir and nonreservoir mammalian species and determine how these differences in host proteins might influence virulence.
The lack of effective antivirals limits the clinical options available to treat filovirus infections of humans. Because VP24 has roles in innate immune evasion and virus assembly, it is a potential therapeutic target. To date, specific information has been available regarding the structure of eVP24, rVP24, Sudan virus (SUDV) VP24, and eVP24 in complex with KPNA5 (21, 37, 38). As it is a nonenzymatic protein, therapeutic targeting may require destabilization of VP24 or disruption of functionally important interactions with host or viral factors. In support of the idea of VP24 as a therapeutic target, phosphorodiamidate morpholino oligomers that target VP24 can protect experimentally infected nonhistone proteins (NHPs) from lethal EBOV challenge (39). Although the eVP24-KPNA5 interface poses challenges due to the fact that it has a large surface area (21), the data in this study highlight potential synergistic effects of disruption of the interaction. Specifically, one could abrogate the viral block to the antiviral effects of IFNs while also decreasing VP24 stability. The reduced levels of VP24 would therefore impact assembly of virus particles and potentially decrease the infectivity of the virus particles that are produced. For these reasons, efforts to target this interaction are warranted.
MATERIALS AND METHODS
Plasmids.
The pCAGGS base plasmid was used for expression of viral proteins in mammalian cells. Point mutations were generated using the overlap PCR method and were verified by sequencing. Sequences were based on the following: eVP24 (NCBI accession number AGB56798.1); rVP24 (NCBI accession number AY769362); bVP24 (NCBI accession number NC_014373 [but codon optimized for mammalian expression]); KPNA1 (NCBI accession number NM_002264.3); KPNA5 (NCBI accession number NP_002260.2); and KPNA6 (NCBI accession number NM_012316.4). For recombinant protein expression in Escherichia coli, eVP24 11-237, bVP24 11-237, rVP24 11-237, and mutant VP24 constructs were subcloned into a modified pET15b vector (Novagen, EMD Millipore, Billerica, MA), while KPNA1 61-512, KPNA5 66-513, and KPNA6 66-510 were subcloned into a modified pET15b vector (Novagen, EMD Millipore, Billerica, MA) containing an AviTag, which contains the target amino acid sequence for biotinylation (Avidity LLC, Aurora, CO).
Recombinant protein expression, purification, and biotinylation.
Proteins were overexpressed as maltose binding protein (MBP) fusion proteins in BL21(DE3) E. coli cells (Novagen, EMD Millipore, Billerica, MA) in LB medium. Cells were harvested and resuspended in buffer containing 25 mM sodium phosphate (pH 7.5), 250 mM NaCl, 20 mM imidazole, and 5 mM 2-mercaptoethanol and were lysed using an EmulsiFlex-C5 homogenizer (Avestin, Ottawa, Canada). Lysates were clarified by centrifugation at 42,000 × g at 10°C for 40 min. Proteins were purified using a series of chromatographic columns, and sample purity was determined by SDS-PAGE.
MBP-Avi-KPNA1, MBP-Avi-KPNA5, and MBP-Avi-KPNA6 were biotinylated using BirA ligase (Avidity LLC, Aurora, CO) and the following reaction conditions: 40 μM MBP-Avi-KPNA1 (or MBP-Avi-KPNA5 or MBP-Avi-KPNA6), 50 mM bicine buffer (pH 8.3), 10 mM ATP, 10 mM magnesium acetate, 50 μM d-biotin, and 1 mg/ml BirA ligase (Avidity LLC, Aurora, CO). The reaction was allowed to proceed for 1 h at room temperature, after which the biotinylated proteins were injected onto a Superdex 200 gel filtration column (GE Healthcare Life Sciences, Pittsburgh, PA). Sample purity was determined by SDS-PAGE.
Biolayer interferometry (BLI) binding studies.
BLI experiments were performed using an Octet RED system (Pall ForteBio LLC, Menlo Park, CA) and buffer containing 10 mM HEPES (pH 7.5), 300 mM NaCl, 2 mM TCEP [tris(2-carboxyethyl)phosphine], 0.5% bovine serum albumin (BSA), and 0.02% Tween 20. Biotinylated KPNA proteins were immobilized on streptavidin sensors for 400 s. Association and dissociation of VP24 proteins were analyzed over a range of concentrations (2-fold serial dilutions) over the course of the experiment. In brief, for each concentration of VP24, protein association was measured for 300 s, followed by dissociation for 600 s in buffer. Steady-state analysis was performed using Octet software (version 8.2.0.7), and the resulting data represent the mean steady-state dissociation constants (KD) and standard deviations across three experiments.
Coimmunoprecipitation assays.
Human embryonic kidney 293T (HEK293T) cells were transfected with the indicated plasmids by using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA), and at 24 h posttransfection, cells were lysed in NP-40 lysis buffer (50 mM Tris [pH 7.5], 280 mM NaCl, 0.5% Nonidet P-40, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, protease inhibitor [cOmplete; Roche, Indianapolis, IN]). Anti-Flag M2 magnetic beads were incubated with lysates for 1 h at 4°C, washed five times in NP-40 lysis buffer, and eluted using 3× Flag peptide (Sigma-Aldrich, St. Louis, MO) at 4°C for 30 min.
Reporter gene assay.
HEK293T cells were transfected with an ISG54 firefly luciferase reporter plasmid, a constitutively active Renilla luciferase reporter plasmid (pRL-tk) (Promega, Madison, WI), and the indicated expression plasmids. At 24 h posttransfection, the cells were treated with 1,000 U/ml human IFN-β (PBL Assay Science, Piscataway, NJ)–Dulbecco's modified Eagle's medium (DMEM)–10% fetal bovine serum. At 24 h posttreatment, cells were lysed and a dual luciferase reporter assay (Promega, Madison, WI) was performed. Firefly luciferase values were normalized to Renilla luciferase values. Statistical significance was assessed using Student's t test.
VP24 half-life analysis.
HEK293T cells were transfected with the indicated plasmids, and at 24 h posttransfection, cells were treated with 100 μg/ml of the translational inhibitor cycloheximide (CHX) for the indicated time periods. Starting at time zero post-CHX treatment, cells were lysed in NP-40 lysis buffer and proteins were resolved by SDS-PAGE. VP24 half-live values were calculated by measuring band intensity using Bio-Rad Image Lab software. Briefly, band intensities for VP24 and beta-tubulin from each time point were measured and compared to those measured at time zero post-cycloheximide treatment. VP24 band intensity was normalized to the beta-tubulin band intensity from each respective time point.
HEK293T cells were transfected with siRNA directed against members of the NPI-1 subfamily of KPNA (si3xKPNA). A 30 nM concentration of ON-TARGETplus SMARTpool (Dharmacon) siRNA directed against KPNA1, KPNA5, and KPNA6 was transfected into 293T cells using RNAiMAX (Qiagen). At 24 h post-siRNA transfection, cells were trypsinized, resuspended, and transfected with the indicated VP24 plasmids. At 24 h post-plasmid transfection, cells were treated with CHX and analyzed as described above.
Antibodies.
Monoclonal mouse anti-Flag M2 antibody, polyclonal rabbit anti-Flag antibody, monoclonal mouse anti-HA antibody, and polyclonal rabbit anti-HA antibody were all purchased from Sigma-Aldrich (St. Louis, MO). Monoclonal mouse anti-KPNA antibodies (against KPNA1, KPNA2, KPNA5, and KPNA6) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant AI109945 to C.F.B. and G.K.A., NIH grant AI120943 to D.W.L., G.K.A., and C.F.B., and NIH T32 Training Grant AI007647 to T.M.S.
We thank Wei Xu and Richard Yu for providing reagents and experimental assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01715-16.
REFERENCES
- 1.Afonso CL, Amarasinghe GK, Banyai K, Bao Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand FX, Briese T, Bukreyev A, Calisher CH, Chandran K, Cheng J, Clawson AN, Collins PL, Dietzgen RG, Dolnik O, Domier LL, Durrwald R, Dye JM, Easton AJ, Ebihara H, Farkas SL, Freitas-Astua J, Formenty P, Fouchier RA, Fu Y, Ghedin E, Goodin MM, Hewson R, Horie M, Hyndman TH, Jiang D, Kitajima EW, Kobinger GP, Kondo H, Kurath G, Lamb RA, Lenardon S, Leroy EM, Li CX, Lin XD, Liu L, Longdon B, Marton S, Maisner A, Muhlberger E, Netesov SV, Nowotny N, et al. 2016. Taxonomy of the order Mononegavirales: update 2016. Arch Virol 161:2351–2360. doi: 10.1007/s00705-016-2880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bausch DG, Rojek A. 10 June 2016. West Africa 2013: re-examining Ebola. Microbiol Spectr 4:(3). doi: 10.1128/microbiolspec.EI10-0022-2016. [DOI] [PubMed] [Google Scholar]
- 3.Towner JS, Sealy TK, Khristova ML, Albarino CG, Conlan S, Reeder SA, Quan PL, Lipkin WI, Downing R, Tappero JW, Okware S, Lutwama J, Bakamutumaho B, Kayiwa J, Comer JA, Rollin PE, Ksiazek TG, Nichol ST. 2008. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog 4:e1000212. doi: 10.1371/journal.ppat.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falzarano D, Feldmann F, Grolla A, Leung A, Ebihara H, Strong JE, Marzi A, Takada A, Jones S, Gren J, Geisbert J, Jones SM, Geisbert TW, Feldmann H. 2011. Single immunization with a monovalent vesicular stomatitis virus-based vaccine protects nonhuman primates against heterologous challenge with Bundibugyo ebolavirus. J Infect Dis 204(Suppl 3):S1082–S1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miranda ME, Ksiazek TG, Retuya TJ, Khan AS, Sanchez A, Fulhorst CF, Rollin PE, Calaor AB, Manalo DL, Roces MC, Dayrit MM, Peters CJ. 1999. Epidemiology of Ebola (subtype Reston) virus in the Philippines, 1996. J Infect Dis 179(Suppl 1):S115–S119. doi: 10.1086/514314. [DOI] [PubMed] [Google Scholar]
- 6.Messaoudi I, Amarasinghe GK, Basler CF. 2015. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 13:663–676. doi: 10.1038/nrmicro3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bamberg S, Kolesnikova L, Moller P, Klenk HD, Becker S. 2005. VP24 of Marburg virus influences formation of infectious particles. J Virol 79:13421–13433. doi: 10.1128/JVI.79.21.13421-13433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han Z, Boshra H, Sunyer JO, Zwiers SH, Paragas J, Harty RN. 2003. Biochemical and functional characterization of the Ebola virus VP24 protein: implications for a role in virus assembly and budding. J Virol 77:1793–1800. doi: 10.1128/JVI.77.3.1793-1800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoenen T, Groseth A, Kolesnikova L, Theriault S, Ebihara H, Hartlieb B, Bamberg S, Feldmann H, Stroher U, Becker S. 2006. Infection of naive target cells with virus-like particles: implications for the function of ebola virus VP24. J Virol 80:7260–7264. doi: 10.1128/JVI.00051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mateo M, Carbonnelle C, Martinez MJ, Reynard O, Page A, Volchkova VA, Volchkov VE. 2011. Knockdown of Ebola virus VP24 impairs viral nucleocapsid assembly and prevents virus replication. J Infect Dis 204(Suppl 3):S892–896. doi: 10.1093/infdis/jir311. [DOI] [PubMed] [Google Scholar]
- 11.Noda T, Halfmann P, Sagara H, Kawaoka Y. 2007. Regions in Ebola virus VP24 that are important for nucleocapsid formation. J Infect Dis 196(Suppl 2):S247–S250. doi: 10.1086/520596. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe S, Noda T, Halfmann P, Jasenosky L, Kawaoka Y. 2007. Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J Infect Dis 196(Suppl 2):S284–S290. doi: 10.1086/520582. [DOI] [PubMed] [Google Scholar]
- 13.Watt A, Moukambi F, Banadyga L, Groseth A, Callison J, Herwig A, Ebihara H, Feldmann H, Hoenen T. 2014. A novel life cycle modeling system for Ebola virus shows a genome length-dependent role of VP24 in virus infectivity. J Virol 88:10511–10524. doi: 10.1128/JVI.01272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebihara H, Takada A, Kobasa D, Jones S, Neumann G, Theriault S, Bray M, Feldmann H, Kawaoka Y. 2006. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog 2:e73. doi: 10.1371/journal.ppat.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volchkov VE, Chepurnov AA, Volchkova VA, Ternovoj VA, Klenk HD. 2000. Molecular characterization of guinea pig-adapted variants of Ebola virus. Virology 277:147–155. doi: 10.1006/viro.2000.0572. [DOI] [PubMed] [Google Scholar]
- 16.Mateo M, Carbonnelle C, Reynard O, Kolesnikova L, Nemirov K, Page A, Volchkova VA, Volchkov VE. 2011. VP24 is a molecular determinant of Ebola virus virulence in guinea pigs. J Infect Dis 204(Suppl 3):S1011–S1020. doi: 10.1093/infdis/jir338. [DOI] [PubMed] [Google Scholar]
- 17.Feagins AR, Basler CF. 2015. Lloviu virus VP24 and VP35 proteins function as innate immune antagonists in human and bat cells. Virology 485:145–152. doi: 10.1016/j.virol.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol 81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mateo M, Reid SP, Leung LW, Basler CF, Volchkov VE. 2010. Ebolavirus VP24 binding to karyopherins is required for inhibition of interferon signaling. J Virol 84:1169–1175. doi: 10.1128/JVI.01372-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, Pappu RV, Leung DW, Basler CF, Amarasinghe GK. 2014. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 16:187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valmas C, Grosch MN, Schumann M, Olejnik J, Martinez O, Best SM, Krahling V, Basler CF, Muhlberger E. 2010. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog 6:e1000721. doi: 10.1371/journal.ppat.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selzer T, Schreiber G. 1999. Predicting the rate enhancement of protein complex formation from the electrostatic energy of interaction. J Mol Biol 287:409–419. doi: 10.1006/jmbi.1999.2615. [DOI] [PubMed] [Google Scholar]
- 24.McBride KM, Reich NC. 2003. The ins and outs of STAT1 nuclear transport. Sci STKE 2003:RE13. [DOI] [PubMed] [Google Scholar]
- 25.Melen K, Fagerlund R, Franke J, Kohler M, Kinnunen L, Julkunen I. 2003. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J Biol Chem 278:28193–28200. doi: 10.1074/jbc.M303571200. [DOI] [PubMed] [Google Scholar]
- 26.Ma J, Cao X. 2006. Regulation of Stat3 nuclear import by importin alpha5 and importin alpha7 via two different functional sequence elements. Cell Signal 18:1117–1126. doi: 10.1016/j.cellsig.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 27.Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. 1997. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J 16:7067–7077. doi: 10.1093/emboj/16.23.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prins KC, Delpeut S, Leung DW, Reynard O, Volchkova VA, Reid SP, Ramanan P, Cardenas WB, Amarasinghe GK, Volchkov VE, Basler CF. 2010. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J Virol 84:3004–3015. doi: 10.1128/JVI.02459-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warfield KL, Bradfute SB, Wells J, Lofts L, Cooper MT, Alves DA, Reed DK, VanTongeren SA, Mech CA, Bavari S. 2009. Development and characterization of a mouse model for Marburg hemorrhagic fever. J Virol 83:6404–6415. doi: 10.1128/JVI.00126-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albariño CG, Wiggleton Guerrero L, Spengler JR, Uebelhoer LS, Chakrabarti AK, Nichol ST, Towner JS. 2015. Recombinant Marburg viruses containing mutations in the IID region of VP35 prevent inhibition of host immune responses. Virology 476:85–91. doi: 10.1016/j.virol.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol 83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, Otwinowski Z, Liu G, Huh J, Basler CF, Amarasinghe GK. 2012. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A 109:20661–20666. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cross RW, Fenton KA, Geisbert JB, Ebihara H, Mire CE, Geisbert TW. 2015. Comparison of the pathogenesis of the Angola and Ravn strains of Marburg virus in the outbred guinea pig model. J Infect Dis 212(Suppl 2):S258–S270. doi: 10.1093/infdis/jiv182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amman BR, Carroll SA, Reed ZD, Sealy TK, Balinandi S, Swanepoel R, Kemp A, Erickson BR, Comer JA, Campbell S, Cannon DL, Khristova ML, Atimnedi P, Paddock CD, Crockett RJ, Flietstra TD, Warfield KL, Unfer R, Katongole-Mbidde E, Downing R, Tappero JW, Zaki SR, Rollin PE, Ksiazek TG, Nichol ST, Towner JS. 2012. Seasonal pulses of Marburg virus circulation in juvenile Rousettus aegyptiacus bats coincide with periods of increased risk of human infection. PLoS Pathog 8:e1002877. doi: 10.1371/journal.ppat.1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swanepoel R, Smit SB, Rollin PE, Formenty P, Leman PA, Kemp A, Burt FJ, Grobbelaar AA, Croft J, Bausch DG, Zeller H, Leirs H, Braack LE, Libande ML, Zaki S, Nichol ST, Ksiazek TG, Paweska JT; International Scientific and Technical Committee for Marburg Hemorrhagic Fever Control in the Democratic Republic of Congo. 2007. Studies of reservoir hosts for Marburg virus. Emerg Infect Dis 13:1847–1851. doi: 10.3201/eid1312.071115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olival KJ, Hayman DT. 2014. Filoviruses in bats: current knowledge and future directions. Viruses 6:1759–1788. doi: 10.3390/v6041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang AP, Bornholdt ZA, Liu T, Abelson DM, Lee DE, Li S, Woods VL Jr, Saphire EO. 2012. The ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog 8:e1002550. doi: 10.1371/journal.ppat.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards MR, Johnson B, Mire CE, Xu W, Shabman RS, Speller LN, Leung DW, Geisbert TW, Amarasinghe GK, Basler CF. 2014. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep 6:1017–1025. doi: 10.1016/j.celrep.2014.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warren TK, Whitehouse CA, Wells J, Welch L, Heald AE, Charleston JS, Sazani P, Reid SP, Iversen PL, Bavari S. 2015. A single phosphorodiamidate morpholino oligomer targeting VP24 protects rhesus monkeys against lethal Ebola virus infection. mBio 6:e02344-14. doi: 10.1128/mBio.02344-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.