

ABSTRACT
During viral infection, accumulation of viral proteins can cause stress in the endoplasmic reticulum (ER) and trigger the unfolded protein response (UPR) to restore ER homeostasis. The inositol-requiring enzyme 1 (IRE1)-dependent pathway is the most conserved of the three UPR signal pathways. Upon activation, IRE1 splices out an intron from the unspliced inactive form of X box binding protein 1 [XBP1(u)] mRNA and produces a transcriptionally potent spliced form [XBP1(s)]. Previous studies have reported that the IRE1/XBP1 pathway is inhibited upon herpes simplex virus 1 (HSV-1) infection; however, the underlying molecular mechanism is still elusive. Here, we uncovered a role of the HSV-1 UL41 protein in inhibiting the IRE1/XBP1 signal pathway. Ectopic expression of UL41 decreased the expression of XBP1 and blocked XBP1 splicing activation induced by the ER stress inducer thapsigargin. Wild-type (WT) HSV-1, but not the UL41-null mutant HSV-1 (R2621), decreased XBP1 mRNA induced by thapsigargin. Nevertheless, infection with both WT HSV-1 and R2621 without drug pretreatment could reduce the mRNA and protein levels of XBP1(s), and additional mechanisms might contribute to this inhibition of XBP1(s) during R2621 infection. Taking these findings together, our results reveal XBP1 as a novel target of UL41 and provide insights into the mechanism by which HSV-1 modulates the IRE1/XBP1 pathway.
IMPORTANCE During viral infection, viruses hijack the host translation apparatus to produce large amounts of viral proteins, which leads to ER stress. To restore ER homeostasis, cells initiate the UPR to alleviate the effects of ER stress. The IRE1/XBP1 pathway is the most conserved UPR branch, and it activates ER-associated protein degradation (ERAD) to reduce the ER load. The IRE1/XBP1 branch is repressed during HSV-1 infection, but little is known about the underlying molecular mechanism. Our results show for the first time that UL41 suppresses the IRE1/XBP1 signal pathway by reducing the accumulation of XBP1 mRNA, and characterization of the underlying molecular mechanism provides new insight into the modulation of UPR by HSV-1.
KEYWORDS: HSV-1, UL41, XBP1, UPR
INTRODUCTION
The endoplasmic reticulum (ER) is an important cellular organelle that controls several critical aspects of cellular processes, such as biosynthesis, assembly, glycosylation, folding, and transport of proteins. Under conditions such as lipid metabolism, differentiation of secretory cells, DNA damage, and chemical insult, as well as viral infection, some misfolded or unfolded proteins accumulate in the lumen of the ER and cause ER stress (1). The ER plays a vital role in the process of viral replication. During viral infection, massive amounts of viral proteins are transported into the ER to induce ER stress. To reduce the protein load, cells activate the unfolded protein response (UPR) signal pathways to restore the homeostasis of the ER (2, 3). The goal of the UPR is to increase the folding capacity and decrease the folding demand of the ER and finally clear the misfolded or unfolded proteins (4). However, under unresolved or intense ER stress, cells initiate the apoptotic pathway (5). The UPR is initiated by three ER transmembrane receptors: pancreatic ER kinase (PKR)-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (6). In resting cells, these three sensors are maintained in an inactive state and are suppressed by the ER-resident chaperone glucose-regulated protein 78 (GRP78) (6, 7). Following the accumulation of unfolded or misfolded proteins in the ER lumen, GRP78 is released from the three sensors and then binds to unfolded proteins. Once GRP78 dissociates from them, the three transducers trigger a complex cascade of signals resulting in activation of the UPR (8, 9).
Under ER stress, PERK undergoes oligomerization and autophosphorylation to form active PERK. Activation of PERK leads to phosphorylation of the subunit of eukaryotic translation initiation factor 2α (eIF2α), resulting in global attenuation of protein translation, and thus relieves the ER stress (10, 11). In response to ER stress, activated ATF6 translocates to the Golgi apparatus, where it is cleaved by site 1 and site 2 proteases and liberates the active N-terminal DNA binding domain of ATF6 [ATF6(N)] (12). ATF6(N) then translocates into the nucleus and induces the expression of genes that improve the ER folding capacity, such as ER chaperones, protein disulfide isomerase (PDI), and X box-binding protein 1 (XBP1) (13).
The IRE1/XBP1 pathway is the most conserved UPR branch in eukaryotic cells (14). IRE1 is a dual-activity enzyme harboring a serine-threonine kinase domain and an endoribonuclease domain (15, 16). Upon activation, IRE1 dimerizes and transphosphorylates itself, and activated IRE1 removes a 26-nucleotide (nt) intron from XBP1 mRNA to form a spliced XBP1 [XBP1(s)], which is subsequently translated into a potent transcription factor. Then, XBP1(s) translocates to the nucleus and induces the expression of genes that enhance the ER protein-folding capacity, phospholipid biosynthesis, and ER-associated protein degradation (ERAD) (17, 18). If the ER stress is too severe to eliminate, activated IRE1 induces apoptosis signal-regulating kinase 1 (ASK 1), c-Jun N-terminal kinase (JNK), and mitogen-activated protein kinase (p38) (19). IRE1 by itself can degrade ER-bound mRNAs, such as the viral mRNAs, through the regulated IRE1-dependent decay pathway to reduce protein translation and release the unfolded protein load in the ER (20).
Herpes simplex virus 1 (HSV-1) is a double-stranded linear DNA virus with a large genome encoding over 80 proteins. During HSV-1 replication, the rapid production of large quantities of viral proteins may induce the UPR, and HSV-1 selectively modulates the UPR to benefit its replication. For example, HSV-1 glycoprotein B (gB) interacts with the PERK luminal domain to resist PERK activation, and another late protein, γ1 34.5, promotes dephosphorylation of eIF2α by recruiting the cellular phosphatase PP1α (21, 22). The IRE1/XBP1 branch is involved in many viral infections. Hepatitis C virus suppresses the IRE1/XBP1 pathway, and influenza A virus induces the IRE1/XBP1 pathway (23, 24). The IRE1/XBP1 branch of the UPR is repressed during HSV-1 infection, but its molecular mechanism remains to be elaborated (25). In this study, we show that the HSV-1 tegument protein UL41, which contains an endoribonuclease activity, inhibits the expression of XBP1 by degrading its mRNA. These findings reveal a novel mechanism for HSV-1 to modulate the IRE1/XBP1 branch of the UPR.
RESULTS
The HSV-1 tegument protein UL41 inhibits the ectopic expression of XBP1.
HSV-1 has been shown to repress the IRE1/XBP1 branch of the UPR. During HSV-1 infection, XBP1(s) mRNA was undetectable, and mRNA of the unspliced inactive form of X box binding protein 1 [XBP1(u)] was rapidly degraded and undetectable in the late stage of infection (25). In our previous study and others, it was reported that the HSV-1 tegument protein UL41 specifically degraded both viral and cellular mRNAs containing an AU-rich element (ARE) in their 3′-untranslated regions, such as tetherin, zinc finger antiviral protein, and cig5 mRNAs (26–34). Additionally, ICP0 possesses the activity of E3 ubiquitin ligase and downregulates the expression of many cellular antiviral proteins, including nuclear factor κB (NF-κB) subunit P50, through the ubiquitin-proteasome pathway (35). To determine whether the tegument protein UL41 or the viral E3 ubiquitin ligase ICP0 participated in reducing the expression of XBP1, HEK293T cells were cotransfected with UL41-hemagglutinin (HA) or ICP0-Flag and XBP1-HA plasmids for 24 h, and then the cells were harvested and subjected to Western blot (WB) analysis. As shown in Fig. 1A and B, HSV-1 UL41, but not the immediate-early protein ICP0, downregulated the ectopic expression of XBP1(u) and XBP1(s).
FIG 1.
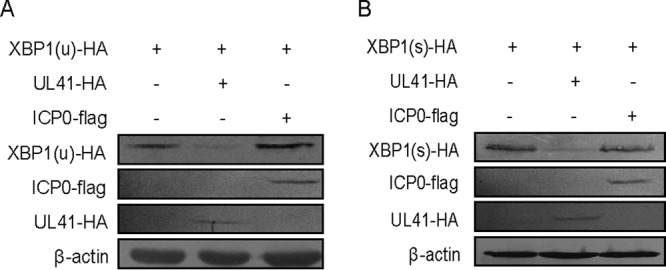
HSV-1 tegument protein UL41 inhibited the ectopic expression of XBP1. (A and B) HEK293T cells were cotransfected with XBP1(u)-HA (500 ng) or XBP1(s)-HA (500 ng) and UL41-HA (500 ng) or ICP0-flag (500 ng) plasmids. At 24 h posttransfection, the cells were harvested and subjected to WB analysis with antibodies against Flag, HA, or β-actin. The data represent the results of one of the triplicate experiments.
Ectopic expression of UL41 decreased the expression of endogenous XBP1 and blocked XBP1 splicing activation induced by thapsigargin.
To investigate whether ectopic expression of UL41 would inhibit the expression of endogenous XBP1, HEK293T cells were transfected with UL41-HA plasmid for 24 h. At 24 h posttransfection, the cells were treated with 20 nM thapsigargin, an ER stress inducer that can disrupt calcium homeostasis through inhibition of the ER Ca2+-ATPase pump. Cells were then harvested at 4 h after thapsigargin treatment and subjected to WB analysis to analyze the XBP1 protein. Our data showed that ectopic expression of UL41 decreased the endogenous expression of XBP1(u) and XBP1(s) induced by thapsigargin in a dose-dependent manner (Fig. 2A). In order to confirm the results, a reporter plasmid, pXBP1u-FLuc, was applied to examine the XBP1 splicing activation (36). Firefly luciferase activity was induced only after XBP1 splicing removed the 26-nt intron, and it simulated the XBP1 splicing-activated event under ER stress. HEK293T cells were cotransfected with pXBP1u-FLuc, pRL-TK, and UL41-HA plasmids. At 24 h posttransfection, the cells were treated with 20 nM thapsigargin for an additional 4 h and subjected to dual-luciferase reporter (DLR) assays. The plasmid pRL-TK was used as an internal control to normalize transfection efficiency. As a result, thapsigargin could successfully increase the luciferase reporter activity, and UL41 significantly inhibited the activity induced by thapsigargin (Fig. 2B). Taken together, these data showed that UL41 decreased the expression of endogenous XBP1 and blocked XBP1 splicing activation.
FIG 2.

Ectopic expression of UL41 decreased the expression of endogenous XBP1 and blocked XBP1 splicing activation induced by thapsigargin. (A, C, D, and E) HEK293T cells were transfected with UL41-HA plasmid (300 ng [+] and 1,000 ng [++]) for 24 h, and then the transfected cells were treated with 20 nM thapsigargin. After 4 h of drug stimulation, cells were harvested and subjected to WB analysis with antibodies against XBP1, HA, or β-actin (A), semiquantitative RT-PCR (C), or qRT-PCR (D and E) analysis. (B) HEK293T cells were cotransfected with pXBP1u-FLuc reporter (250 ng), Renilla luciferase plasmid pRL-TK (150 ng), and pCMV-HA control vector or UL41-HA plasmid. At 24 h posttransfection, the cells were treated or not with 20 nM thapsigargin for an additional 4 h, followed by cell lysis. Firefly luciferase activity was determined by a dual-luciferase assay. (F and G) HEK293T cells were transfected with UL41-HA plasmid (300 ng [+] and 1000 ng [++]) for 24 h, and then the cells were harvested and subjected to WB analysis with antibodies against IRE1, HA, or β-actin (F) or semiquantitative RT-PCR analysis (G). The data represent the results of one of the triplicate experiments. The error bars represent mean standard deviations (SD) of three independent experiments. Statistical analysis was performed using Student's t test. *, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001.
It was reported that UL41 is an endoribonuclease that selectively degrades viral and cellular mRNAs (28, 34, 37). Therefore, it is very likely that UL41 decreases XBP1 expression via its RNase activity to degrade XBP1 mRNA. To confirm this hypothesis, UL41-HA plasmid was transfected into HEK293T cells for 24 h, and the transfected cells were treated with 20 nM thapsigargin for an additional 4 h; then, cells were harvested and subjected to semiquantitative reverse transcription (RT)-PCR (Fig. 2C) and quantitative RT (qRT)-PCR analysis (Fig. 2D and E). As a result, UL41 downregulated the XBP1(u) and XBP1(s) mRNAs in a dose-dependent manner. Collectively, these results demonstrated that the HSV-1 tegument protein UL41 reduced the expression of endogenous XBP1 via its RNase activity to degrade XBP1 mRNA. To determine whether UL41 specifically targeted XBP1, HEK293T cells were transfected with UL41-HA plasmid for 24 h, and then the cells were harvested and subjected to WB and semiquantitative RT-PCR analyses. As shown in Fig. 2F and G, UL41 failed to affect the expression of IRE1, indicating that UL41 blocked XBP1 splicing activation by specifically repressing the expression of XBP1, but not IRE1.
HSV-1 infection reduced the accumulation of XBP1 mRNA induced by thapsigargin via UL41.
The above-mentioned results demonstrated that HSV-1 UL41 protein reduced the expression of XBP1 by degrading its mRNA. We wondered whether XBP1(u) mRNA and XBP1(s) mRNA induced by thapsigargin were downregulated during HSV-1 infection. The cells were treated with thapsigargin to promote high expression of XBP1(s), and then the drug-induced XBP1 was examined to investigate the direct effect of UL41 on XBP1 during HSV-1 infection. HeLa cells were treated with a high concentration of thapsigargin (100 nM) for 4 h and then infected with either WT HSV-1 or UL41-null mutant HSV-1 (R2621) at a multiplicity of infection (MOI) of 5 or 10. At 4 h postinfection, cells were harvested and subjected to semiquantitative RT-PCR (Fig. 3A) and qRT-PCR (Fig. 3B and C) analyses to test the level of XBP1 mRNA. As shown in Fig. 3, we found that thapsigargin treatment resulted in a dramatic increase of XBP1(s) mRNA. WT HSV-1, but not R2621, markedly reduced the accumulation of thapsigargin-induced XBP1 mRNA. In conclusion, these results suggested UL41 reduced the accumulation of XBP1 mRNA via its RNase activity.
FIG 3.

HSV-1 infection reduced the accumulation of XBP1 mRNA induced by thapsigargin via UL41. (A, B, and C) HeLa cells were pretreated or not with 100 nM thapsigargin; after 4 h, the medium was removed and the cells were mock infected or infected with WT HSV-1 or R2621 at MOI of 5 and 10. At 4 h postinfection, cells were harvested and subjected to semiquantitative RT-PCR (A) or qRT-PCR (B and C) analysis. The data represent the results of one of the triplicate experiments. The error bars indicate SD. **, 0.001 < P < 0.01; ***, P < 0.001.
HSV-1 infection suppressed the endogenous expression of XBP1(s).
To investigate whether XBP1 was downregulated during HSV-1 infection without drug stimulation, HeLa cells were infected with WT HSV-1 or R2621 at MOI of 5 and 10. Then, cells were harvested at 10 h postinfection, and semiquantitative RT-PCR (Fig. 4A) and qRT-PCR (Fig. 4B and C) were performed to analyze the XBP1 mRNA. As a result, the mRNA levels of XBP1(u) and XBP1(s) were significantly decreased during WT HSV-1 infection. Interestingly, R2621 infection failed to degrade XBP1(u) mRNA but reduced the accumulation of XBP1(s) mRNA at an MOI of 10. Similar results were obtained using WB analysis. As shown in Fig. 4D, WT HSV-1 markedly abrogated the expressions of XBP1(u) and XBP1(s), while R2621 decreased the expression of XBP1(s) but not XBP1(u). In short, these results suggested that UL41 dampened the IRE1/XBP1 branch by reducing XBP1 mRNA accumulation. Surprisingly, we found that, unlike those of XBP1(u), XBP1(s) mRNA and protein levels were decreased under R2621 infection, indicating that additional mechanisms might contribute to the inhibition of XBP1 splicing activation. As the activation of XBP1(s) is mediated by the only enzyme of activated IRE1, HSV-1 might express another protein to effect the activation of IRE1, which results in the suppression of XBP1(s) during both WT HSV-1 and R2621 infections.
FIG 4.

HSV-1 infection suppressed the endogenous expression of XBP1(s). (A, B, and C) HeLa cells were infected with WT-HSV-1 or R2621 at an MOI of 5 or 10. At 10 h postinfection, cells were harvested and subjected to semiquantitative RT-PCR (A) or qRT-PCR (B and C) analysis. (D) HeLa cells were infected with WT-HSV-1 or R2621 at an MOI of 5 or 10. At 20 h postinfection, cells were harvested and subjected to WB analysis with antibodies against UL42, XBP1, or β-actin. The data represent the results of one of the triplicate experiments. The error bars indicate SD. *, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001.
DISCUSSION
The ER is a cellular organelle that plays vital roles in viral replication and maturation. Viruses must utilize the ER to complete efficient replication, including viral entry, viral protein synthesis and modification, genome replication, and viral assembly (38, 39). Therefore, virus infection causes ER stress and activation of the UPR. Previous studies have revealed that HSV-1 could modulate the PERK arm of the UPR through gB and γ1 34.5 (21, 40). Moreover, XBP1(s) is undetectable, and the expression level of XBP1(u) is downregulated upon HSV-1 infection, but the underlying molecular mechanism is unclear (25). In the present study, we focused on the IRE1/XBP1 branch of the UPR. We demonstrated for the first time that XBP1 was a novel target of the HSV-1 tegument protein UL41 and that UL41 reduced the expression of XBP1 by reducing its mRNA accumulation. The virion host shutoff protein UL41 is a broad-specificity endoribonuclease that triggers rapid degradation of host mRNAs to facilitate viral replication (28, 37). UL41 associates with the cap-binding initiation factor complex eIF4F and cleaves mRNA in regions of translation initiation (41). UL41 could cleave single-stranded RNA at the 3′ side of U and C residues in vitro and specifically target the ribosome-associated mRNA in vivo (37, 42).
The IRE1/XBP1 pathway is the most conserved branch among the three UPR signal pathways, and activated IRE1 could splice the XBP1(u) mRNA to form XBP1(s) under ER stress. In the present study, we found that R2621 infection did not affect the accumulation of XBP1(s) mRNA in HeLa cells pretreated with thapsigargin, but the expression of XBP1(s) was decreased under R2621 infection alone. HSV-1 might express another viral protein that suppressed the activation of IRE1; therefore, R2621 infection alone could also inhibit the expression of XBP1(s). In this study, cells pretreated with a high concentration of thapsigargin expressed the abundance of XBP1(s), and subsequent infection with WT HSV-1 or R2621 could reveal the direct effect of HSV-1 UL41 on the expression of XBP1, avoiding interference of IRE1 in the signal pathway.
HSV-1 can avoid cellular responses that are likely detrimental to viral replication by suppressing the IRE1/XBP1 pathway. The ERAD proteins that are encoded by XBP1(s) target genes would degrade the viral proteins. XBP1(s) has also been reported to enhance beta interferon production in dendritic cells (43). IRE1 could cleave viral mRNA via its endoribonuclease activity into single-stranded fragments that lack markers of self, activating the retinoic acid-inducible gene I (RIG-I) pathway and triggering an inflammatory response (44). These negative impacts provide good reasons for HSV-1 to block the IRE1/XBP1 pathway.
In summary, we have shown here for the first time that the HSV-1 tegument protein UL41 suppresses the IRE1/XBP1 signal pathway by reducing the accumulation of XBP1 mRNA, revealing a strategy by which HSV-1 modulates the UPR.
MATERIALS AND METHODS
Cell culture, viruses, antibodies, and reagents.
Human embryonic kidney (HEK293T) and HeLa cells were cultured in Dulbecco's modified minimal essential medium (DMEM) (Gibco-BRL) containing 10% fetal bovine serum (FBS) (Gibco-BRL) in a humidified atmosphere containing 5% CO2 at 37°C as described previously (45). The WT HSV-1 (strain F) virus and UL41-null mutant virus (R2621) were propagated in Vero cells, and the titers were determined as described previously (45). Mouse anti-HA and anti-Flag monoclonal antibodies (MAbs), rabbit polyclonal anti-IRE1, rabbit polyclonal anti-XBP1, and mouse anti-β-actin MAbs were purchased from Abmart (Shanghai, China), Beyotime Biotechnology (Shanghai, China), Proteintech (Wuhan, China), and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. Rabbit polyclonal anti-UL42 was made by GL Biochem Ltd. (Shanghai, China). Thapsigargin was purchased from Cayman Chemical (Ann Arbor, MI, USA).
Plasmid construction.
All enzymes used for cloning procedures were purchased from Vazyme (Nanjing, China). To construct UL41-HA and ICP0-Flag plasmids, the UL41 and ICP0 genes were amplified from the HSV-1 genome as described in our previous study (46) and cloned into the pCMV-HA or pCMV-Flag vector (Beyotime, Shanghai, China). To construct XBP1(u)-HA and XBP1(s)-HA plasmids, the XBP1(u) and XBP1(s) genes were cloned into the pCMV-HA vector (Beyotime, Shanghai, China). The Renilla luciferase plasmid pRL-TK (expressing thymidine kinase [TK]) was purchased from Promega (Madison, WI, USA). The pXBP1u-FLuc plasmid was a gift from Yi-Ling Lin (Institute of Biomedical Sciences, Academia Sinica).
Transfection and DLR assay.
HEK293T cells were transfected by standard calcium phosphate precipitation, whereas HeLa cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturers' recommendations. Luciferase assays were performed with a dual-specific luciferase assay kit (Promega) as previously described (47, 48).
WB analysis.
WB analysis was performed as previously described (35).
RNA isolation and qRT-PCR.
Total RNA was extracted using TRIzol (Invitrogen, CA, USA) according to the manufacturer's manual. Samples were digested with DNase I and subjected to reverse transcription. The cDNA was used as a template for qRT-PCR to detect accumulation of the indicated mRNA, and 18S rRNA was used as an internal reference as previously described (48). The nucleotide sequences of the primers are available on request.
Statistical analysis.
Comparisons between different groups were analyzed using Student's t test. All differences were considered statistically significant at a P value of <0.05.
ACKNOWLEDGMENTS
We thank Yi-Ling Lin for the pXBP1u-FLuc plasmid and Bernard Roizman for the R2621 virus.
Work in the Zheng laboratory relevant to this article was supported by grants from the National Natural Science Foundation of China (81371795 and 81571974) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (YX13400214).
REFERENCES
- 1.Schroder M. 2008. Endoplasmic reticulum stress responses. Cell Mol Life Sci 65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harding HP, Calfon M, Urano F, Novoa I, Ron D. 2002. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol 18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 3.Zhang K, Kaufman RJ. 2004. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem 279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 4.Schroder M, Kaufman RJ. 2005. The mammalian unfolded protein response. Annu Rev Biochem 74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 5.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufman RJ. 2002. Orchestrating the unfolded protein response in health and disease. J Clin Invest 110:1389–1398. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Hendershot LM. 2004. ER chaperone functions during normal and stress conditions. J Chem Neuroanat 28:51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Brush MH, Weiser DC, Shenolikar S. 2003. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol 23:1292–1303. doi: 10.1128/MCB.23.4.1292-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan SW, Egan PA. 2005. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J 19:1510–1512. [DOI] [PubMed] [Google Scholar]
- 10.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. 2005. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell 16:5493–5501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang W, Hinnebusch AG. 1996. Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol Cell Biol 16:6603–6616. doi: 10.1128/MCB.16.11.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. 2000. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6:1355–1364. doi: 10.1016/S1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 14.Hetz C, Martinon F, Rodriguez D, Glimcher LH. 2011. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev 91:1219–1243. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
- 15.Sidrauski C, Walter P. 1997. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90:1031–1039. doi: 10.1016/S0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 16.Szegezdi E, Logue SE, Gorman AM, Samali A. 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee AH, Iwakoshi NN, Glimcher LH. 2003. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. 2004. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Hetz C, Glimcher LH. 2009. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell 35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. 2009. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol 81:3377–3390. doi: 10.1128/JVI.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He B, Chou J, Liebermann DA, Hoffman B, Roizman B. 1996. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J Virol 70:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. 2004. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J Biol Chem 279:17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 24.Hassan IH, Zhang MS, Powers LS, Shao JQ, Baltrusaitis J, Rutkowski DT, Legge K, Monick MM. 2012. Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J Biol Chem 287:4679–4689. doi: 10.1074/jbc.M111.284695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burnett HF, Audas TE, Liang G, Lu RR. 2012. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 17:473–483. doi: 10.1007/s12192-012-0324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zenner HL, Mauricio R, Banting G, Crump CM. 2013. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J Virol 87:13115–13123. doi: 10.1128/JVI.02167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su C, Zhang J, Zheng C. 2015. Herpes simplex virus 1 UL41 protein abrogates the antiviral activity of hZAP by degrading its mRNA. Virol J 12:203. doi: 10.1186/s12985-015-0433-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everly DN Jr, Feng P, Mian IS, Read GS. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J Virol 76:8560–8571. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng P, Everly DN Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J Virol 75:10272–10280. doi: 10.1128/JVI.75.21.10272-10280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read GS. 2013. Virus-encoded endonucleases: expected and novel functions. RNA 4:693–708. doi: 10.1002/wrna.1188. [DOI] [PubMed] [Google Scholar]
- 31.Schek N, Bachenheimer SL. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J Virol 55:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esclatine A, Taddeo B, Evans L, Roizman B. 2004. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc Natl Acad Sci U S A 101:3603–3608. doi: 10.1073/pnas.0400354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen G, Wang K, Wang S, Cai M, Li ML, Zheng C. 2014. Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J Virol 88:12163–12166. doi: 10.1128/JVI.01380-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esclatine A, Taddeo B, Roizman B. 2004. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. Proc Natl Acad Sci U S A 101:18165–18170. doi: 10.1073/pnas.0408272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Wang K, Wang S, Zheng C. 2013. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J Virol 87:12935–12948. doi: 10.1128/JVI.01952-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu CY, Hsu YW, Liao CL, Lin YL. 2006. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J Virol 80:11868–11880. doi: 10.1128/JVI.00879-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J Virol 80:9341–9345. doi: 10.1128/JVI.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He B. 2006. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ 13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 39.Inoue T, Tsai B. 2013. How viruses use the endoplasmic reticulum for entry, replication, and assembly. Cold Spring Harb Perspect Biol 5:a013250. doi: 10.1101/cshperspect.a013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng G, Feng Z, He B. 2005. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J Virol 79:1379–1388. doi: 10.1128/JVI.79.3.1379-1388.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Page HG, Read GS. 2010. The virion host shutoff endonuclease (UL41) of herpes simplex virus interacts with the cellular cap-binding complex eIF4F. J Virol 84:6886–6890. doi: 10.1128/JVI.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shiflett LA, Read GS. 2013. mRNA decay during herpes simplex virus (HSV) infections: mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J Virol 87:94–109. doi: 10.1128/JVI.01557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu F, Yu X, Wang H, Zuo D, Guo C, Yi H, Tirosh B, Subjeck JR, Qiu X, Wang XY. 2011. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur J Immunol 41:1086–1097. doi: 10.1002/eji.201040831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lencer WI, DeLuca H, Grey MJ, Cho JA. 2015. Innate immunity at mucosal surfaces: the IRE1-RIDD-RIG-I pathway. Trends Immunol 36:401–409. doi: 10.1016/j.it.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xing J, Wang S, Lin F, Pan W, Hu CD, Zheng C. 2011. Comprehensive characterization of interaction complexes of herpes simplex virus type 1 ICP22, UL3, UL4, and UL205. J Virol 85:1881–1886. doi: 10.1128/JVI.01730-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xing J, Wu F, Pan W, Zheng C. 2010. Molecular anatomy of subcellular localization of HSV-1 tegument protein US11 in living cells. Virus Res 153:71–81. doi: 10.1016/j.virusres.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528–3540. doi: 10.1128/JVI.06713-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL. 2011. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol 85:11079–11089. doi: 10.1128/JVI.05098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]