

ABSTRACT
The Flavivirus genus contains several arthropod-borne viruses that pose global health threats, including dengue viruses (DENV), yellow fever virus (YFV), and Zika virus (ZIKV). In order to understand how these viruses replicate in human cells, we previously conducted genome-scale RNA interference screens to identify candidate host factors. In these screens, we identified ribosomal proteins RPLP1 and RPLP2 (RPLP1/2) to be among the most crucial putative host factors required for DENV and YFV infection. RPLP1/2 are phosphoproteins that bind the ribosome through interaction with another ribosomal protein, RPLP0, to form a structure termed the ribosomal stalk. RPLP1/2 were validated as essential host factors for DENV, YFV, and ZIKV infection in two human cell lines: A549 lung adenocarcinoma and HuH-7 hepatoma cells, and for productive DENV infection of Aedes aegypti mosquitoes. Depletion of RPLP1/2 caused moderate cell-line-specific effects on global protein synthesis, as determined by metabolic labeling. In A549 cells, global translation was increased, while in HuH-7 cells it was reduced, albeit both of these effects were modest. In contrast, RPLP1/2 knockdown strongly reduced early DENV protein accumulation, suggesting a requirement for RPLP1/2 in viral translation. Furthermore, knockdown of RPLP1/2 reduced levels of DENV structural proteins expressed from an exogenous transgene. We postulate that these ribosomal proteins are required for efficient translation elongation through the viral open reading frame. In summary, this work identifies RPLP1/2 as critical flaviviral host factors required for translation.
IMPORTANCE Flaviviruses cause important diseases in humans. Examples of mosquito-transmitted flaviviruses include dengue, yellow fever and Zika viruses. Viruses require a plethora of cellular factors to infect cells, and the ribosome plays an essential role in all viral infections. The ribosome is a complex macromolecular machine composed of RNA and proteins and it is responsible for protein synthesis. We identified two specific ribosomal proteins that are strictly required for flavivirus infection of human cells and mosquitoes: RPLP1 and RPLP2 (RPLP1/2). These proteins are part of a structure known as the ribosomal stalk and help orchestrate the elongation phase of translation. We show that flaviviruses are particularly dependent on the function of RPLP1/2. Our findings suggest that ribosome composition is an important factor for virus translation and may represent a regulatory layer for translation of specific cellular mRNAs.
KEYWORDS: flavivirus, ribosomal proteins, translation
INTRODUCTION
Flaviviruses, particularly those transmitted by Aedes spp. mosquitoes, pose emerging and reemerging global health threats (1–4). Dengue viruses (DENV-1 to -4), the most prevalent of all arthropod-borne viruses, infect approximately 390 million people per year (1); yellow fever virus (YFV), which causes life-threatening disease (5) has reemerged this year in Africa (6); Zika virus (ZIKV) is currently responsible for a pandemic in the Americas that has caused grave concern because of associations with birth defects and Guillain-Barré syndrome (3).
The flavivirus genome is a positive-strand, ∼11-kb RNA molecule that contains a single open reading frame (ORF) coding for a polyprotein which is co- and posttranslationally cleaved into three structural proteins, capsid (C), premembrane (prM), and envelope (E), and seven nonstructural (NS) proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. The 5′ end is modified with a type I cap structure, and the 3′ end lacks a poly(A) tail. The genome also contains structured cis-acting RNA elements that are required for efficient viral protein and RNA synthesis (7–9).
As is true for all viruses, flaviviruses absolutely require host translation machinery to synthesize viral proteins. Flavivirus translation initiation is thought to occur primarily by a mechanism analogous to cellular cap-dependent translation; however, it has also been reported that DENV-2 initiates translation by a cap-independent mechanism under conditions where cap-dependent translation is inhibited (10). The viral genome is assumed to engage the eukaryotic initiation factor 4F (eIF4F) complex via the 5′ cap structure and poly(A)-binding protein (PABP), which interacts with the 3′ untranslated region (UTR) (11). The 43S preinitiation complex is then recruited and scans the 5′ UTR until the start codon is located with the assistance of an RNA stem-loop in the capsid-coding region known as the cHP (8). Dissociation of initiation factors and joining with the 60S ribosomal subunit forms the 80S ribosome, which commences the elongation phase.
While the general mechanisms of translation have been studied extensively, the specific roles of many host proteins in translation are unclear. This is the case for most of the approximately 80 ribosomal proteins (RPs), most of which interact with rRNA (12, 13). While some RPs are important for basic ribosomal functions, recent evidence suggests that several RPs play roles in mRNA-specific translation (14–20). Supporting the idea of specialized roles, many RPs are differentially expressed in mammalian tissues (19, 20), and mice bearing a heterozygotic or homozygotic knockout for specific RP genes display tissue-specific phenotypes during development (13). For instance, RPL38 is required for translation of specific HOX mRNAs, and this requirement is connected to the presence of a common functional element in the 5′ UTRs of some HOX mRNAs (14, 15). In addition to cellular mRNAs, specific RPs have been implicated as translational enhancers for viruses, such as vesicular stomatitis virus, measles virus, rabies virus (16), and hepatitis C virus (HCV) (21), suggesting that these viruses have evolved a dependency on ribosomal functions conferred by these RPs.
Previous genome-scale small interfering RNA (siRNA) screens performed by our laboratory identified host factors required for DENV-2 (New Guinea C strain) (22; N. J. Barrows et al., unpublished data) and YFV (17D strain) infection (23). Many of the identified host factors are components of the translation machinery, including RPs. Other research groups have also performed genome-scale screens to identify host factors for flaviviruses by using siRNA or CRISPR and identified components of the translation machinery, but follow-up experiments of these components were limited (24–28). Among the top candidate proteins identified as host factors for flavivirus infection of human cells in our screens were RPLP1 and RPLP2 (RPLP1/2), which form a stable heterodimer that is tethered to the ribosome by a third RP, RPLP0, to form a conserved structure known as the ribosomal stalk. The stalk acts in recruitment of elongation factor eEF2, which functions in GTP-dependent ribosomal translocation (29). However, previous studies have reported little (∼20% reduction) to no change in global translation when RPLP1/2 are depleted in cells (30–32).
Here, we show that RPLP1/2 are essential for DENV-2, YFV, and ZIKV infection. RPLP1/2 were also required for DENV-2 infection of Aedes aegypti mosquitoes, the main vector for these viruses. Investigation of the underlying mechanism revealed that RPLP1/2 promoted both the accumulation of viral protein early after infection and the accumulation of DENV-2 structural proteins in a cell-based heterologous expression assay. In contrast to viral translation, RPLP1/2 depletion resulted in milder and cell-type-specific positive or negative effects on global cellular protein synthesis. Taken together, our observations suggest that RPLP1/2 are ribosomal proteins required for flavivirus translation.
RESULTS
The RPLP1/2 heterodimer is required for DENV-2 and YFV replication.
Previous genome-scale screens from our laboratory identified RPLP1/2 in the top 0.05% of candidate host factors necessary for infection of HuH-7 cells by DENV-2 (Barrows et al., unpublished) and YFV (23). To validate the siRNA screen data, we knocked down RPLP1/2 in A549 and HuH-7 cells by using multiple independent siRNAs and infected the cells with either DENV-2 (New Guinea C) or YFV (17D) at a multiplicity of infection (MOI) of 1 for 24 h (Fig. 1). In accordance with previous reports (31), knockdown of RPLP1/2 reduced cell proliferation by about 2-fold without affecting viability as measured in a trypan blue assay (unpublished data). We noted that knockdown of either RPLP1 or RPLP2 resulted in codepletion of the other binding partner (Fig. 1A and B), in agreement with previous observations (31). Although the cells were depleted of most RPLP1/2, a fractional pool of ribosomes containing RPLP1/2 probably remains.
FIG 1.

The RPLP1/2 heterodimer and RPLP0 are required for efficient DENV-2 and YFV infection of A549 and HuH-7 cells. Cells were transfected with either a nonsilencing control siRNA (NSC) or one of five independent siRNAs used to deplete RPLP1/2, three targeting RPLP1 (siP1_1, siP1_2, and siP1_6) and two targeting RPLP2 (siP2_1 and siP2_4). After 48 h, cells were infected at an MOI of 1 and infection was assessed after 24 h. (A and B) Western blotting results show knockdown of RPLP1/2 with independent siRNAs in A549 (A) and HuH-7 (B) cells. (C) Representative images showing A549 cells infected with DENV-2 (New Guinea C). Nuclei were Hoechst stained (blue), and the viral E protein was stained with 4G2 antibody (green). (D and E) Quantification of infection rates for DENV-2 and YFV (17D) are shown for A549 (D) and HuH-7 (E) cells. (F) A549 cells were transfected with the indicated siRNAs against RPLP0 and infected with YFV 48 h later at an MOI of 1. Western blotting results show knockdown of RPLP0 with two independent siRNAs. (G) Rates of infection are shown for cells transfected with NSC or siRNAs targeting RPLP0. The error bars represent standard deviations of three biological replicates. Statistical significance was assessed by a two-tailed Student's t test between NSC and experimental siRNAs. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
After infection, cells were stained for immunofluorescence using a pan-flavivirus E protein antibody (4G2) and analyzed by high-content imaging to determine infection rates (Fig. 1C). Depletion of RPLP1/2 caused a dramatic reduction in the percentage of infected cells for both viruses and in both cell lines, in comparison to control siRNA (Fig. 1D and E). Because RPLP0 bridges the RPLP1/2 heterodimer to the ribosome, we additionally tested whether RPLP0 is required for virus replication. Knockdown of RPLP0 with two independent siRNAs strongly reduced YFV infection (Fig. 1F and G). This suggests that flaviviruses depend on RPLP1/2 bound to ribosomes to effectively replicate.
In addition to measuring infection rates by immunofluorescence, we determined the titers of virus in cell supernatants under the different experimental conditions. Measurements of infectious DENV (Fig. 2A) and YFV (Fig. 2B) showed that levels of secreted virus were reduced by up to 2 orders of magnitude in A549 cells due to RPLP1/2 knockdown. Results from HuH-7 cells showed a 15- to 30-fold reduction for DENV and 10- to 50-fold reduction for YFV. These data confirmed that RPLP1/2 are required for efficient production of infectious DENV and YFV in both cell lines analyzed.
FIG 2.

The RPLP1/2 heterodimer is required for efficient production of infectious DENV-2 and YFV. (A) Titers for supernatants from HuH-7 or A549 cells infected with DENV-2 were determined in Vero cells. (B) The same experiments as show in panel A, except with YFV. Statistical significance was assessed by a two-tailed Student's t test between NSC and experimental siRNAs. *, P < 0.05; **, P < 0.01, ***, P < 0.001.
Exogenous expression of RPLP1/2 rescues DENV-2 infection.
To rule out the possibility that off-target effects of the siRNAs contributed to reduction of virus infection, we performed rescue experiments using an expression construct resistant to siRNA siP1_1 (Fig. 3A). HuH-7 cells were transfected with RPLP1 siRNA and subsequently transfected with siRNA-resistant RPLP1 plasmid before DENV-2 infection. The cells transfected with nonsilencing control siRNA (NSC) and empty vector had 35% infection, while transfection with siRNA against RPLP1 and empty vector reduced DENV-2-infected cells to 3% (Fig. 3B and C). Transfection of siRNA-resistant RPLP1 vector rescued DENV-2 infection to 30% (Fig. 3B and C), further solidifying RPLP1/2 as critical host factors.
FIG 3.

Exogenous expression of RPLP1/2 rescues DENV-2 infection under conditions of endogenous RPLP1/2 knockdown. HuH-7 cells were transfected with the indicated siRNAs (NSC or siP1) and subsequently transfected with either empty vector (EV) plasmid or the siRNA-resistant RPLP1 (P1) expression plasmid. Cells were then infected with DENV-2 at an MOI of 1. (A) Western blotting results, showing RPLP1/2 levels after transfection with the indicated siRNA/plasmid combinations. (B) Representative immunofluorescence images of virus infection under different conditions. Nuclei were stained with Hoechst (blue), and the viral E protein was stained with 4G2 antibody (green). (C) Quantification of virus infection rates. The error bars represent standard deviations of three biological replicates. Statistical significance was assessed by a two-tailed Student's t test between the indicated conditions. ***, P < 0.001; ****, P < 0.0001.
RPLP1/2 are required for DENV-2 infection of Aedes aegypti mosquitoes.
RPLP1/2 are conserved genes that have clear homologs in the main vectors for DENV-2, mosquitoes of the Aedes genus. Within the ORF, human (CR542209.1) and A. aegypti (DQ440047.1) RPLP1 genes share 63.39% nucleotide sequence identity, whereas human (CR542212.1) and A. aegypti (DQ440065.1) RPLP2 genes have 69.37% identity. We therefore tested whether RPLP1/2 are required for DENV-2 infection of A. aegypti. Mosquitoes were injected with double-stranded RNA (dsRNA) targeting LacZ (negative control), RPLP1, or RPLP2 and orally infected with DENV-2 (ST strain) by blood meal 3 days later. After 6 days of infection, mosquitoes were homogenized for measurements of viral titers. Both dsRNAs targeting RPLP1/2 were effective at reducing the corresponding mRNA levels by at least 50%, and the RPLP2 dsRNA also reduced RPLP1 mRNA levels (Fig. 4A). No difference was observed in survival of the mosquitoes injected with the different dsRNAs, but the percentage of mosquitoes that fed from the blood meal was reduced in RPLP1 and RPLP2 knockdown mosquitoes (Table 1). For at least 21 mosquitoes that fed on the DENV-2 blood meal and survived, titers were determined for each treatment group. This analysis showed a reduction of approximately 3-fold in the number of mosquitoes with detectable virus and a significant reduction in the average titer for the RPLP1/2 knockdown groups compared to the negative-control group (Fig. 4B). These data indicate that RPLP1/2 are important for productive DENV-2 infection of A. aegypti.
FIG 4.

RPLP1/2 are required for DENV-2 infection of Aedes aegypti mosquitoes. Mosquitoes were injected with dsRNA targeting RPLP1, RPLP2, or LacZ (control) and 3 days later were offered a blood meal containing DENV-2 at 1 × 107 PFU/ml. Six days after oral infection, mosquitoes were homogenized and virus titers were determined in a cell-based plaque assay (yielding the number PFU per mosquito). Each data point represents an individual mosquito. (A) RNA levels of RPLP1/2 9 days after dsRNA injection. Statistical significance was assessed by a t test. *, P < 0.05. (B) The DENV-2 titer in each mosquito injected with dsRNA targeting LacZ, RPLP1, or RPLP2. The black lines represent the means and error bars represent standard errors of the means. Statistical significance was assessed by using the ranked nonparametric Mann-Whitney U test. *, P < 0.05; **, P < 0.01. N indicates the number of infected mosquitoes analyzed per condition.
TABLE 1.
Survival and blood feeding of control and RPLP1/2 knockdown mosquitoes
| dsRNA target | No. of mosquitoes injected | No. of mosquitoes fed blood meal | Survival of blood-fed mosquitoes |
|---|---|---|---|
| LacZ | 100 | 56 | 55/56 |
| RPLP1 | 100 | 34 | 32/34 |
| RPLP2 | 100 | 44 | 42/44 |
The RPLP1/2 heterodimer is required for replication of ZIKV.
Given the urgent need to increase our understanding of ZIKV, we asked whether RPLP1/2 are also required for ZIKV infection. We analyzed ZIKV isolates of the American lineage (isolate MEX_I_7) (33) and African lineage (isolate 41525) (34). To this end, we knocked down RPLP1/2 in A549 and HuH-7 cells and then infected the cells at an MOI of 1. Similar to our results with DENV-2 and YFV, both lineages of ZIKV were incompetent for replication in RPLP1/2-depleted cells (Fig. 5A). We observed reductions in infection rates of up to 50-fold for ZIKV isolate 41525 and 19-fold for ZIKV isolate MEX_I_7 (Fig. 5B and C), with some siRNAs reducing signal to background levels. Viral titers in supernatants were reduced by 2 orders of magnitude in A549 cells (Fig. 5B) due to the knockdown. In HuH-7 cells, titers were reduced ∼20-fold for ZIKV 41525 and by 2 orders of magnitude for ZIKV MEX_I_7 (Fig. 5C). Thus, ZIKV was also found to be strongly dependent on RPLP1/2 for infectivity.
FIG 5.

RPLP1/2 are host factors for ZIKV. A549 and HuH- 7 cells were transfected with the indicated siRNAs and infected 48 h later with ZIKV (isolate 41525 or MEX_I_7) at an MOI of 1. (A) Representative images of infected A549 cells are shown. Nuclei were Hoechst stained (blue), and the viral E protein was stained with 4G2 antibody (green). (B and C) Quantification of infected A549 (B) and HuH-7 (C) cells is shown (top graphs) as is quantifications of infectious virus in the supernatants of A549 (B) or HuH-7 (C) cells infected with ZIKV (41525 or MEX_I_7). Error bars represent standard deviations of three biological replicates. Statistical significance was assessed by a two-tailed student's t test between NSC and experimental siRNAs, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Effects of RPLP1/2 knockdown on other positive-strand RNA viruses.
To understand whether the requirement for RPLP1/2 is generalizable to other positive-strand RNA viruses, we tested the effects of RPLP1/2 knockdown on replication of HCV (JFH1 strain) (35), a hepacivirus within the Flaviviridae, and coxsackievirus B3 (CBV3; strain 20) (36), an enterovirus within the Picornaviridae. Notably, both of these viruses use internal ribosome entry sites (IRES) to drive viral translation, and HCV IRES activity was previously reported to be unaffected by RPLP1/2 levels (37). Because RPLP1/2 are both efficiently depleted when either of them is knocked down (Fig. 1A and B), we used siRNAs against RPLP2 only. For HCV, we used HuH-7.5 cells, since the parental HuH-7 cells are less permissive for replication (38). HCV and CBV3 infection rates were reduced approximately 2-fold when RPLP1/2 were depleted (Fig. 6A to D). Infection rates with HCV and CBV3 were compared side by side with that for DENV-2, which was inhibited about 25-fold in HuH-7 and 7-fold in HuH-7.5 cells. We additionally determined the titers of viruses in supernatants and found that, consistent with experiments shown above, infectious DENV was reduced at least 10-fold in RPLP1/2-depleted HuH-7 cells (Fig. 6C) and up to 10-fold in HuH-7.5 cells (Fig. 6D). In contrast, HCV and CBV3 titers were unaffected by RPLP1/2 knockdown (Fig. 6C and D). Thus, DENV-2 infection was particularly sensitive to RPLP1/2 depletion compared to the other positive-strand RNA viruses tested.
FIG 6.

Effects of RPLP1/2 knockdown on replication of CBV3 and HCV. HuH-7 or HuH-7.5 cells were transfected with siRNAs and then infected with the indicated viruses at an MOI of 1 for 8 h (CBV3), 24 h (DENV-2), or 72 h (HCV). (A) Representative images showing HuH-7 cells infected with DENV-2 or CBV3. (B) Representative images of HuH-7.5 cells infected with DENV-2 or HCV. Nuclei were Hoechst stained (blue). Shown in green are the DENV-2 E protein, CBV3 VP1 protein, or HCV core protein. (C and D) Quantification of infection rates are shown for HuH-7 (C) and HuH-7.5 cells (D). Quantifications of virus yields are shown for DENV-2 in HuH-7 (C) or HuH-7.5 (D) cells, for CBV3 in HuH-7 cells (C), and for HCV (D) in HuH-7.5 cells. Graphs show means and standard deviations of percentages of infected cells or viral yields from three biological replicates. Statistical significance was assessed by a two-tailed Student's t test between NSC and experimental siRNAs. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
The RPLP1/2 heterodimer is not required for global cellular translation.
We analyzed the effect of RPLP1/2 depletion on global translation in HuH-7 cells and A549 cells to determine whether the observed decreases of flavivirus infection could be explained by a global impairment of cellular translation. RPLP1/2 were knocked down (Fig. 7A), and then cells were metabolically labeled with [35S]methionine for 30 min. As a translation inhibition control, we also treated cells with cycloheximide (CHX). Incorporated label was analyzed by SDS-PAGE (Fig. 7B) and liquid scintillation counting (Fig. 7C). The outcome of these experiments showed opposite trends for the two cell lines. When RPLP1/2 were depleted, incorporation of labeled [35S]methionine was reduced by ∼50% in HuH-7 cells, but it was increased by approximately ∼80% in A549 cells (Fig. 7C). It is not clear why RPLP1/2 knockdown causes different effects on cellular translation in these cell lines. However, these observations are in agreement with previous reports that showed variable effects on cellular translation upon RPLP1/2 depletion (30–32). Importantly, these data indicate that the potent reductions we observed in flavivirus infectivity upon RPLP1/2 knockdown cannot be explained by a generalized inhibition of cellular translation.
FIG 7.

Depletion of RPLP1/2 results in cell-line-specific effects on global translation. Cells were transfected with siRNAs and 48 h later were incubated with methionine-free medium for 30 min before addition of [35S]methionine for 30 min. Cells were subsequently lysed, and proteins were precipitated using TCA. CHX was used to control for background signal after TCA precipitation. (A) Western blotting results, showing knockdown of RPLP1/2 in HuH-7 and A549 cells. Only antibody against RPLP1 was used for the Western blot assay with HuH-7 cells. (B) Autoradiography of 35S-labeled proteins fractionated by SDS-PAGE. (C) Liquid scintillation counting of incorporated 35S-labeled proteins. For each cell line, the NSC results was set to 100%. The graphs show mean values of two independent experiments for HuH-7 cells and three independent experiments for A549 cells. Error bars represent standard deviation measurements. Statistical significance was assessed by a two-tailed Student's t test between NSC and other conditions. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
RPLP1/2 are required for accumulation of DENV-2 proteins early after infection.
We next used a recombinant DENV-2 virus (New Guinea C strain) engineered to express Renilla luciferase (RLUC) (39) to directly test whether RPLP1/2 promote early viral protein accumulation, as would be expected if these proteins are important for translation of viral RNA. Control and RPLP1/2-depleted A549 cells (Fig. 8A) were infected at an MOI of 1, and viral RNA levels and luciferase accumulation were assessed at 4 h postinfection (hpi). There was no effect of RPLP1/2 knockdown on viral RNA levels compared to the negative control (Fig. 8B). In contrast, we observed a strong reduction of RLUC levels at 4 hpi in RPLP1/2 knockdown cells (Fig. 8C), indicating an early defect in the virus life cycle. To ascertain whether RNA replication contributed to RLUC levels, we performed the same assay in the presence of NITD008, a potent NS5 inhibitor (40), and observed similar results (Fig. 8C), suggesting that RNA replication did not substantially contribute to RLUC levels at 4 hpi. These data are consistent with a requirement for RPLP1/2 in early DENV translation.
FIG 8.
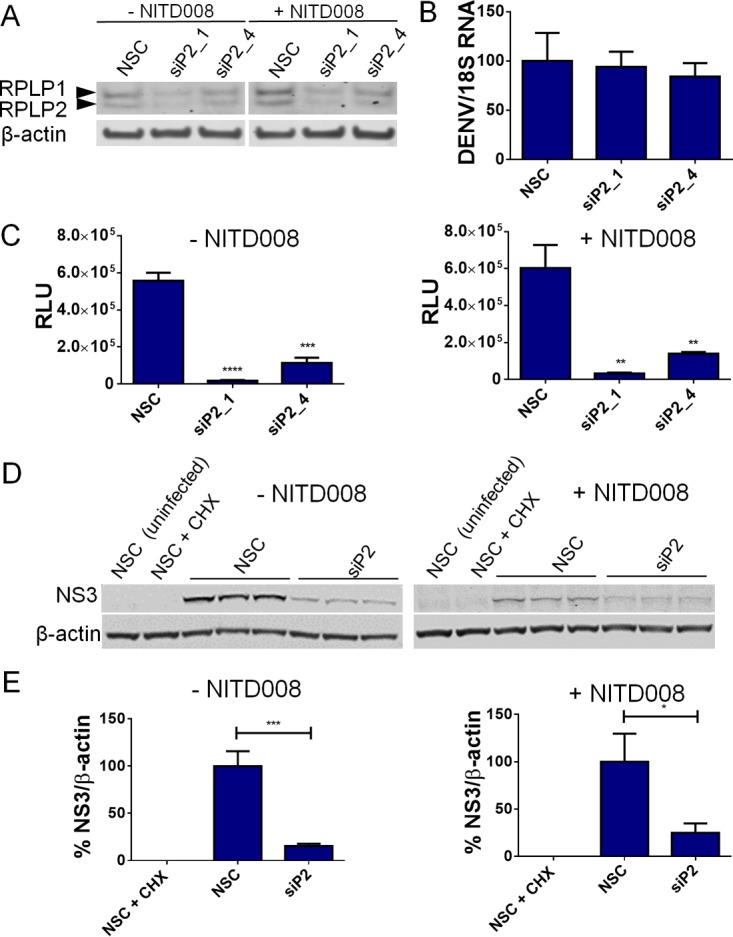
RPLP1/2 are required for early viral protein accumulation. (A) Knockdown of RPLP1/2 is shown by Western blotting of A549 cells with or without NITD008 treatment. (B) Quantification of DENV-2 RNA by RT-qPCR normalized to 18S rRNA is shown under control and RPLP1/2 knockdown conditions. (C) Luciferase measurements from infected cells harvested 4 hpi are shown for each siRNA transfection condition in the presence or absence or NITD008 treatment. (D) A549 cells were transfected with NSC siRNA or siRNA against RPLP2 and then infected with DENV-2 for 6 h at an MOI of 10 in the presence or absence of NITD008. NS3 accumulation was detected by Western blotting, and results for triplicate samples are shown for NSC and siP2 conditions. CHX-treated and uninfected samples served as controls for background signal. (E) Quantification of NS3 levels was calculated by normalizing results to those for β-actin. The NSC condition result was set to 100%. Error bars represent standard deviation measurements of three independent wells. Statistical significance was assessed by a two-tailed Student's t test between NSC and experimental siRNA conditions. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We next examined NS3 accumulation in control and RPLP1/2-depleted A549 cells infected with wild-type DENV-2. We chose to examine NS3 levels because of the high sensitivity achieved with the anti-NS3 antibody used (see Materials and Methods). DENV-2 NS3 protein accumulation was analyzed at 6 hpi in control and RPLP1/2-depleted cells infected at an MOI of 10 (Fig. 8D). We used CHX treatment to control for background signal. Quantitative Western blot analysis revealed a reduction of ∼85% in NS3 levels in cells transfected with RPLP2 siRNA compared to results with negative-control siRNA (Fig. 8D and E). We also performed experiments in which cells were treated with NITD008 to restrict viral RNA synthesis. In the presence of NITD008, RPLP2 depletion similarly reduced NS3 levels (∼75%) (Fig. 8D and E). Together with data from the luciferase reporter DENV-2 assay, these observations show that RPLP1/2 promote an early stage of DENV-2 infection and suggest that efficient DENV-2 translation requires RPLP1/2.
RPLP1/2 are required for accumulation of DENV-2 structural proteins independently of virus infection.
We next tested whether RPLP1/2 are required for accumulation of DENV-2 proteins under conditions in which viral entry is bypassed. To address this, we isolated stable HeLa and HEK293 cell lines with cytomegalovirus (CMV) promoter-driven transgenes that express DENV-2 C-prM-E structural proteins in a tetracycline-inducible manner (Fig. 9A). In these cells, the E protein is cleaved from C-prM by cellular signalase. In contrast, the capsid protein is not efficiently cleaved from prM, because this cleavage requires NS3-mediated processing of capsid on the cytosolic face of the endoplasmic reticulum (41). Cells were transfected with control or RPLP2 siRNAs, expression of C-prM-E RNA was measured by quantitative reverse transcription-PCR (RT-qPCR), and structural proteins were measured by Western blotting with C-prM and E antibodies (Fig. 9B and C). We observed slight or no reduction in C-prM-E RNA levels upon knockdown of RPLP1/2 in HeLa or HEK-293 cells, respectively (Fig. 9B and C). For C-prM, we observed two protein bands at 33 and 37 kDa (Fig. 9A). Based on published data (42), these bands are consistent with the unglycosylated and glycosylated forms of C-prM protein, respectively. In contrast to RNA levels, the 37-kDa band was reduced in RPLP1/2-depleted cells in comparison to control siRNA-transfected cells in both cell types. We also observed reduction of the 33-kDa band with RPLP1/2 depletion in HEK-293 cells (Fig. 9C), but there was no consistent reduction of the 33-kDa band in HeLa cells (Fig. 9B). For E protein, we observed a 4- to 5-fold reduction in protein levels due to RPLP1/2 knockdown in both HeLa and HEK-293 cells (Fig. 9B and C). As RPLP1/2 have been implicated in translation elongation (29, 43), these data raise the possibility that ribosomal elongation through structural protein coding sequences may be impaired in cells with reduced RPLP1/2 levels, with the strongest effects on accumulation of full-length E protein.
FIG 9.

RPLP1/2 knockdown impairs accumulation of DENV-2 structural proteins expressed in stable cell lines. Tetracycline-inducible HeLa and HEK-293 cells expressing C-prM-E were transfected with the indicated siRNAs, and 48 h later tetracycline was added to the medium for 24 h. Cell lysates were harvested for Western blot analysis 24 h after addition of tetracycline. (A) Representative Western blots showing expression of C-prM and E proteins under NSC or siP2_4 transfection conditions (performed in triplicate). The arrows indicate cleaved E protein, the 37-kDa C-prM minority species, and the 33-kDa C-prM majority species. Samples from uninduced cells are also indicated (-tet). (B) Quantifications of C-prM-E RNA by RT-qPCR normalized to 18S rRNA and protein bands in the Western blot assay from HeLa cells, normalized to results with β-actin. The NSC conditions were set to 100%. Graphs show means and standard deviations from quantification of two independent assays performed in triplicate. (C) The same experiment in shown in panel B, except HEK-293 cells were used. Graphs show means from the quantification of three independent assay results. Statistical significance was assessed by a two-tailed Student's t test between NSC and experimental siRNA conditions. *, P < 0.05; ****, P < 0.0001.
DISCUSSION
RPs have emerged as pivotal regulators controlling translation of viral and cellular RNAs. Here, we show that ribosomal stalk proteins are crucial host factors for replication of multiple flaviviruses in human cells. RPLP1/2 were also required for DENV-2 infection of A. aegypti mosquitoes, highlighting a conserved role for these host factors across different species. RPLP1/2 were not essential for protein synthesis in human cells, consistent with previous reports (30–32). Two lines of evidence implicate RPLP1/2 as drivers of flavivirus translation: (i) the early accumulation of DENV-2-expressed luciferase and NS3 was reduced in RPLP1/2 knockdown cells after infection, and (ii) RPLP1/2 depletion reduced expression levels of structural proteins in heterologous cell-based assays that bypass viral entry. Taken together, these results are consistent with an essential role for RPLP1/2 in translation of viral RNA.
Translation elongation is generally thought to be less prone to regulation than the initiation phase, although recent studies have challenged this view (44–48). The observation that DENV structural proteins expressed from a chromosomally encoded, CMV promoter-driven transgene were reduced by RPLP1/2 knockdown suggests that translation elongation through the structural protein coding region could be defective when RPLP1/2 are limiting. The fact that E protein levels were more strongly reduced by RPLP1/2 knockdown than the 33-kDa C-prM protein could reflect ribosomal stalling within the E protein-coding region (49). We also observed that accumulation of the 37-kDa C-prM protein species was reduced in both cell lines when RPLP1/2 were depleted. It is possible that this is due to reduced posttranslational modification of prM as a consequence of protein misfolding, which can be caused by a possible altered translation elongation (50, 51).
Diverse viruses have evolved to hijack specific RPs to achieve optimal viral protein synthesis. For example, RPL40 has been shown to promote efficient translation initiation for vesicular stomatitis virus, measles virus, and rabies virus, but is not required for global protein synthesis (16). RPL18 knockdown was shown to inhibit an early stage of DENV-2 infection without affecting global cellular translation or cell viability (52). RPS25 appears to be broadly required for IRES-mediated translation initiation of HCV, human T-cell leukemia virus 1, Cricket paralysis virus, and poliovirus, as well as ribosome shunting in adenovirus infection (21, 53, 54). For HCV, RPs of the small subunit were found to be disproportionately required for translation and virus replication compared to RPs of the large subunit (55), suggesting that the rate-limiting step of HCV translation is at the stage of 40S subunit recruitment. Interestingly, this appears to contrast with DENV-2 (Barrows et al., unpublished) and YFV (23), for which we have observed an overrepresentation of RPs from the large subunit in host factors that are required for infection. This suggests that elongation or joining of the 60S subunit during initiation is the rate-limiting step for flavivirus translation.
RPLP1/2 participate in formation of the ribosomal stalk, which has been implicated in recruitment of translation elongation factors. The stalk proteins have been shown to interact with human eEF2 (29) and eEF1α in other species (56), and eEF2's ribosome-dependent GTPase activity has been functionally linked to RPLP1/2. Despite being implicated as constitutive drivers of translation elongation, there are a few previous studies that reported contrasting roles for RPLP1/2 in translation and/or infectivity of other RNA viruses in different model systems. L-A virus showed enhanced propagation in yeast strains lacking RPLP1/2 homologs, and this effect was hypothesized to be linked to increased translation (57). Foot-and-mouth disease virus IRES activity was elevated in cells with depleted RPLP1/2 but only in the presence of viral Lb protease (37). In parallel, the HCV IRES was unaffected by RPLP1/2 knockdown (37), suggesting that the reduction in HCV infection that we observed after RPLP1/2 knockdown is likely not due to defective HCV IRES function. These examples illustrate that protein synthesis may be enhanced for some viruses in the context of RPLP1/2 depletion, contrasting with effects on flaviviruses.
While some RPs are important for basic ribosomal functions and/or ribosome biogenesis, other RPs have been shown to regulate translation of specific cellular mRNAs through promoting translation, primarily at the initiation stage. We speculate that RPLP1/2 are important for translation of a unique subset of cellular mRNAs that share currently undefined features with flavivirus genomes. 5′ and 3′ untranslated regions of mRNAs have been shown to play key roles in translational regulation, especially during initiation (58). Features in the ORF, such as the cHP in DENV, have also been described to regulate translation initiation (8), but they appear to be less common. In contrast, translation elongation can be regulated by features contained within the ORF or nascent polypeptide (45, 47, 48). For example, codon usage (47), RNA structure (59, 60), and certain nascent polypeptide sequences can alter the rate of ribosomal transit (45). It will be of interest to identify those features of flaviviral and cellular RNAs that confer a dependency on function of the ribosomal stalk proteins.
Ribosomes with different RP compositions may help tailor protein synthesis to a given tissue or environmental condition (61). As some disorders have been linked to RP expression levels or gene mutations (62, 63), uncovering how each RP affects translational status of diverse cellular mRNAs should increase our grasp of underlying disease mechanisms. Relatedly, understanding how RPs interface with viruses could spur development of novel therapeutic strategies that target viral translation.
MATERIALS AND METHODS
Cell culture and viruses.
Vero, A549, HuH-7, and HuH-7.5 cells (38) (kindly provided by Charles Rice, Rockefeller University) were grown in complete Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator at 37°C with 5% CO2. C6/36 cells were grown in RPMI supplemented with 10% fetal bovine serum, nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml and propagated at 28°C with 5% CO2. Tetracycline-inducible cell lines were established with the Flp-In T-REx system (Thermo Fisher Scientific) according to the manufacturer's protocol. After introduction of transgenes, HEK-293 Flp-In T-REx cells were grown in medium with 100 μg/ml of hygromycin B and 15 μg/ml of blasticidin, and HeLa Flp-In T-Rex cells (64) (kindly provided by Elena Dobrikova, Duke University) were grown in medium with 100 μg/ml of hygromycin B and 2 μg/ml of blasticidin. Preparation of DENV-2 (New Guinea C) and YFV (17D) stocks has been previously described (22). The Renilla luciferase reporter–DENV-2 construct (39) was produced by RNA transfection as described previously and passaged once in C6/36 cells. ZIKV isolates were kindly provided by Scott Weaver (UTMB) and Nikos Vasilakis (UTMB) and produced in C6/36 (MEX_I_7) or Vero (41525) cells. HCV (JFH1 strain) (35) was produced in HuH-7.5 cells, and coxsackievirus B3 (strain 20) (36) was produced in HeLa cells. The supernatants were collected at 8 hpi for CBV3, 24 hpi for DENV-2 and ZIKV 41525, and 72 hpi for ZIKV MEX_I_7 and HCV. Flavivirus titers were determined via focus-forming assays in Vero cells as previously described (22). HCV and CBV3 titers were determined based on the 50% tissue culture infective dose (TCID50) using the Reed and Muench method (65) for HuH-7.5 or HeLa cells, respectively.
Cloning of expression constructs.
RPLP1 open reading frames were amplified by RT-PCR from HuH-7 cells and cloned into pcDNA5/FRT/TO plasmid (Thermo Fisher Scientific) by using BamHI and NotI. The primers used to amplify RPLP1 were as follows: forward (5′-GAGGAT CCACCA TGGCCT CTGTCT CCGAGC TCGCCC GCATCT ACTC-3′) and reverse (5′-GAGCGG CCGCTT AGTCAA AAAGAC CAAAGC CCATGT C-3′). The RPLP1 construct was mutated by overlap PCR to make it resistant to siP1_1 siRNA. Primers used in overlap PCR were forward (5′-TCCAGC TGAGGA GAAGAA AGTGGA AGCTAA AAAAGA AGAATC CGAGG-3′) and reverse (5′-CATCAT CAGACT CCTCGG ATTCTT CTTTTT TAGCTT CCAC-3′). The C-prM-E construct, corresponding to nucleotides 97 to 2421 (AF038403.1), was cloned in pcDNA5/FRT/TO using NotI and XhoI restriction sites.
Western blotting.
Samples were lysed in RIPA buffer (Cell Signaling Technologies) or lysis buffer [400 mM KOAc, 25 mM HEPES (pH 7.2), 15 mM Mg(OAc)2, 1% (vol/vol) IGEPAL ca-630, 1× protease inhibitor (Roche)]. Proteins were fractionated under denaturing conditions on 4-to-12% acrylamide gels (Novex, Thermo Fisher Scientific). Antibodies used were anti-RPLP1 (Ab121190; Abcam), anti-RPLP2 (Ab154958; Abcam), anti-mouse beta-actin (sc-47778; Santa Cruz Biotechnology), anti-DENV-2 NS3 (GTX124252; GeneTex), anti-DENV-2 E (GTX127277; GeneTex), and anti-DENV-2 C (GTX103343; GeneTex).
Transfections.
Plasmid transfections were done using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer's instructions, and medium was changed 5 h after transfection. siRNA transfections were done using RNAiMAX reagent following the manufacturer's instructions. All siRNAs used were purchased from Qiagen. RPLP1 and RPLP2 were knocked down by forward transfection using 10 nM siRNA for experiments shown in Fig. 1, 4, and 5 and using 30 nM siRNA for all other experiments.
Immunofluorescence staining and high-content imaging.
To assess percentages of infected cells, 5 × 104 cells were plated per well in 24-well plates. The next day, cells were transfected with siRNAs against RPLP1, RPLP2, or nonsilencing control siRNA (siAllStars; Qiagen). Two days later, cells were infected with virus for 8 h (CBV3), 24 h (DENV-2, YFV, and ZIKV), or 72 h (HCV). After infection, cells were fixed with cold methanol and then stained using 4G2 antibody (DENV-2, YFV, and ZIKV) (66), CBV3 VP1 antibody (Dako), or HCV core C7-50 antibody (Abcam). Secondary Alexa Fluor 488 antibody (Thermo Fisher Scientific) and Hoechst 33342 (Sigma) were used to visualize infected cells. Cells were imaged, and infection rates were quantified using a high-content imaging microscope (Opera Phenix [Perkin-Elmer] or ArrayScan VTI [Thermo Fisher Scientific]).
Rescue using siRNA-resistant expression constructs.
For the rescue assays, cells were plated as described above and 1 day later transfected with 30 nM RPLP1 siRNA. After 24 h, 800 ng of RPLP1 (resistant to siP1_1) plasmid was transfected using 2 μl Lipofectamine 2000 per well. After 24 h, aliquots of cells were split to a new plate and infected the following day with DENV-2 at an MOI of 1 for 24 h before fixation and staining as described above.
Metabolic labeling.
To assess bulk translation in RPLP1/2 knockdown cells, metabolic labeling assays with [35S]methionine were performed. A549 or HuH-7 cells were plated at 3 × 105 cells per well in 6-well plates. The following day, cells were transfected with siRNAs and after 48 h incubated in methionine-free medium (Thermo Fisher Scientific) for 20 min. Cells were then labeled with medium containing 0.2 mCi of [35S]methionine for 30 m, washed with phosphate-buffered saline (PBS) three times, and lysed in 0.2 ml lysis buffer. We treated one sample with CHX (0.2 mM) 5 min prior and during labeling to control for background signal. Proteins were precipitated with trichloroacetic acid (TCA), homogenized in 1× sample loading buffer, and analyzed by SDS-PAGE or liquid scintillation counting.
Luciferase reporter virus infections and monitoring early NS3 accumulation.
A549 cells were plated in 24-well plates at 5 × 104 cells per well. One day later, cells were transfected with siRNA as described above. After 48 h, cells were trypsinized and counted, and equal numbers of cells were plated for each infection condition. Cells were treated with 20 μM NITD008 for 2 h before infection with an MOI of 1, and NITD008 was retained in the medium during the course of infection. After 4 h, cells were washed five times with PBS and then lysed with Renilla lysis buffer (Promega). Luciferase assays were performed using an EnSpire plate reader (PerkinElmer). For NS3 analysis, A549 cells were plated in 6-well plates at 3 × 105 cells/well and transfected the following day with siRNAs. After 48 h, cells were infected with DENV-2 at an MOI of 10 in the presence or absence of NITD008 or CHX, as indicated. After 6 h of infection, cells were lysed in 70 μl lysis buffer per well and analyzed by Western blotting using NS3 antibody.
Viral RNA quantification by RT-qPCR.
RNA was extracted using the ReliaPrep kit (Promega) and reverse transcribed using the High-Capacity cDNA kit (Thermo Fisher Scientific). qPCR was performed with SYBR mix on a StepOne Plus instrument (Applied Biosystems) to measure DENV-2 RNA and 18S rRNA. The ΔΔCT method was used to calculate relative expression levels. The primers used for DENV-2 were forward (5′-GAAATG GGTGCC AACTTC AAGGCT-3′) and reverse (5′-TCTTTG TGCTGC ACTAGA GTGGGT-3′), which amplify nucleotides 5755 to 5892 of the genome (AF038403.1). For amplification of C-prM-E RNA, the primers used were forward (5′-ACTGTA CAACAG CTGACA AAGA-3′) and reverse (5′-TGCGTC TCCTGT TCAAGA TG-3′). For 18S amplification the primers used were forward (5′-GTAACC CGTTGA ACCCCA TT-3′) and 18S reverse (5′-CCATCC AATCGG TAGTAG CG-3′).
Mosquito experiments.
A colony of A. aegypti mosquitoes collected in Singapore and established in 2014 was used. Larvae were fed a mix of fish food (TetraMin Crisps Pro) and liver powder (MP Biomedicals), while adults were held in a rearing cage (Bioquip) supplemented with 10% sucrose solution. Mosquitoes were maintained in the insectary at 28°C and 50% humidity on a 12-h:12-h dark:light cycle. To deplete RPLP1 (UniProt accession number AAEL003530) and RPLP2 (accession number AAEL014583), dsRNA targeting RPLP1 and RPLP2 were produced from PCR-amplified fragments and using the following two primer sets, with all primers being flanked on the 5′ side with T7 promoter: dsRNA-RPLP1 forward (5′-TACTTC CGTTTT TGCGAC CT-3′) and dsRNA-RPLP1 reverse (5′-TTCAGC TTTGTT GAGAGC CA-3′); dsRNA-RPLP2 forward (5′-TGAACG TCCAAA CAAAAT GC-3′) and dsRNA-RPLP2 reverse (5′-GATTTG CCCTTCAGCTCG T-3′). dsRNA targeting LacZ was produced from an amplicon containing the LacZ sequence (67) and the following set of primers, which were also flanked on the 5′ side with the T7 promoter: dsRNA-LacZ forward (5′-TACCCG TAGGTA GTCACG CA-3′) and dsRNA-LacZ reverse (5′-TACGAT GCGCCC ATCTAC AC-3′). dsRNA was generated and purified using the MEGAscript T7 kit (Thermo Fisher Scientific) and the EZNA total RNA kit I (Omega Bio-Tek). RNA samples adjusted to 3 μg/μl were annealed by heating to 95°C and slowly cooling. dsRNAs were injected (69-nl aliquots) into the thorax of cold-anesthetized 3- to 5-day-old mosquitoes by using a glass capillary mounted onto a Nanoject II injector (Drummond). Sequence identity between human and mosquito RPLP1/2 genes was calculated using the Muscle alignment tool (version 3.8.31).
To quantify the mRNA levels of RPLP1 and RPLP2, total RNA was extracted from 10 mosquitoes at 9 days post-dsRNA injection by using the EZNA total RNA kit I. Reverse transcription was performed with an iScript cDNA synthesis kit (Bio-Rad), and gene expression was quantified using the SensiFAST SYBR No-ROX kit (Bioline) with the following sets of primers: RPLP1 forward (5′-ACCGGG ATTACG TTGGAA CC-3′) and RPLP1 reverse (5′-CGAATG TGGTGC TGTTAG CG-3′); RPLP2 forward (5′-GACGAC ATGGGA TTCGGT C-3′) and RPLP2 reverse (5′-TATTTG GCGGAT TTTGGG CG-3′). Amplification was conducted in a CFX96 Touch real-time PCR detection system (Bio-Rad). Actin gene (UniProt accession number AAEL011197) quantification was used for normalization and quantified following the same protocol as described above with the primers actin forward (5′-GAACA CCCAG TCCTG CTGAC A-3′) and actin reverse (5′-TGCGT CATCT TCTCA CGGTT AG-3′).
Mosquitoes were infected with the DENV-2 strain ST (68) propagated in Vero cells. Three days after dsRNA injection, 24-h-starved mosquitoes were offered a blood meal comprised of 40% washed erythrocytes from specific-pathogen-free pig's blood (PWG Genetics), 5% 5 mM ATP (Thermo Fisher Scientific), 5% human serum (Sigma), and a 50% volume of RPMI containing virus diluted to 107 PFU/ml. Blood titers were validated in a plaque assay as described previously (69). The infectious blood meal was maintained at 37°C using a hemotek feeder system (Discovery Workshops) covered by a stretched pig's intestine. Mosquitoes were allowed to feed for 2 h. Engorged females were selected and maintained with access to sugar in the insectary. Six days post-oral infection, individual mosquitoes were homogenized in 500 μl of RPMI, filtered through a 0.22-μm filter (Sartorius) and determined the titer of by plaque assay (69).
ACKNOWLEDGMENTS
We thank our colleagues from the Bradrick and Garcia-Blanco laboratory, University of Texas Medical Branch, and Duke University for their support. We thank Gaddiel Galarza-Muñoz (UTMB) and Andrew Routh (UTMB) for comments on the manuscript. We have no conflicts of interest to disclose.
R.K.C., M.A.G.-B., and S.S.B. conceived and designed the experiments. R.K.C., S.S.B., and Y.-F.L. performed the experiments, except for the experiment involving mosquitoes, which was performed by B.W. and J.P. R.K.C., B.W., X.X., Y.-F.L., P.-Y.S., J.P., M.A.G.-B., and S.S.B. analyzed and interpreted the data. R.K.C., M.A.G.-B., and S.S.B. wrote the paper.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilder-Smith A, Byass P. 2016. The elusive global burden of dengue. Lancet Infect Dis 16:629–631. doi: 10.1016/S1473-3099(16)00076-1. [DOI] [PubMed] [Google Scholar]
- 3.Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, Shi PY, Vasilakis N. 2016. Zika virus: history, emergence, biology, and prospects for control. Antiviral Res 130:69–80. doi: 10.1016/j.antiviral.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burki T. 2016. Yellow fever in Africa: a disaster waiting to happen. Lancet Infect Dis 16:896–897. doi: 10.1016/S1473-3099(16)30224-9. [DOI] [PubMed] [Google Scholar]
- 5.Garske T, Van Kerkhove MD, Yactayo S, Ronveaux O, Lewis RF, Staples JE, Perea W, Ferguson NM, Yellow Fever Expert C. 2014. Yellow fever in Africa: estimating the burden of disease and impact of mass vaccination from outbreak and serological data. PLoS Med 11:e1001638. doi: 10.1371/journal.pmed.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green A. 2016. Yellow fever continues to spread in Angola. Lancet 387:2493. doi: 10.1016/S0140-6736(16)30835-2. [DOI] [PubMed] [Google Scholar]
- 7.Friebe P, Harris E. 2010. Interplay of RNA elements in the dengue virus 5′ and 3′ ends required for viral RNA replication. J Virol 84:6103–6118. doi: 10.1128/JVI.02042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clyde K, Harris E. 2006. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J Virol 80:2170–2182. doi: 10.1128/JVI.80.5.2170-2182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI, Gamarnik AV. 2005. Long-range RNA-RNA interactions circularize the dengue virus genome. J Virol 79:6631–6643. doi: 10.1128/JVI.79.11.6631-6643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edgil D, Polacek C, Harris E. 2006. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J Virol 80:2976–2986. doi: 10.1128/JVI.80.6.2976-2986.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polacek C, Friebe P, Harris E. 2009. Poly(A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J Gen Virol 90:687–692. doi: 10.1099/vir.0.007021-0. [DOI] [PubMed] [Google Scholar]
- 12.Khatter H, Myasnikov AG, Natchiar SK, Klaholz BP. 2015. Structure of the human 80S ribosome. Nature 520:640–645. doi: 10.1038/nature14427. [DOI] [PubMed] [Google Scholar]
- 13.Shi Z, Barna M. 2015. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu Rev Cell Dev Biol 31:31–54. doi: 10.1146/annurev-cellbio-100814-125346. [DOI] [PubMed] [Google Scholar]
- 14.Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, Barna M. 2011. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145:383–397. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue S, Tian S, Fujii K, Kladwang W, Das R, Barna M. 2015. RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature 517:33–38. doi: 10.1038/nature14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee AS, Burdeinick-Kerr R, Whelan SP. 2013. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc Natl Acad Sci U S A 110:324–329. doi: 10.1073/pnas.1216454109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen-Lefebvre AT, Leprun G, Morin V, Vinuelas J, Coute Y, Madjar JJ, Gandrillon O, Gonin-Giraud S. 2013. V-erbA generates ribosomes devoid of RPL11 and regulates translational activity in avian erythroid progenitors. Oncogene 33:1581–1589. doi: 10.1038/onc.2013.93. [DOI] [PubMed] [Google Scholar]
- 18.Jiang N, Hu L, Liu C, Gao X, Zheng S. 2015. 60S ribosomal protein L35 regulates beta-casein translational elongation and secretion in bovine mammary epithelial cells. Arch Biochem Biophys 583:130–139. doi: 10.1016/j.abb.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Bortoluzzi S, d'Alessi F, Romualdi C, Danieli GA. 2001. Differential expression of genes coding for ribosomal proteins in different human tissues. Bioinformatics 17:1152–1157. doi: 10.1093/bioinformatics/17.12.1152. [DOI] [PubMed] [Google Scholar]
- 20.Ishii K, Washio T, Uechi T, Yoshihama M, Kenmochi N, Tomita M. 2006. Characteristics and clustering of human ribosomal protein genes. BMC Genomics 7:37. doi: 10.1186/1471-2164-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landry DM, Hertz MI, Thompson SR. 2009. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 23:2753–2764. doi: 10.1101/gad.1832209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sessions OM, Barrows NJ, Souza-Neto JA, Robinson TJ, Hershey CL, Rodgers MA, Ramirez JL, Dimopoulos G, Yang PL, Pearson JL, Garcia-Blanco MA. 2009. Discovery of insect and human dengue virus host factors. Nature 458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Sommer C, Barrows NJ, Bradrick SS, Pearson JL, Garcia-Blanco MA. 2012. G protein-coupled receptor kinase 2 promotes Flaviviridae entry and replication. PLoS Negl Trop Dis 6:e1820. doi: 10.1371/journal.pntd.0001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, Carette JE. 2016. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS. 2016. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535:164–168. doi: 10.1038/nature18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, Guo Z, Green S, Kowalik TF, Brass AL. 2016. Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, Montgomery RR, Lev S, Mason PW, Koski RA, Elledge SJ, Xavier RJ, Agaisse H, Fikrig E. 2008. RNA interference screen for human genes associated with West Nile virus infection. Nature 455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yasunaga A, Hanna SL, Li J, Cho H, Rose PP, Spiridigliozzi A, Gold B, Diamond MS, Cherry S. 2014. Genome-wide RNAi screen identifies broadly-acting host factors that inhibit arbovirus infection. PLoS Pathog 10:e1003914. doi: 10.1371/journal.ppat.1003914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bargis-Surgey P, Lavergne JP, Gonzalo P, Vard C, Filhol-Cochet O, Reboud JP. 1999. Interaction of elongation factor eEF-2 with ribosomal P proteins. Eur J Biochem 262:606–611. doi: 10.1046/j.1432-1327.1999.00434.x. [DOI] [PubMed] [Google Scholar]
- 30.Perucho L, Artero-Castro A, Guerrero S, Ramon y Cajal S, LLeonart ME, Wang ZQ. 2014. RPLP1, a crucial ribosomal protein for embryonic development of the nervous system. PLoS One 9:e99956. doi: 10.1371/journal.pone.0099956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Azorin F, Remacha M, Ballesta JP. 2008. Functional characterization of ribosomal P1/P2 proteins in human cells. Biochem J 413:527–534. doi: 10.1042/BJ20080049. [DOI] [PubMed] [Google Scholar]
- 32.Artero-Castro A, Perez-Alea M, Feliciano A, Leal JA, Genestar M, Castellvi J, Peg V, Ramon YCS, Lleonart ME. 2015. Disruption of the ribosomal P complex leads to stress-induced autophagy. Autophagy 11:1499–1519. doi: 10.1080/15548627.2015.1063764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guerbois M, Fernandez-Salas I, Azar SR, Danis-Lozano R, Alpuche-Aranda CM, Leal G, Garcia-Malo IR, Diaz-Gonzalez EE, Casas-Martinez M, Rossi SL, Del Rio-Galvan SL, Sanchez-Casas RM, Roundy CM, Wood TG, Widen SG, Vasilakis N, Weaver SC. 2016. Outbreak of Zika virus infection, Chiapas State, Mexico, 2015, and first confirmed transmission by Aedes aegypti mosquitoes in the Americas. J Infect Dis 214:1349–1356. doi: 10.1093/infdis/jiw302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ladner JT, Wiley MR, Prieto K, Yasuda CY, Nagle E, Kasper MR, Reyes D, Vasilakis N, Heang V, Weaver SC, Haddow A, Tesh RB, Sovann L, Palacios G. 2016. Complete genome sequences of five Zika virus isolates. Genome Announc 4:e00377-15. doi: 10.1128/genomeA.00377-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tracy S, Chapman NM, Tu Z. 1992. Coxsackievirus B3 from an infectious cDNA copy of the genome is cardiovirulent in mice. Arch Virol 122:399–409. doi: 10.1007/BF01317202. [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Azorin F, Remacha M, Martinez-Salas E, Ballesta JP. 2008. Internal translation initiation on the foot-and-mouth disease virus IRES is affected by ribosomal stalk conformation. FEBS Lett 582:3029–3032. doi: 10.1016/j.febslet.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 38.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou G, Xu HY, Qing M, Wang QY, Shi PY. 2011. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res 91:11–19. doi: 10.1016/j.antiviral.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SB, Goh A, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KA, Garrett C, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A 106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amberg SM, Nestorowicz A, McCourt DW, Rice CM. 1994. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: in vitro and in vivo studies. J Virol 68:3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markoff L. 1989. In vitro processing of dengue virus structural proteins: cleavage of the pre-membrane protein. J Virol 63:3345–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veit G, Oliver K, Apaja PM, Perdomo D, Bidaud-Meynard A, Lin ST, Guo J, Icyuz M, Sorscher EJ, Hartman Iv JL, Lukacs GL. 2016. Ribosomal stalk protein silencing partially corrects the ΔF508-CFTR functional expression defect. PLoS Biol 14:e1002462. doi: 10.1371/journal.pbio.1002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richter JD, Coller J. 2015. Pausing on polyribosomes: make way for elongation in translational control. Cell 163:292–300. doi: 10.1016/j.cell.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR. 2015. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep 11:13–21. doi: 10.1016/j.celrep.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorochowski TE, Ignatova Z, Bovenberg RA, Roubos JA. 2015. Trade-offs between tRNA abundance and mRNA secondary structure support smoothing of translation elongation rate. Nucleic Acids Res 43:3022–3032. doi: 10.1093/nar/gkv199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu CH, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y. 2015. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell 59:744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gamble CE, Brule CE, Dean KM, Fields S, Grayhack EJ. 2016. Adjacent codons act in concert to modulate translation efficiency in yeast. Cell 166:679–690. doi: 10.1016/j.cell.2016.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolin SL, Walter P. 1988. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J 7:3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodnina MV, Wintermeyer W. 2016. Protein elongation, co-translational folding and targeting. J Mol Biol 428:2165–2185. doi: 10.1016/j.jmb.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 51.Tsai CJ, Sauna ZE, Kimchi-Sarfaty C, Ambudkar SV, Gottesman MM, Nussinov R. 2008. Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J Mol Biol 383:281–291. doi: 10.1016/j.jmb.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cervantes-Salazar M, Angel-Ambrocio AH, Soto-Acosta R, Bautista-Carbajal P, Hurtado-Monzon AM, Alcaraz-Estrada SL, Ludert JE, Del Angel RM. 2015. Dengue virus NS1 protein interacts with the ribosomal protein RPL18: this interaction is required for viral translation and replication in Huh-7 cells. Virology 484:113–126. doi: 10.1016/j.virol.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 53.Hertz MI, Landry DM, Willis AE, Luo G, Thompson SR. 2013. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol Cell Biol 33:1016–1026. doi: 10.1128/MCB.00879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olivares E, Landry DM, Caceres CJ, Pino K, Rossi F, Navarrete C, Huidobro-Toro JP, Thompson SR, Lopez-Lastra M. 2014. The 5′ untranslated region of the human T-cell lymphotropic virus type 1 mRNA enables cap-independent translation initiation. J Virol 88:5936–5955. doi: 10.1128/JVI.00279-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang JY, Su WC, Jeng KS, Chang TH, Lai MM. 2012. Attenuation of 40S ribosomal subunit abundance differentially affects host and HCV translation and suppresses HCV replication. PLoS Pathog 8:e1002766. doi: 10.1371/journal.ppat.1002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ito K, Honda T, Suzuki T, Miyoshi T, Murakami R, Yao M, Uchiumi T. 2014. Molecular insights into the interaction of the ribosomal stalk protein with elongation factor 1α. Nucleic Acids Res 42:14042–14052. doi: 10.1093/nar/gku1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krokowski D, Tchorzewski M, Boguszewska A, McKay AR, Maslen SL, Robinson CV, Grankowski N. 2007. Elevated copy number of L-A virus in yeast mutant strains defective in ribosomal stalk. Biochem Biophys Res Commun 355:575–580. doi: 10.1016/j.bbrc.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 58.Hinnebusch AG, Ivanov IP, Sonenberg N. 2016. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352:1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen C, Zhang H, Broitman SL, Reiche M, Farrell I, Cooperman BS, Goldman YE. 2013. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat Struct Mol Biol 20:582–588. doi: 10.1038/nsmb.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mao Y, Liu H, Liu Y, Tao S. 2014. Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in Saccharomyces cerevisiae. Nucleic Acids Res 42:4813–4822. doi: 10.1093/nar/gku159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mauro VP, Edelman GM. 2002. The ribosome filter hypothesis. Proc Natl Acad Sci U S A 99:12031–12036. doi: 10.1073/pnas.192442499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Artero-Castro A, Kondoh H, Fernandez-Marcos PJ, Serrano M, Ramon y Cajal S, Lleonart ME. 2009. Rplp1 bypasses replicative senescence and contributes to transformation. Exp Cell Res 315:1372–1383. doi: 10.1016/j.yexcr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 63.McCann KL, Baserga SJ. 2013. Genetics. Mysterious ribosomopathies. Science 341:849–850. doi: 10.1126/science.1244156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaiser C, Dobrikova EY, Bradrick SS, Shveygert M, Herbert JT, Gromeier M. 2008. Activation of cap-independent translation by variant eukaryotic initiation factor 4G in vivo. RNA 14:2170–2182. doi: 10.1261/rna.1171808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 66.Henchal EA, Gentry MK, McCown JM, Brandt WE. 1982. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg 31:830–836. [DOI] [PubMed] [Google Scholar]
- 67.Fraiture M, Baxter RHG, Steinert S, Chelliah Y, Frolet C, Quispe-Tintaya W, Hoffmann JA, Blandin SA, Levashina EA. 2009. Two mosquito LRR proteins function as complement control factors in the TEP1-mediated killing of Plasmodium. Cell Host Microbe 5:273–284. doi: 10.1016/j.chom.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 68.Schreiber MJ, Holmes EC, Ong SH, Soh HSH, Liu W, Tanner L, Aw PPK, Tan HC, Ng LC, Leo YS, Low JGH, Ong A, Ooi EE, Vasudevan SG, Hibberd ML. 2009. Genomic epidemiology of a Dengue virus epidemic in urban Singapore. J Virol 83:4163–4173. doi: 10.1128/JVI.02445-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, Tan HC, Sessions OM, Ward AM, Gubler DJ, Harris E, Garcia-Blanco MA, Ooi EE. 2015. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350:217–221. doi: 10.1126/science.aab3369. [DOI] [PMC free article] [PubMed] [Google Scholar]