

ABSTRACT
Interleukin-1β (IL-1β), an inflammatory cytokine and IL-1 receptor ligand, has diverse activities in the brain. We examined whether IL-1 signaling contributes to the encephalitis observed in mouse adenovirus type 1 (MAV-1) infection, using mice lacking the IL-1 receptor (Il1r1−/− mice). Il1r1−/− mice demonstrated reduced survival, greater disruption of the blood-brain barrier (BBB), higher brain viral loads, and higher brain inflammatory cytokine and chemokine levels than control C57BL/6J mice. We also examined infections of mice defective in IL-1β production (Pycard−/− mice) and mice defective in trafficking of Toll-like receptors to the endosome (Unc93b1−/− mice). Pycard−/− and Unc93b1−/− mice showed lower survival (similar to Il1r1−/− mice) than control mice but, unlike Il1r1−/− mice, did not have increased brain viral loads or BBB disruption. Based on the brain cytokine levels, MAV-1-infected Unc93b1−/− mice had a very different inflammatory profile from infected Il1r1−/− and Pycard−/− mice. Histological examination demonstrated pathological findings consistent with encephalitis in control and knockout mice; however, intranuclear viral inclusions were seen only in Il1r1−/− mice. A time course of infection of control and Il1r1−/− mice evaluating the kinetics of viral replication and cytokine production revealed differences between the mouse strains primarily at 7 to 8 days after infection, when mice began succumbing to MAV-1 infection. In the absence of IL-1 signaling, we noted an increase in the transcription of type I interferon (IFN)-stimulated genes. Together, these results indicate that IL-1 signaling is important during MAV-1 infection and suggest that, in its absence, increased IFN-β signaling may result in increased neuroinflammation.
IMPORTANCE The investigation of encephalitis pathogenesis produced by different viruses is needed to characterize virus and host-specific factors that contribute to disease. MAV-1 produces viral encephalitis in its natural host, providing a good model for studying factors involved in encephalitis development. We investigated the role of IL-1 signaling during MAV-1-induced encephalitis. Unexpectedly, the lack of IL-1 signaling increased the mortality and inflammation in mice infected with MAV-1. Also, there was an increase in the transcription of type I IFN-stimulated genes that correlated with the observed increased mortality and inflammation. The findings highlight the complex nature of encephalitis and suggests that IL-1 has a protective effect for the development of MAV-1-induced encephalitis.
KEYWORDS: IL-1, adenovirus, blood-brain barrier, encephalitis, innate immunity
INTRODUCTION
Neuroinvasive viral infections are a significant cause of morbidity and mortality worldwide, and infections caused by emerging and reemerging viruses in the central nervous system (CNS) are becoming more frequent. Members of at least 11 virus families cause encephalitis, including DNA viruses, retroviruses, and RNA viruses (1–3). Encephalitis is characterized by the recruitment of inflammatory cells, the secretion of cytokines and chemokines, and alteration of localization and levels of tight junction proteins, leading to disruption of the blood-brain barrier (BBB) (1, 2). The BBB is composed of endothelial cells and basal lamina surrounded by pericytes and astrocytes that regulate the access of molecules and cells to the brain parenchyma, limiting the entry of inflammatory cells and invading microorganisms into the brain, thus maintaining CNS homeostasis (1, 3).
Mouse adenovirus type 1 (MAV-1; also known as MAdV-1) causes both acute and persistent infection in mice and leads to a dose-dependent encephalitis in susceptible mice (4–6). MAV-1 infects monocytes (including macrophages and microglia), astrocytes, and endothelial cells (4, 7; S. L. Ashley, C. D. Pretto, M. T. Stier, P. Kadiyala, L. Castro-Jorge, T.-H. Hsu, R. Doherty, K. E. Carnahan, M. G. Castro, P. R. Lowenstein, and K. R. Spindler, unpublished data). MAV-1 disrupts the BBB by altering expression of endothelial tight junction proteins, altering other critical components of the BBB, and disrupting BBB permeability (8). Increased proinflammatory cytokine production, BBB permeability, and leukocyte invasion are common events following brain injury (9), neurodegenerative diseases (10), and viral infections (1). One important player that can drive the neuroinflammatory process is interleukin-1β (IL-1β), a proinflammatory cytokine highly expressed within the CNS during neuroinflammatory diseases, including viral encephalitis (11, 12).
The expression and activation of IL-1β is controlled through the combination of two distinct signals. Signal 1 induces mRNA production in an NF-κB-dependent manner, resulting in increased IL-1β gene transcription and translation. Signal 2 results in IL-1β protein maturation, by cleavage of the IL-1β proform into mature IL-1β, generally by caspase-1 (13). Signal 1 is frequently induced by a Toll-like receptor (TLR) ligand, and signal 2 is mediated by inflammasome assembly (14). Canonical inflammasomes are protein complexes comprised of nucleotide oligomerization domain-like receptors (NLRs), adaptor molecules such as apoptosis-associated speck-like protein containing C-terminal caspase recruitment domain (ASC), and the effector, caspase-1 (15, 16).
IL-1β signals through the IL-1R1 receptor and the MyD88 pathway, leading to downstream NF-κB activation and expression of genes that regulate the immune response to infection (17, 18). IL-1R1 receptor signaling mediates inflammatory responses via two cytokine species, IL-1α and IL-1β (19). IL-1α and IL-1β generate a vast spectrum of biological responses in the central nervous, hematologic, and metabolic systems. IL-1β regulates cellular infiltration to sites of viral infection and, depending on the virus type infecting the CNS, it has been associated with either protection from or enhancement of disease (20–22). During herpes simplex virus 1 (HSV-1)-induced encephalitis, IL-1β works synergistically with tumor necrosis factor alpha (TNF-α) to protect against encephalitis, whereas during Sindbis virus encephalitis and human immunodeficiency virus (HIV) encephalitis, IL-1β increases pathogenesis (21, 23, 24). West Nile virus (WNV) infection is exacerbated in mice deficient in IL-1R1 due to increased neuroinflammation and an inability to fully activate CD4+ T and dendritic cells within the CNS (25). In contrast, during Theiler's murine encephalomyelitis virus infection, IL-1 elevates pathogenic responses, whereas the lack of IL-1 signaling leads to viral persistence due to insufficient T cell activation (26).
Internalized human adenovirus (HAdV) DNA induces maturation of pro-IL-1β in murine macrophages and differentiated human THP-1 and is dependent on the NACHT-, LRR-, and PYD-domain-containing protein 3 (NALP3) and ASC, components of the inflammasome (27). ASC protein is encoded by the Pycard gene and is composed of a pyrin domain (PYD) and a caspase-recruitment domain (CARD). ASC interacts with multiple pattern recognition receptors (PRRs), such as NACHT-, LRR-, and PYD domain-containing proteins (NLRPs), NLR caspase recruitment domain-containing protein, and absent in melanoma 2 (AIM2), to form caspase 1-activating platforms (inflammasomes) (16, 28). ASC inflammasomes play a central role in innate and adaptive immunity to influenza virus (29) and are essential for IL-1β production and development of effective host immunity to WNV (30). ASC-deficient mice exhibit enhanced WNV replication and reduced survival. The immune response to acute MAV-1 infection involves innate and adaptive immunity (31–35), but the role of inflammasomes in MAV-1 infection is unknown. HAdV activates TLR9, the pattern recognition receptor for nonmethylated CpG-rich DNA, in human peripheral blood mononuclear cells and plasmacytoid dendritic cells (pDCs) (36, 37). TLR9 is critical for HAdV-induced type I interferon (IFN) production in pDCs and induction of proinflammatory cytokines in primary murine macrophages (38). TLR9 and other endosome-associated TLRs (TLR3 and TLR7 and, in mice, also TLR11, TLR12, and TLR13) (39) traffic from the endoplasmic reticulum (ER) to the endolysosomes, where they respond to ligands. UNC93B is a transmembrane protein localized to the ER that is required for this trafficking (40, 41). Mice with a nonfunctional UNC93B fail to respond to TLR3, TLR7, or TLR9 ligands, and mice and humans deficient in UNC93B are highly susceptible to viral infection (42–44).
Although MAV-1 infection results in IL-1β production (31, 45, 46), the specific signaling pathways associated with IL-1 production have not been characterized. Therefore, the objective of this study was to examine the in vivo role of IL-1 signaling in the context of DNA virus-induced encephalitis, using MAV-1 as a model. To address this, we used mice with mutations that knock out Unc93b1 (involved in TLR3, TLR7, TLR9, TLR11, TLR12, and TLR13 trafficking and thus signaling), Pycard (involved in IL-1β production), or Il1r1 (involved in IL-1β signaling). These mice had increased mortality and increased neuroinflammation compared to infected control mice. Infected mice that lacked IL-1R1 had higher CNS viral loads and greater disruption of the BBB than control mice. They also had higher levels of inflammatory cytokines in the brain than control mice. IL-1 signaling was critical for the control of viral replication and immunopathology in the CNS of mice infected with MAV-1.
RESULTS
Mice deficient in IL-1 production or signaling or in endosomal TLR expression have reduced survival after sublethal MAV-1 challenge.
IL-1 promotes antiviral immunity outside the CNS and is associated with CNS injury during autoimmune diseases and viral infections (20, 26, 47). The role of IL-1 during MAV-1 viral encephalitis is unknown. To characterize the in vivo physiological relevance and the importance of IL-1 signaling in immune-mediated protection from intraperitoneal (i.p.) MAV-1 infection, we compared survival in wild-type (WT; C57BL/6J) mice to mice that are deficient in an inflammasome intermediate component and thus have a genetic deficiency in IL-1β production (Pycard−/− mice). We also tested mice that lack the IL-1R1 receptor and thus are deficient in IL-1 signaling (Il1r1−/− mice). To determine whether the endosomal TLRs (TLR-3, 7, 9) have a role in the production of “signal 1” for IL-1β production during MAV-1 infection, we also evaluated Unc93b1−/− mice. All knockout strains were highly susceptible to MAV-1 infection with 103 PFU of virus (Fig. 1). Although the infectious i.p. dose of 103 PFU resulted in 38% mortality in WT mice at 21 days postinfection (dpi), mortality was 72% in Pycard−/− mice, 80% in Unc93b1−/− mice, and 84% in Il1r1−/− mice (Fig. 1). Thus, an absence of IL-1R1 signaling resulted in increased mortality after i.p. inoculation.
FIG 1.

Pycard−/−, Unc93b1−/−, and Il1r1−/− mice are more susceptible to MAV-1 infection than were WT (C57BL/6J) mice. Survival analysis after i.p. inoculation with 103 PFU of MAV-1 was performed. WT (n = 47) mice are represented by closed squares and a solid line, Pycard−/− (n = 18) mice are represented by open circles and a dotted-dashed line, Unc93b1−/− (n = 31) mice are represented by open triangles and a dotted line, and Il1r1 −/− (n = 19) mice are represented by open squares and a dashed line. The data shown are pooled from independent experiments. Asterisks indicate a statistically significant difference of Pycard −/−, Unc93b1 −/−, and Il1r1 −/− mice compared to WT mice by the Mantel-Cox log-rank test (**, P = 0.0056; ***, P = 0.0001; ****, P < 0.0001).
IL-1 signaling modulates MAV-1 replication and BBB permeability.
To examine what factors contribute to higher mortality following MAV-1 infection of knockout mice compared to WT mice, we infected all mouse strains with 103 PFU of MAV-1 and measured viral loads in brains 7 dpi, when mice started showing signs of disease. DNA viral loads were quantitated by quantitative real-time PCR (qPCR) and were significantly higher in the brains of Il1r1−/− mice compared to WT mice (Fig. 2A). There was no significant difference between Pycard−/− and Unc93b1−/− mice compared to WT mice. Quantitation of viral loads by qPCR correlates well with quantitation of infectious virus by plaque assay (L. A. Castro-Jorge and K. R. Spindler, unpublished data).
FIG 2.

Sodium fluorescein uptake and viral load in Pycard−/−, Unc93b1−/−, and Il1r1−/− mice infected with MAV-1. (A) Viral loads after i.p. MAV-1 infection of WT (C57BL/6J), Pycard−/−, Unc93b1−/−, and Il1r1−/− mice were measured by qPCR in brain samples at 7 dpi. The limit of detection of the assay was 104 viral genome copies/100 ng of total DNA. (B) BBB permeability was assessed by sodium fluorescein uptake assay. The same mice from panel A were injected i.p. with sodium fluorescein 10 min prior to euthanasia at 7 dpi. Brain uptake of sodium fluorescein was normalized to sodium fluorescein in the serum, and the values were then normalized to mock-infected mice. WT (C57BL/6J) (n = 33) mice are represented by closed squares, Pycard−/− (n = 23) mice are represented by open circles, Unc93b1−/− (n = 14) mice are represented by open triangles, and Il1r1−/− (n = 20) mice are represented by open squares. The data shown are pooled from four independent experiments. Horizontal bars denote arithmetic means. Asterisks indicate a statistically significant difference of Il1r1−/− mice compared to WT mice by Mann-Whitney test (**, P < 0.005). (C) Correlation between viral load measured by qPCR with ng of sodium fluorescein per g of brain from the mice from panels A and B. Each sample point is an individual mouse brain. The linear regression equation and Spearman correlation value (rs) are shown in the graph. Correlation was also seen between viral load and sodium fluorescein for each mouse strain individually (data not shown).
Because Il1r1−/− mice had lower survival and higher viral loads in the brain compared than WT mice, we compared whether BBB permeability was altered in the Il1r1−/−, Pycard−/−, and Unc93b1−/− mice. Sodium fluorescein is a small molecule (376 Da) that is only able to access and stain brain tissue when the BBB is compromised. We administered sodium fluorescein i.p. at 7 dpi to WT, Pycard−/−, Unc93b1−/−, and Il1r1−/− mice, and we quantitated the sodium fluorescein in the right brain hemispheres. The amount of sodium fluorescein uptake in the brains of Il1r1−/− mice was significantly higher than in WT mice, but there was no statistical difference between Pycard−/− and Unc93b1−/− mice compared to WT mice (Fig. 2B). As seen previously (8), the sodium fluorescein levels in brains correlated with MAV-1 viral loads in all mouse strains (Fig. 2C). These results suggest that the lower survival and higher viral loads found in the brains of MAV-1-infected Il1r1−/− mice were due to a BBB dysfunction caused by defective IL-1 signaling.
Inflammatory cytokine levels are increased in the absence of IL-1 production and signaling.
Since proinflammatory cytokines and chemokines play a significant role in leukocyte trafficking into the CNS, we analyzed the protein levels of inflammatory cytokines and chemokines in the brains of MAV-1 infected Pycard−/−, Unc93b1−/−, and Il1r1−/− mice compared to WT controls from 6 to 8 dpi. Compared to WT mice, the levels of IFN-γ, IL-1β, and IL-10 were higher in Unc93b1−/− mice at 8 dpi (P < 0.05) (Fig. 3A, B, and C), whereas there was no difference in Pycard−/− and Il1r1−/− mice for these cytokines. The increase in the levels of inflammatory cytokines, such as IL-1β, was concomitant with an increase in anti-inflammatory cytokine, IL-10. The increase in IL-10 may counteract the proinflammatory effects of IL-1β. The IL-6 levels were higher in Unc93b1−/− and Il1r1−/− mice compared to WT mice at 8 dpi (Fig. 3D). There were no differences in levels of IL-1α or KC among any of the four strains of mice (data not shown). At 8 dpi, Pycard−/− and Il1r1−/− mice had higher levels of MCP-1 and RANTES compared to WT mice, whereas there was no difference in Unc93b1−/− mice (Fig. 3E and F). This suggests that during MAV-1-induced encephalitis, TLR3, TLR7, TLR9, TLR11, TLR12, and TLR13 are not the exclusive pattern recognition receptors activated by MAV-1, since Unc93b1−/− mice had higher levels of IL-1β.
FIG 3.

Cytokine production and MAV-1 replication in brains from WT (C57BL/6J), Pycard−/−, Unc93b1−/−, and Il1r1−/− mice at 6, 7, and 8 dpi. WT, Pycard−/−, Unc93b1−/−, and Il1r1−/− mice were infected with 103 PFU of MAV-1. At 6, 7, and 8 dpi, the brains were collected, and viral loads (G) and the concentrations of cytokines and chemokines (A to F) were determined. Cytokine and chemokine protein levels were assessed by ELISA in panels A to F. Mock data for all strains were similar and are shown as a single point (open circles). (G) Viral loads were assessed by qPCR for viral DNA, and the limit of detection of the assay (LOD) was 104 viral genome copies/100 ng of DNA. Brain viral loads were determined as in Fig. 2. WT (C57BL/6J) mice are represented by filled squares, Pycard−/− mice are represented by circles, Unc93b1−/− mice are represented by triangles, and Il1r1−/− mice are represented by open squares. Data points represent individual mice. Horizontal bars denote arithmetic means. The asterisks indicate statistical significance from WT as calculated by Mann-Whitney test (*, P < 0.05; **, P < 0.005).
MAV-1 viral loads were assayed in the same mice, and as in the experiment described in Fig. 2A, only the Il1r1−/− mice had significantly higher brain virus levels compared to WT mice (Fig. 3G). In summary, the lack of IL-1 production and signaling during MAV-1 infection increased the levels of inflammatory cytokines in infected brains, but only the lack of IL-1 signaling increased MAV-1 viral loads in infected brains significantly.
Histopathology in the absence of IL-1 signaling.
Microscopic findings in the brains of MAV-1-infected mice consisted of focal or multifocal acute vasculitis, predominantly in the meninges, but also affecting small diameter blood vessels embedded deep within the parenchyma of the pons, midbrain, and thalamus. Affected vessels were surrounded by edema, free red blood cells, and lymphoid cuffs (Fig. 4A to D). Some vessel walls were effaced by infiltrates of macrophages, lymphocytes, and rare neutrophils. Small capillaries lost their endothelial lining, contained fibrin admixed with inflammatory cells, and were surrounded by free red blood cells and small clusters of neutrophils. The severity and distribution of the lesions were quantified by blinded scoring of histological sections of brains (Fig. 4G). Consistent with brain inflammatory cytokine levels (Fig. 3), substantial cellular inflammation was present in the brains of Il1r1−/− mice infected with MAV-1; however, this was not statistically different from WT mice (Fig. 4G). There were also histopathological findings in the Pycard−/− and Unc93b1−/− mice that did not score differently relative to WT mice. In all MAV-1-infected mouse strains, the endothelium was variably hypertrophic, and there was perivascular edema and leakage of red blood cells. In contrast, large basophilic intranuclear viral inclusion bodies were seen in endothelial, glial, and neuronal cells only in the brains of the Il1r1−/− mice (Fig. 4D and F). These histopathologic findings confirm that defective IL-1 signaling increased brain inflammation and pathology in MAV-1-infected mice of all four strains compared to mock-infected mice.
FIG 4.

Representative images of hematoxylin/eosin-stained slides from the midbrains of WT, Pycard−/−, Unc93b1−/−, and Il-1r1−/− mice infected with MAV-1 and histological scores of brain damage. Brain sections are from mice infected with 103 PFU and euthanized at 8 dpi. (A) A Unc93b1−/− mouse shows mild diffuse perivascular edema (black arrowheads). (B) A WT (C57BL/6J) mouse shows mildly hypertrophic endothelial cells (white arrow) and leakage of red blood cells and fluid into the perivascular parenchyma (black arrow). (C) A Pycard−/− mouse shows destruction of vascular integrity with loss of endothelial cells and scattered microhemorrhages (black arrow), fibrin (white star), and clusters of neutrophils in the parenchyma (white arrowheads). (D) A Il1r1−/− mouse shows marked vasculitis with loss of vascular tunics, perivascular hemorrhages (black arrows), and clusters of neutrophils, macrophages, and few lymphocytes in the parenchyma (white arrowheads). (E) Mock infected Il1r1−/− mouse. (F) Image (×400) of an Il1r1−/− mouse shows a blood vessel with a large intranuclear viral inclusion body in an endothelial cell (yellow arrow). Scale bars: A to E, 100 μm; F, 20 μm. (G) Histological scores of brain damage for WT, Pycard−/−, Unc93b1−/−, and Il1r1−/− mice. Data points represent individual mice, and the horizontal lines indicate arithmetic mean values.
IL-1 signaling does not limit MAV-1 neuroinvasion but may play a role in viral clearance.
To better understand the impact of the lack of IL-1 signaling on MAV-1 replication in the brain at various times during infection, viral loads were measured by qPCR in the brains of Il1r1−/− and WT mice harvested at 2 to 8 dpi. MAV-1 is typically detected in the CNS of mice between 3 and 5 days after i.p. inoculation (5). Consistent with this, infected WT and Il1r1−/− mice had detectable viral loads in the brain by 3 dpi (Fig. 5). Viral loads in the brains were similar in Il1r1−/− and WT mice through 6 dpi. In Il1R1−/−mice, MAV-1 levels were statistically significantly higher at 8 dpi. As seen in Fig. 2, the MAV-1 levels were also statistically significantly higher in Il1r1−/− mice at 7 dpi, where a higher number of animals was used than in Fig. 5 (n = 23 in Fig. 2, n = 5 in Fig. 5). Thus, during initial infection MAV-1 replicated at similar levels in mice brains with or without IL-1 signaling. However, when presenting with encephalitic signs, Il1r1−/− mice succumbed to infection, and this correlated with higher brain viral loads than in WT mice.
FIG 5.
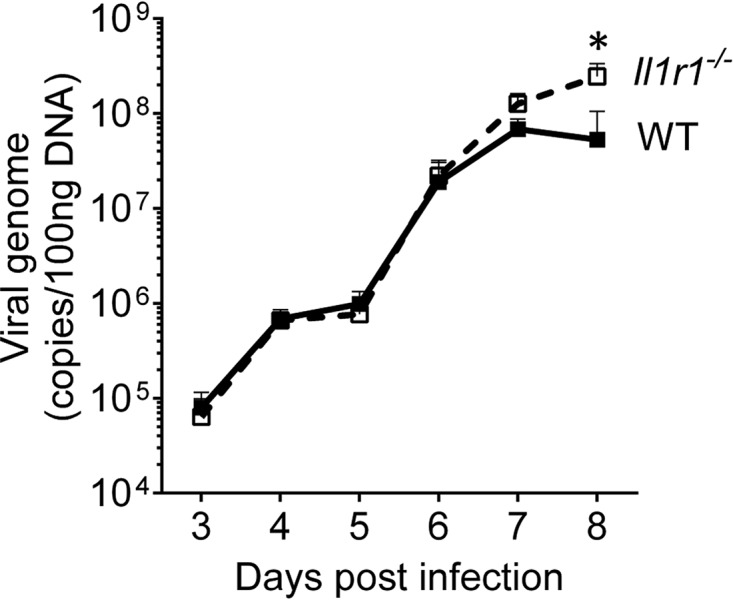
Kinetics of MAV-1 replication in brains from WT (C57BL/6J) and Il1r1 −/− mice from 2 to 8 dpi. WT (C57BL/6J) mice are represented by closed squares and a solid line, and Il1r1 −/− mice are represented by open squares and a dashed line. Viral loads were assessed by qPCR for viral DNA. The LOD of the assay was 104 viral genome copies/100 ng of DNA. Data are shown as means ± the standard errors of the mean (SEM) for n = 5 mice per time point per strain. The asterisk indicates statistical significance as calculated by two-way ANOVA with Sidak's multiple-comparison test between strains (*, P < 0.05).
Serum cytokine responses in Il1r1−/− mice.
To define additional effects of IL-1 signaling deficiency during MAV-1 infection, we assessed the levels of cytokines and chemokines in the sera and brains of uninfected and infected WT and Il1r1−/− mice over a time course. We first measured serum levels of 21 pro- and anti-inflammatory cytokines and chemokines 2 to 8 dpi using Milliplex assays. We did not observe any differences in the basal serum cytokine and chemokine levels between uninfected WT and Il1r1−/− mice, except for IL-10, which was present at higher levels in Il1r1−/− mouse sera (P < 0.05; data not shown). However, after infection, serum cytokine levels peaked earlier or higher in infected Il1r1−/− mice compared to WT mice (Fig. 6A to D). Serum IFN-γ levels peaked on day 6 for both mouse strains; however, Il1r1−/− mice had higher peak levels of IFN-γ than WT mice (Fig. 6A). Serum chemokine levels also peaked earlier, on day 6, in Il1r1−/− mice versus day 7 in WT mice for IP-10 (CXCL10), KC (CXCL1), and MCP-1 (CCL2) (Fig. 6B to D). Il1r1−/− mice had significantly lower serum levels of IP-10 and KC at 7 dpi (Fig. 6B and C) than WT mice. In contrast, Il1r1−/− mice had significantly higher serum levels of MCP-1 at 6 dpi (P < 0.05) (Fig. 6D). Serum levels of IL-1α, IL-1β, IL-2, MIP-1α (CCL3), MIP-1β (CCL4), MIG (CXCL9), and RANTES (CCL5) were not significantly different between WT and Il1r1−/− mice at any time point after infection (data not shown). The levels of LIX (CXCL5) exceeded the reliable limit of detection (LOD) and the levels of IL-4, IL-15, IL-17, and vascular endothelial growth factor (VEGF) were below the limit of detection of the assay in serum for both strains.
FIG 6.

Kinetics of cytokine protein levels in serum and brains of WT (C57BL/6J) and Il1r1 −/− mice from 2 to 8 days postinfection. WT (C57BL/6J) mice are represented with closed squares and solid lines, and Il1r1 −/− mice are represented by open squares and dashed lines. Cytokine protein levels were assessed in serum by Milliplex multi-analyte profiling (A to D) and in brains by ELISA (E to H). Data are shown as the means ± the SEM for n = 5 mice per time point per strain. Mock data for both strains were similar and are shown as a single point (open circles). The asterisks indicate statistical difference between the strains at the indicated time points as calculated by two-way ANOVA with Sidak's multiple-comparison test between two strains of mice on a given day (*, P < 0.05; ***, P < 0.0005).
Inflammatory cytokine expression in brains is increased in the absence of IL-1R1 signaling.
We analyzed the protein levels and mRNA profiles of inflammatory cytokines and chemokines in the brains of MAV-1-infected Il1r1−/− and WT control mice from 2 to 8 dpi. We first analyzed mRNA levels from 59 genes by using a NanoString hybridization assay (Table 2) and then assayed the protein levels of selected genes by enzyme-linked immunosorbent assay (ELISA). The protein (Fig. 6A to D and Fig. 7A to C) and mRNA levels (Fig. 7D to H) of cytokines (Fig. 6) and chemokines (Fig. 7) increased in infected mouse brains at 6 dpi, with peak levels either at 7 or 8 dpi, depending on the mouse strain.
TABLE 2.
Genes evaluated by NanoString RNA hybridization analysis
 |
Nucleotide numbers indicate where the probe anneals in gene sequence deposited in GenBank.
FIG 7.

Kinetics of chemokine production in brains of WT (C57BL/6J) and Il1r1−/− mice from 2 to 8 days postinfection. WT (C57BL/6J) mice are represented with closed squares and solid lines, and Il1r1−/− mice are represented by open squares and dashed lines. Cytokine protein levels were assessed by ELISA (A to C) and RNA levels by NanoString (D to H). Data are shown as means ± the SEM for n = 5 mice per time point per strain. Mock ELISA data for both strains were similar and are shown as a single point (open circles) for ELISA. The asterisks indicate statistical significance as calculated by two-way ANOVA with Sidak's multiple-comparison test between two strains of mice on a given day (*, P < 0.05; **, P < 0.005; ***, P < 0.0005). For all graphs, statistics were performed on the sample values (rather than the fold change for panels D to H).
Overall, the protein levels of the analyzed chemokines correlated with mRNA levels in brains, and when there were differences between WT and Il1r1−/− mice, levels were higher in Il1r1−/− mice (Fig. 7). IFN-γ, IL-1α, IL-6, and TNF-α protein levels were higher in Il1r1−/− mice than WT mice (Fig. 6E to H). Il1r1−/− mice also had higher levels of proinflammatory chemokine mRNAs and proteins compared to WT mice (Fig. 7). IP-10, MCP-1, and RANTES mRNA and protein levels were higher in Il1r1−/− mice than WT mice; this difference reached a peak on day 8 (P < 0.05) (Fig. 7). Because of the increase in IL-1, IL-6, and TNF-α, we also evaluated the mRNA levels of MIP-1α and MIP-1β in infected mouse brains, since MIP-1 molecules can induce the secretion of IL-1, IL-6, and TNF-α from murine peritoneal macrophages (48). MIP-1α and MIP-1β mRNA levels started to increase in brains 6 dpi; at day 8, Il1r1−/− mice had significantly higher levels than WT mice (Fig. 7G and H). In summary, consistent with the histology results, the lack of IL-1 signaling during MAV-1 infection led to increased levels of inflammatory cytokine and chemokine gene expression and protein production in infected brains.
Higher transcription levels of type I IFN-stimulated genes in infected Il1r1−/− mice.
To better understand the mechanism behind increased inflammation in brains of Il1r1−/− mice, we evaluated the type 1 IFN response. Type I IFNs are crucial regulators of noncanonical inflammasome activation and pyroptosis and can suppress IL-1α and IL-1β transcription and translation in various cell types (49). On the other hand, IL-1 can regulate type I IFN production and effector functions (50). We evaluated the mRNA levels of several genes related to innate immunity by using NanoString. IFN-β1 mRNA levels increased in brains of both mouse strains at day 5, and Il1r1−/− mice had higher levels of IFN-β1 than WT mice at 8 dpi (P < 0.0005) (Fig. 8A). Since TLR signaling triggers the induction of type I IFN (IFN-α/β), providing a crucial mechanism of antiviral defense, we also evaluated the mRNA levels of IFN-stimulated genes IRF3, IRF7, TLR2, TLR3, TLR4, TLR7, and TLR9 by using NanoString. There were statistical differences between WT and Il1r1−/− mice only in the levels of IRF-7, TLR2, and TLR3 mRNAs, which were higher in Il1r1−/− mice at 8 dpi (P < 0.05) (Fig. 8B to D). IRF-3 and IRF-7 are the main inducers of IP-10 production through TLR3 signaling (51). Upon infection, Il1r1−/− mice had significantly higher mRNA levels of IP-10 and IRF-7 compared to WT mice (Fig. 7D and 8B), but there was no difference in IRF3 levels (data not shown). In addition, IFIT1, which also induces IP-10 production, had higher transcription levels after MAV-1 infection in Il1r1−/− mice compared to WT mice (Fig. 8E). STING and cGAS are intracellular sensors that activate the IFN pathway, and HAdV DNA is sensed by cGAS (37). We observed an increase in STING and cGAS mRNA levels after infection with MAV-1 that did not differ between WT and Il1r1−/− mice (Fig. 8F and G). In summary, in the absence of IL-1 signaling during MAV-1 infection, there was an increase in the mRNA levels of IFN-stimulated genes.
FIG 8.

Kinetics of mRNA levels of IFN-β1 (A), IRF-7 (B), TLR2 (C), TLR3 (D), IFIT1 (E), STING (F), and cGAS (G) in the brains of WT (C57BL/6J) and Il1r1−/− mice from 2 to 8 dpi. WT (C57BL/6J) mice are represented by closed squares and solid lines, and Il1r1−/− mice are represented by open squares and dashed lines. Cytokine gene RNA levels were determined by NanoString. Data are shown as means ± the SEM for n = 5 mice per time point per strain. The asterisks indicate statistical significance as calculated by two-way ANOVA with Sidak's multiple-comparison test between two strains of mice on a given day (*, P < 0.05; **, P < 0.005; ***, P < 0.0005), performed on the sample values rather than fold change.
DISCUSSION
In this study, we investigated the role of IL-1 signaling in MAV-1-induced encephalitis, evaluating whether IL-1 contributes to the encephalitis and BBB disruption observed in MAV-1-infected C57BL/6J mice. Using Il1r1−/− mice lacking the IL-1 receptor, we demonstrated the importance of IL-1 signaling during MAV-1 infection. Mice that lack this receptor had lower survival, higher disruption of the BBB, higher viral loads, and higher levels of inflammatory cytokines and chemokines in the brain compared to control mice. MAV-1-infected Il1r1−/− mice had inflammatory profiles similar to Pycard−/− mice, which alone might suggest that the inflammasome-dependent production of IL-1 that occurred during MAV-1 infection was dependent on ASC (which is encoded by Pycard). However, Il1r1−/− mice had greater BBB disruption and higher viral loads than Pycard−/− mice; this distinction between disease phenotypes in the two strains suggests that instead there may be another pathway involved in IL-1 production that is operating in the infected Pycard−/− mice, which did not recapitulate all the phenotypes of Il1r1−/− mice. In addition, we did not test IL-1α knockout mice, and it is possible that IL-1α has a protective role during MAV-1 infection, since Il1r1−/− mice had a poorer outcome compared to Pycard−/− mice. To better understand the mechanism by which the lack of IL-1 signaling in Il1r1−/− mice caused a poorer outcome compared to WT mice, we assayed the transcription levels of several IFN-stimulated genes. The mRNA levels of IFN-β1, TLR2, TLR3, IRF-7, and IFIT1 were elevated after MAV-1 infection in Il1r1−/− mouse brains. One possibility is that type I IFN-stimulated genes contribute to pathology during the lack of IL-1 signaling.
Previous studies have associated IL-1 signaling with either protection or enhancement of disease. In the context of MAV-1-induced encephalitis our results indicate that IL-1 signaling has a protective role, as observed for WNV infection (11, 25). Il1r1−/− and Pycard−/− MAV-1-infected mice had lower survival than WT mice. We observed a more pronounced effect on MAV-1 pathogenesis in Il1r1−/− mice and higher brain viral loads in brains compared to the other mouse strains. This is different from WNV infection in mouse models, where not only Il1r1−/− mice but also Pycard−/− mice have higher brain viral loads (11, 25, 30). During HSV-1-induced encephalitis, IL-1β also has a protective role against encephalitis (21). However, for Sindbis virus encephalitis and HIV encephalitis, IL-1 has an opposite effect: it increases pathogenesis (21, 23, 24). The IL-1 protective role found here for MAV-1 infections was surprising since IL-1β, along with TNF-α and IL-6, increases BBB permeability to viruses (52). However, we observed that MAV-1 infection of mice lacking IL-1R1 resulted in higher disruption of BBB (as evidenced by sodium fluorescein uptake) compared to control mice, confirming the protective role of IL-1 signaling.
Chemokines also have an effect on BBB disruption, as demonstrated for MCP-1 (CCL2) during HIV-1 infection, where MCP-1 disrupts adherens junctions and transiently opens the endothelial cell barrier (53). Chemokines also affect BBB integrity by enhancing migration of immune cells to the infected brain areas. The chemokine receptors CXCR3, CCR2, and CCR5 are crucial for leukocyte trafficking to the brain during SFV and WNV encephalitis (54). The results of our study suggest that these chemokine receptors may contribute to BBB disruption in MAV-1 infection, since we found high levels of their ligands in infected Il1r1−/− mice, accompanied by higher BBB disruption than in WT mice. For example, we found higher levels of IP-10 (CXCL10), MCP-1, MIP-1α and -1β (CCL3 and CCL4), and RANTES (CCL5), which are ligands of CXCR3, CCR2, and CCR5, in Il1r1−/− mice than WT mice. Il1r1−/− mice also had higher levels of KC (CXCL1), the ligand of CXCR2. We did not characterize the infiltrating leukocyte population in Il1r1−/− MAV-1-infected mice. However, the histopathology analysis indicated that there were infiltrates of macrophages, lymphocytes, and few neutrophils. IP-10 (CXCL10), MCP-1 (CCL2), MIP-1β (CCL4), and RANTES (CCL5) attract mainly monocytes, macrophages, T cells, and NK cells, and thus the higher increase in these chemokine levels in Il1r1−/− mice is consistent with them contributing to the observed infiltration.
To better characterize the IL-1 pathway involved in MAV-1-induced BBB disruption, we used Unc93b1−/− and Pycard−/− mice. UNC93B is required for TLR3, TLR7, TLR9, TLR11, TLR12, and TLR13 endosomal localization (39), and thus signaling through these receptors is impaired in Unc93b1−/− mice (43). For IL-1β to be in its active form, it needs two signals (13). The first signal can be provided by TLR stimulation and triggers the synthesis of the IL-1β precursor (55). The second signal leads to inflammasome assembly, and the adaptor protein ASC must become linearly ubiquitinated and phosphorylated for inflammasome assembly to occur (28). If the absence of IL-1β signaling was responsible for the poorer outcome observed in MAV-1-infected Il1r1−/− mice, we expected that Unc93b1−/− and Pycard−/− mice would also have lower survival than WT mice, because the former could be defective in IL-1β production. Indeed, these mice did have poorer survival when infected with MAV-1, an observation consistent with an IL-1β defect contributing to the reduction in survival. However, the infected Unc93b1−/− and Pycard−/− mice did produce IL-1β. In addition, not all the MAV-1-induced pathogenesis findings observed in Il1r1−/− mice were replicated in Unc93b1−/− and Pycard−/− mice, such as increased BBB disruption and viral load, and the presence of intranuclear viral inclusion bodies. One possibility is that IL-1β, which is present in the Pycard mice during MAV-1 infection, can be produced by a non-ASC pathway (14) and provides some protective effects in Pycard−/− mice. Alternatively, perhaps IL-1α produced in Unc93b1−/− and Pycard−/− mice can act in a protective manner like IL-1β with respect to BBB disruption and viral replication. This could be addressed by examining MAV-1-induced encephalitis in IL-1α and IL-1β knockout mice.
To get a more detailed picture of the IL-1 signaling effects during MAV-1-induced-encephalitis, we compared the systemic cytokine response (in serum) to the local response (in brain). A study with WNV showed that inflammatory responses in the periphery are impaired in WNV-infected mice that have a mutation in Pycard that renders them ASC deficient compared to inflammatory responses in the brains of the same mice (30). However, we observed that in Il1r1−/− mice the systemic cytokine response peaked earlier than in WT mice and decreased on days 7 and 8, when viral loads were still increasing, in contrast to a decrease in viral loads in WT mice at those times. This suggests that the lack of IL-1 signaling resulted in reduced systemic cytokines that led to continued viral replication, possibly due to insufficient T cell activation (26). The brain inflammatory response was much higher in Il1r1−/− mice compared to WT mice, with higher levels of IL-6 and TNF-α, which, along with IL-1β, have been correlated with increased BBB disruption (52). Astrocytes become activated subsequent to IL-1β production and consequently have an enhanced ability to sustain neuronal survival, reestablish the BBB, and reestablish homeostasis (56). It is possible that some of these positive functions of IL-1β might be operating during MAV-1 infection. We hypothesize that peak levels of cytokines in MAV-1-infected brains occurred only at 7 to 8 dpi because at this time virus replication reached a sufficient level to stimulate this inflammatory response. We first detected viral DNA here (Fig. 5) at 3 dpi and detect replicated infectious virus (5) at 3 to 5 dpi. Thus, 7 to 8 dpi would correspond to ∼2 rounds of viral infection. It would be interesting to determine whether cytokines are produced from the same sources throughout the infection or whether there is a difference in cytokine-producing cells in the initial infection versus subsequent replication.
The transcription levels of type I IFN-stimulated genes were higher in Il1r1−/− mice than in WT mice. Both IL-1 and type I IFN signaling pathways can cause harm when dysregulated or activated in an inappropriate context, and they also can regulate each other (50). IL-1 can antagonize type I IFN responses by regulating transcription and translation of IFN-β via induction of prostaglandin E2 (57). IFN-β mRNA and protein levels are upregulated in the lungs of Il1r1−/− mice upon infection by Mycobacterium tuberculosis (57). Similarly, we found that in MAV-1-infected Il1r1−/− mice, type I IFN-stimulated gene levels were upregulated in the brains relative to mock-infected mice. It could be that in the context of MAV-1 infection IL-1 also regulates the type I IFN response; alternatively, increased IFN-stimulated genes could be a result of Il1r1−/− mice having higher viral loads. These possibilities could be addressed by evaluating protein levels of IFN-stimulated genes in the type I IFN pathway.
In summary, we used knockout mice to determine whether IL-1 has a protective role in mouse infections by MAV-1. We used mice deficient in IL-1, TLR3, TLR7, and TLR9 signaling. All the knockout mice had survival significantly worse than control mice. However, the Il1r1−/− mice had the most severe disease phenotype, more inflammatory cytokine production, and increased transcription of type I IFN-stimulated genes compared to WT control mice. We conclude that IL-1 protects mice from some of the pathogenic effects of MAV-1 infection.
MATERIALS AND METHODS
Virus and mouse experiments.
WT MAV-1 was originally obtained from S. Larsen (58, 59), and virus was grown and passaged in 3T6 fibroblasts as described previously (60). Lysates from infected 3T6 cells were centrifuged to remove cellular debris, and titers of virus stocks were determined by plaque assay on 3T6 cells (60). Conditioned media used for mock infections was isolated and prepared from uninfected 3T6 cells in a similar manner.
C57BL/6J male mice were obtained from Jackson Laboratory. Il1r1−/− and Pycard−/− mice (61, 62) on the C57BL/6 background were obtained from Gabriel Nuñez (University of Michigan). C57BL/6J-Unc93b13d (Unc93b1−/−) mice (43) were obtained from David Irani (University of Michigan). The knockout mice were bred in-house, and both sexes were used in experiments. No differences based on sex were noted. Mice 4 to 5 weeks old were infected via i.p. injection with 103 PFU in a volume of 100 μl. Virus was diluted in 10-fold serial dilutions in endotoxin-free Dulbecco phosphate-buffered saline (DPBS; Lonza). Mock-infected mice were injected with conditioned media diluted similarly in DPBS. Mice were monitored twice daily for signs of disease (e.g., ruffled fur, hunched posture, seizures, inability to feed) and were euthanized by CO2 asphyxiation if moribund or at the indicated time points. Organs were harvested, snap-frozen on dry ice, and stored at −70°C until processed. All animal work complied with relevant federal and University of Michigan policies. Mice were housed in microisolator cages and provided with food and water ad libitum.
Histological analysis.
C57BL/6J, Pycard−/−, Unc93b1−/−, and Il1r1−/− mice were either mock infected or infected with MAV-1, perfused with 10% formalin (3.7% formaldehyde in PBS) after euthanasia, and organs were collected for histopathology. Organs (thymus, lung, heart, brain, liver, kidney, and spleen) were immersion fixed in 10% neutral buffered formalin for 24 h, embedded in paraffin, and sectioned at 5 μm. Sections were stained with hematoxylin and eosin. The University of Michigan Comprehensive Cancer Center Research Histology and Immunoperoxidase Laboratory performed sectioning and staining. Slides were randomized and blinded for evaluation by a board-certified pathologist. Five coronal sections from each brain representing the olfactory bulb, cortex/ventral striatum, hippocampus/thalamus, midbrain, and pons/cerebellum were evaluated. Points for each animal were summed to generate a total histology score. A single point was assigned to each of the following microscopic findings: perivascular edema, parenchymal microhemorrhages, vasculitis with loss or hypertrophy of endothelial cells, perivascular or meningeal vascular cuffing, and multifocal distribution of lesions affecting two or more brain regions.
Isolation of DNA and RNA.
DNA for the measurement of viral load was extracted from 20 mg of brain or 10 mg of spleen using the PureLink Genomic DNA minikit (Thermo Fisher Scientific). Total RNA used for gene transcription was extracted from brains as follows. Approximately 50 mg of each brain was homogenized using sterile glass beads in a Mini-Beadbeater (Biospec Products) for 30 s in 1 ml of TRIzol (Invitrogen). RNA was then isolated from the homogenates according to the manufacturer's protocol and stored at −70°C until use.
Quantitation of viral loads in tissues.
MAV-1 viral loads were measured in brains and spleens using qPCR as previously described (63). MAV-1 early region 1A (E1A) primers and probe detect a 59-bp region of the MAV-1 E1A gene (Table 1). A 5-ng portion of extracted DNA was added to reaction mixturess containing TaqMan Universal PCR mix (Applied Biosystems), forward and reverse primers (each at 200 nM final concentration), and probe (40 nM final concentration) in a 10-μl reaction volume. Real-time PCR was performed on an ABI Prism 7500 sequence detector (Applied Biosystems) and consisted of 40 cycles of 15 s at 90°C and 60 s at 60°C. Standard curves generated using known amounts of plasmid containing the MAV-1 EIA gene were used to convert cycle threshold values to copy numbers of E1A DNA. Results were standardized to the amount of input DNA. Each sample was assayed in duplicate. Viral loads determined by qPCR correlated with values determined by plaque assay (Castro-Jorge and Spindler, unpublished).
TABLE 1.
Primers and probes used for real-time PCR analysis
| Target | Oligonucleotide | Sequence (5′–3′) |
|---|---|---|
| MAV-1 E1A | Forward primer | GCACTCCATGGCAGGATTCT |
| Reverse primer | GGTCGAAGCAGACGGTTCTTC | |
| Probe | TACTGCCACTTCTGC | |
| IFN-γ | Forward primer | AAAGAGATAATCTGGCTCTGC |
| Reverse primer | GCTCTGAGACAATGAACGCT | |
| IFN-α2 | Forward primer | TTGAAGGTCCTGGCACAG |
| Reverse primer | GAGGTTCAAGGTCTGCTGA | |
| IFN-β | Forward primer | AGCTCCAAGAAAGGACGAACAT |
| Reverse primer | GCCCTGTAGGTGAGGTTGATCT | |
| IL-1β | Forward primer | GCAACTGTTCCTGAACTCAACT |
| Reverse primer | ATCTTTTGGGGTCCGTCAACT | |
| TNF-α | Forward primer | CCACCACGCTCTTCTGTCTAC |
| Reverse primer | AGGGTCTGGGCCATAGAACT | |
| GAPDH | Forward primer | TGCACCACCAACTGCTTAG |
| Reverse primer | GGATGCAGGGATGATGTTC |
Analysis of cytokine and chemokine gene expression.
Cytokine gene expression was quantified from RNA extracted from brains of mock-infected or MAV-1-infected C57BL/6J and Il1r1−/− mice using a NanoString nCounter analysis system and qRT-PCR. Details of the nCounterTM technology (NanoString Technologies, Seattle, WA) have been reported previously (64). Briefly, 60 genes associated with IL-1β/innate immune pathways and 3 reference genes were evaluated by NanoString RNA hybridization analysis (Table 2). Each gene probe in the 63 gene set was manufactured with specificity to a 100-base region of target gene mRNA and linked to a biotinylated capture probe; a complementary reporter probe was also included and tagged with a specific fluorescent barcode, resulting in two sequence-specific probes for each target transcript. Probes were hybridized to 100 ng of total RNA for 16 h at 65°C and applied to the nCounterTM Preparation Station for automated removal of excess probe and immobilization of probe-transcript complexes on a streptavidin-coated cartridge. Data were collected using a GEN2 digital analyzer by counting the individual barcodes.
To confirm NanoString results, we performed reverse transcription-qPCR analysis. Portions (2 μg) of RNA were reverse transcribed using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) according to the manufacturer's instructions. cDNA corresponding to 50 ng of RNA equivalent was used in each qPCR, and each sample was analyzed in triplicate. The target genes evaluated are detailed in Table 1. Quantitation was performed by normalizing target gene mRNA levels to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels, and infected sample values are expressed relative to the mean of mock values, set to 1 for each gene. To calculate the statistical significance of between-group differences, we used ΔCT values, and for data presentation, we used 2−ΔΔCT values.
Analysis of cytokine and chemokine protein levels.
The levels of cytokines and chemokines were measured in mouse serum by the Milliplex mouse cytokine/chemokine panel I kit (Millipore) by the University of Michigan Cancer Center Immunology Core. In each serum sample we measured GM-SCF, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12p70, IL-15, IL-17A, IP-10, KC, LIX, MCP-1, MIG, MIP-1α, MIP-1β, RANTES, TNF-α, and VEGF-A. The assay was performed according to the manufacturer's instructions. Plates were read using a MAGPIX plate reader and analyzed using xPONENT Software (Merck Millipore).
The levels of cytokines and chemokines in mouse brains were measured by ELISA at the University of Michigan Cancer Center Immunology Core. We measured the IFN-γ, IL-1α, IL-1β, IL-6, IL-10, IP-10, KC, MCP-1, RANTES, and TNF-α. Approximately 50 mg of each brain was homogenized at a 10% concentration with extraction buffer solution (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% Igepal, and 1× protease inhibitor cocktail [Thermo Scientific]) using sterile glass beads in a Mini-Beadbeater (Biospec Products) for 30 s three times, with intervals of 10 s on ice. Samples were incubated on ice for 30 min and then centrifuged at 2,000 × g for 10 min at 4°C. Samples were transferred to a new 1.5-ml tube and centrifuged again at 20,000 × g for 20 min at 4°C to pellet debris. Samples were stored at −70°C until use. Before use, samples were thawed on ice and centrifuged at 20,000 × g for 5 min at 4°C. Cytokine measurement (R&D and Peprotech) and protein quantification by Pierce BCA protein assay kit (Thermo Scientific) were performed according to the manufacturer's instructions.
BBB permeability assay.
Mice were injected i.p. with 100 μl of 10% sodium fluorescein (Sigma) in DPBS 10 min prior to euthanasia. Mice were euthanized by CO2 asphyxiation. Cardiac blood was collected, and mice were transcardially perfused with 30 ml of ice-cold PBS. Brains were then snap-frozen until used for quantitation. Sodium fluorescein levels in brain and serum were determined as previously described (8) using the right brain hemisphere. Fluorescence levels were measured on a Bio-Tek multidetection microplate reader with a 485-nm excitation and a 530-nm emission. Standards were used to calculate the sodium fluorescein content of brain and serum samples. Brain values were normalized to their respective serum dye values to allow comparisons among mice. The amount of sodium fluorescein in each infected mouse brain is represented as a fold change from the average uptake in the brains of mock-infected mice of the respective strain.
Data analysis.
Statistical analyses were performed using Prism 6 (GraphPad Software, Inc.) and JMP 11 (SAS Software). Log-transformed values for viral load data were used for statistical comparisons. Differences between two groups were analyzed using Mann–Whitney rank sum test. Comparisons made between groups at multiple time points were analyzed using two-way analysis of variance (ANOVA), followed by Sidak's multiple-comparison tests. P values of <0.05 were considered statistically significant.
Analysis and normalization of the raw NanoString data were conducted using nSolver Analysis Software v2.5 (NanoString Technologies). Raw counts were normalized to internal levels of three mouse reference genes: GAPDH, GUSB, and HPRT. Normalized data were log2 transformed for statistical comparisons. RNAs from mock-infected mice served as controls and were used to calculate fold change.
ACKNOWLEDGMENTS
We thank Gabriel Nuñez for the Il1r1−/− and Pycard−/− mice and David Irani for Unc93b1−/− mice. We also thank Jason Weinberg, Beth Moore, and Danielle Goodman for comments on the manuscript and Mary McCarthy and Megan Procario for helpful discussions about data analysis. We thank Natalie Thompson for technical assistance.
This study was supported by NIH R01 AI23762 and NIH R01 091721 (K.R.S.) and Brazil Science without Borders (Ciências sem Fronteiras) (2548/13-5) (L.A.C.-J.). Research reported here (histological sectioning and staining, as well as cytokine analysis) was supported by the National Cancer Institute of the National Institutes of Health under award P30CA046592.
REFERENCES
- 1.Spindler KR, Hsu TH. 2012. Viral disruption of the blood-brain barrier. Trends Microbiol 20:282–290. doi: 10.1016/j.tim.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salimi H, Cain MD, Klein RS. 2016. Encephalitic arboviruses: emergence, clinical presentation, and neuropathogenesis. Neurotherapeutics 13:514–534. doi: 10.1007/s13311-016-0443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou J, Baker LA, Zhou L, Klein RS. 2016. Viral interactions with the blood-brain barrier: old dog, new tricks. Tissue Barriers 4:e1142492. doi: 10.1080/21688370.2016.1142492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guida JD, Fejer G, Pirofski LA, Brosnan CF, Horwitz MS. 1995. Mouse adenovirus type 1 causes a fatal hemorrhagic encephalomyelitis in adult C57BL/6 but not BALB/c mice. J Virol 69:7674–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spindler KR, Fang L, Moore ML, Hirsch GN, Brown CC, Kajon A. 2001. SJL/J mice are highly susceptible to infection by mouse adenovirus type 1. J Virol 75:12039–12046. doi: 10.1128/JVI.75.24.12039-12046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kring SC, King CS, Spindler KR. 1995. Susceptibility and signs associated with mouse adenovirus type 1 infection of adult outbred Swiss mice. J Virol 69:8084–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kajon AE, Brown CC, Spindler KR. 1998. Distribution of mouse adenovirus type 1 in intraperitoneally and intranasally infected adult outbred mice. J Virol 72:1219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gralinski LE, Ashley SL, Dixon SD, Spindler KR. 2009. Mouse adenovirus type 1-induced breakdown of the blood-brain barrier. J Virol 83:9398–9410. doi: 10.1128/JVI.00954-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corps KN, Roth TL, McGavern DB. 2015. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol 72:355–362. doi: 10.1001/jamaneurol.2014.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amor S, Puentes F, Baker D, Van Der Valk P. 2010. Inflammation in neurodegenerative diseases. Immunology 129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M. 2012. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 8:e1003039. doi: 10.1371/journal.ppat.1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basu A, Krady JK, Levison SW. 2004. Interleukin-1: a master regulator of neuroinflammation. J Neurosci Res 78:151–156. doi: 10.1002/jnr.20266. [DOI] [PubMed] [Google Scholar]
- 13.Walsh JG, Muruve DA, Power C. 2014. Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- 14.Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 15.Tschopp J, Martinon F, Burns K. 2003. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 16.Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 17.Muzio M, Ni J, Feng P, Dixit VM. 1997. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello CA. 1996. Biologic basis for interleukin-1 in disease. Blood 87:2095–2147. [PubMed] [Google Scholar]
- 19.Dinarello CA. 1988. Biology of interleukin 1. FASEB J 2:108–115. [PubMed] [Google Scholar]
- 20.Schmitz N, Kurrer M, Bachmann MF, Kopf M. 2005. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79:6441–6448. doi: 10.1128/JVI.79.10.6441-6448.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sergerie Y, Rivest S, Boivin G. 2007. Tumor necrosis factor-alpha and interleukin-1β play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis 196:853–860. doi: 10.1086/520094. [DOI] [PubMed] [Google Scholar]
- 22.Durrant DM, Daniels BP, Klein RS. 2014. IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during West Nile virus encephalitis. J Immunol 193:4095–4106. doi: 10.4049/jimmunol.1401192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prow NA, Irani DN. 2008. The inflammatory cytokine, interleukin-1β, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J Neurochem 105:1276–1286. doi: 10.1111/j.1471-4159.2008.05230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng H, Erdmann N, Whitney N, Dou H, Gorantla S, Gendelman HE, Ghorpade A, Zheng J. 2006. HIV-1-infected and/or immune activated macrophages regulate astrocyte SDF-1 production through IL-1β. Glia 54:619–629. doi: 10.1002/glia.20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durrant DM, Robinette ML, Klein RS. 2013. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med 210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim BS, Jin YH, Meng L, Hou W, Kang HS, Park HS, Koh CS. 2012. IL-1 signal affects both protection and pathogenesis of virus-induced chronic CNS demyelinating disease. J Neuroinflammation 9:217. doi: 10.1186/1742-2094-9-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. 2008. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 28.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J. 1999. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem 274:33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 29.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M, Verma S. 2013. Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J Virol 87:3655–3667. doi: 10.1128/JVI.02667-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charles PC, Guida JD, Brosnan CF, Horwitz MS. 1998. Mouse adenovirus type-1 replication is restricted to vascular endothelium in the CNS of susceptible strains of mice. Virology 245:216–228. doi: 10.1006/viro.1998.9180. [DOI] [PubMed] [Google Scholar]
- 32.Coutelier JP, Coulie PG, Wauters P, Heremans H, vander Logt JT. 1990. In vivo polyclonal B-lymphocyte activation elicited by murine viruses. J Virol 64:5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inada T, Uetake H. 1980. Cell-mediated immunity to mouse adenovirus infection. Blocking of macrophage migration inhibition and T cell-mediated cytolysis of infected cells by anti-S antigen or anti-alloantigen serum. Microbiol Immunol 24:525–535. [DOI] [PubMed] [Google Scholar]
- 34.Moore ML, Brown CC, Spindler KR. 2003. T cells cause acute immunopathology and are required for long-term survival in mouse adenovirus type 1-induced encephalomyelitis. J Virol 77:10060–10070. doi: 10.1128/JVI.77.18.10060-10070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore ML, McKissic EL, Brown CC, Wilkinson JE, Spindler KR. 2004. Fatal disseminated mouse adenovirus type 1 infection in mice lacking B cells or Bruton's tyrosine kinase. J Virol 78:5584–5590. doi: 10.1128/JVI.78.11.5584-5590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iacobelli-Martinez M, Nemerow GR. 2007. Preferential activation of Toll-like receptor nine by CD46-utilizing adenoviruses. J Virol 81:1305–1312. doi: 10.1128/JVI.01926-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendrickx R, Stichling N, Koelen J, Kuryk L, Lipiec A, Greber UF. 2014. Innate immunity to adenovirus. Hum Gene Ther 25:265–284. doi: 10.1089/hum.2014.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerullo V, Seiler MP, Mane V, Brunetti-Pierri N, Clarke C, Bertin TK, Rodgers JR, Lee B. 2007. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol Ther 15:378–385. doi: 10.1038/sj.mt.6300031. [DOI] [PubMed] [Google Scholar]
- 39.Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM. 2013. UNC93B1 mediates differential trafficking of endosomal TLRs. eLife 2:e00291. doi: 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. 2007. The interaction between the ER membrane protein UNC93B and TLR3, -7, and -9 is crucial for TLR signaling. J Cell Biol 177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim Y-M, Brinkmann MM, Paquet M-E, Ploegh HL. 2008. UNC93B1 delivers nucleotide-sensing Toll-like receptors to endolysosomes. Nature 452:234–238. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 42.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 43.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 7:156–164. doi: 10.1038/nrm1901. [DOI] [PubMed] [Google Scholar]
- 44.Lafferty EI, Wiltshire SA, Angers I, Vidal SM, Qureshi ST. 2015. Unc93b1-dependent endosomal Toll-like receptor signaling regulates inflammation and mortality during coxsackievirus B3 infection. J Innate Immun 7:315–330. doi: 10.1159/000369342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charles PC, Chen X, Horwitz MS, Brosnan CF. 1999. Differential chemokine induction by the mouse adenovirus type-1 in the central nervous system of susceptible and resistant strains of mice. J Neurovirol 5:55–64. doi: 10.3109/13550289909029746. [DOI] [PubMed] [Google Scholar]
- 46.McCarthy MK, Procario MC, Twisselmann N, Wilkinson JE, Archambeau AJ, Michele DE, Day SM, Weinberg JB. 2015. Proinflammatory effects of interferon gamma in mouse adenovirus 1 myocarditis. J Virol 89:468–479. doi: 10.1128/JVI.02077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. 1996. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson's disease. Neurosci Lett 211:13–16. doi: 10.1016/0304-3940(96)12706-3. [DOI] [PubMed] [Google Scholar]
- 48.Fahey TJ, Tracey KJ, Tekamp-Olson P, Cousens LS, Jones WG, Shires GT, Cerami A, Sherry B. 1992. Macrophage inflammatory protein 1 modulates macrophage function. J Immunol 148:2764–2769. [PubMed] [Google Scholar]
- 49.Malireddi RK, Kanneganti TD. 2013. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front Cell Infect Microbiol 3:77. doi: 10.3389/fcimb.2013.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mayer-Barber KD, Yan B. 6 June 2016. Clash of the cytokine titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol doi: 10.1038/cmi.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 52.Miner JJ, Diamond MS. 2016. Mechanisms of restriction of viral neuroinvasion at the blood-brain barrier. Curr Opin Immunol 38:18–23. doi: 10.1016/j.coi.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roberts TK, Eugenin EA, Lopez L, Romero IA, Weksler BB, Couraud P-O, Berman JW. 2012. CCL2 disrupts the adherens junction: implications for neuroinflammation. Lab Invest 92:1213–1233. doi: 10.1038/labinvest.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michlmayr D, Lim JK. 2014. Chemokine receptors as important regulators of pathogenesis during arboviral encephalitis. Front Cell Neurosci 8:264. doi: 10.3389/fncel.2014.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He Y, Franchi L, Núñez G. 2013. TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol 190:334–339. doi: 10.4049/jimmunol.1202737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. 2004. Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem 89:1092–1100. doi: 10.1111/j.1471-4159.2004.02420.x. [DOI] [PubMed] [Google Scholar]
- 57.Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, Sher A. 2014. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ball AO, Beard CW, Villegas P, Spindler KR. 1991. Early region 4 sequence and biological comparison of two isolates of mouse adenovirus type 1. Virology 180:257–265. doi: 10.1016/0042-6822(91)90030-F. [DOI] [PubMed] [Google Scholar]
- 59.Larsen SH, Margolskee RF, Nathans D. 1979. Alignment of the restriction map of mouse adenovirus FL with that of human adenovirus 2. Virology 97:406–414. doi: 10.1016/0042-6822(79)90351-9. [DOI] [PubMed] [Google Scholar]
- 60.Cauthen AN, Welton AR, Spindler KR. 2007. Construction of mouse adenovirus type 1 mutants. Methods Mol Med 130:41–59. [DOI] [PubMed] [Google Scholar]
- 61.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Núñez G, Schlueter D, Flavell RA, Sutterwala FS, Sher A. 2010. Caspase-1 independent IL-1β production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ozören N, Masumoto J, Franchi L, Kanneganti T-D, Body-Malapel M, Ertürk I, Jagirdar R, Zhu L, Inohara N, Bertin J, Coyle A, Grant EP, Núñez G. 2006. Distinct roles of TLR2 and the adaptor ASC in IL-1β/IL-18 secretion in response to Listeria monocytogenes. J Immunol 176:4337–4342. doi: 10.4049/jimmunol.176.7.4337. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen Y, McGuffie BA, Anderson VE, Weinberg JB. 2008. Gammaherpesvirus modulation of mouse adenovirus type 1 pathogenesis. Virology 380:182–190. doi: 10.1016/j.virol.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kulkarni MM. 2011. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol Chapter 25:Unit 25B.10. doi: 10.1002/0471142727.mb25b10s94. [DOI] [PubMed] [Google Scholar]