

Natural selection drove the evolution of all living beings for millions of years until mankind acquired the ability to interfere with this mechanism by counteracting the natural hazards that tend to eliminate the weakest humans and select the strongest ones for the continuation of the species. Medicine, which has acquired the ability to cure many common diseases that were previously lethal, is an important result of the attempt to oppose natural selection. Medical progress was initially very slow, but with the advent of the scientific revolution in the 18th century, this has continued to get faster and has played a role in doubling the life expectancy in Western countries since the beginning of the 19th century.1
Strenuous research, brilliant insights and, much more rarely, serendipity, were at the basis of medical progress. Importantly, some major advances in medicine derived from a widening of knowledge in other disciplines. For example, the development of the microscope (the “occhiolino” or “little eye” of Galileo Galilei) in the early 17th century was the prerequisite for recognizing blood cells and starting to identify blood disorders. Thus, a major advance in optical science made the birth of hematology possible.
Platelets, because of their small size and the limited resolution of early microscopes, escaped identification for a long time, and when, in 1735, the German physician and poet Paul Gottlieb Werlhof provided the first detailed description of ‘morbus maculosus haemorrhagicus’,2 now known as immune thrombocytopenia (ITP), these blood cells were unknown (Figure 1). The discovery of platelets had to wait until 1882, when the Italian pathologist Giulio Bizzozzero, also thanks to a major technical improvement in microscope technology (correction of chromatic aberration), described in detail these small elements and the relationship between platelet adhesion and aggregation, and the subsequent fibrin formation and deposition.3 A year after the brilliant insight of Bizzozero, the link between thrombocytopenia and ITP was identified by Brohm.4 So, 150 years after its description, we finally had an explanation for the ‘morbus maculosus haemorrhagicus’. Also subsequent advances in knowledge of ITP were made thanks to ingenious intuition and laborious research. The intuition of Kaznelson, in 1916, that the spleen was responsible for platelet destruction led to the identification of splenectomy as an effective treatment for this disease.5 A few decades later, in 1951, Harrington observed a child with transient purpura born to a mother with ITP and suspected that the passage of a humoral factor from mother to child was responsible for platelet destruction.6 In the same year, Evans hypothesized that the ITP had an immune genesis,7 paving the way to immunosuppressants as an effective treatment for this disorders.8 Finally, the search for the humoral substance responsible for causing the platelet count to rise in response to thrombocytopenia, which had involved many groups of researchers for many decades, led in 1994 to the purification and cloning of thrombopoietin (TPO).9 This achievement opened the way to the development of TPO mimetics, which have proven effective not only in ITP, but also in other forms of thrombocytopenia and in bone marrow aplasia.
Figure 1.

The long history of immune thrombocytopenia (ITP). After nearly 300 years of research, we now know the main pathogenetic mechanisms of ITP and have several effective treatments. However, further investigation is required to make the diagnosis of this disease easier and more reliable, to better understand the pathogenic mechanism in the individual patient and personalize accordingly the therapeutic approach.
Can we assume that, after three centuries of investigation, the history of ITP is over and there is no longer a need to allocate resources to the study of this disease? We believe that the answer is “surely not”! Just think that the diagnosis of ITP is still a process of exclusion because we have no sensitive and specific laboratory tests for this condition. As a consequence, both patients with ITP and those with other forms of isolated thrombocytopenia are at risk of misdiagnosis and unnecessary treatments. For instance, patients with inherited thrombocytopenias (ITs) are often misdiagnosed with ITP and many of them receive useless immunosuppressant drugs or even splenectomy.10 Another goal yet to be achieved is personalization of treatment. We currently have different treatment options, each of them effective in a variable proportion of patients, but we are still unable to predict to which one(s) each patient will respond. Moreover, we are unable to recognize subjects whose disease will go into remission spontaneously and therefore might benefit from treatment as limited as possible. Finally, we are also still unable to identify patients requiring immediate treatment because they are at risk of clinically relevant bleeding episodes, since the degree of thrombocytopenia is not always effective in this respect. These uncertainties are primarily derived from the fact that the pathogenic mechanisms of ITP, probably different in different patients, are not yet fully known. So, as recognized by the “EHA Roadmap for European Hematology Research” recently published in this Journal,11 both basic research and clinical studies are required to further improve care for ITP patients.
While advances in the field of ITP occurred slowly over centuries, knowledge of ITs have been increasing exponentially in recent years due to the huge improvement in technologies for gene sequencing. Up to 20 years ago, only a few ITs were well defined from a clinical point of view and only for 4 of them was the genetic defect known. Now, we know more than 30 diseases caused by mutations in different genes, with half of these disorders identified in the last five years (Figure 2). However, nearly 50% of patients with ITs have forms that do not fit the criteria for any known disorder,12 and identification of these ‘new’ disorders is required for several reasons. First of all, we recently realized that nearly 50% of patients with known ITs are at risk of because of acquiring additional illnesses during life, such as bone marrow aplasia, kidney failure or hematologic malignancies, which endanger patients’ lives much more than thrombocytopenia itself. Of note, the 2016 revision of the WHO classification of myeloid neoplasms and acute leukemias introduced a new category of diseases defined as ‘Myeloid neoplasms with germ line predisposition and pre-existing platelet disorders’, which includes neoplasms developing in patients with ITs caused by mutations in the genes RUNX1, ANKRD26 and ETV6. We do not know whether some of the yet unknown forms of IT expose patients to risks other than those deriving from thrombocytopenia, and only their identification and characterization will allow us to answer this important question. Another reason to keep searching for new diseases is that affected subjects could benefit from treatments that are effective in some known ITs. For instance, it is possible that, as already shown for Wiskott-Aldrich syndrome,13 ANKRD26-related thrombocytopenia and MYH9-related disease,14 TPO mimetics increase the number of platelets even in some yet unknown forms of IT and can therefore be used instead of platelet transfusions to prepare patients to surgery.
Figure 2.
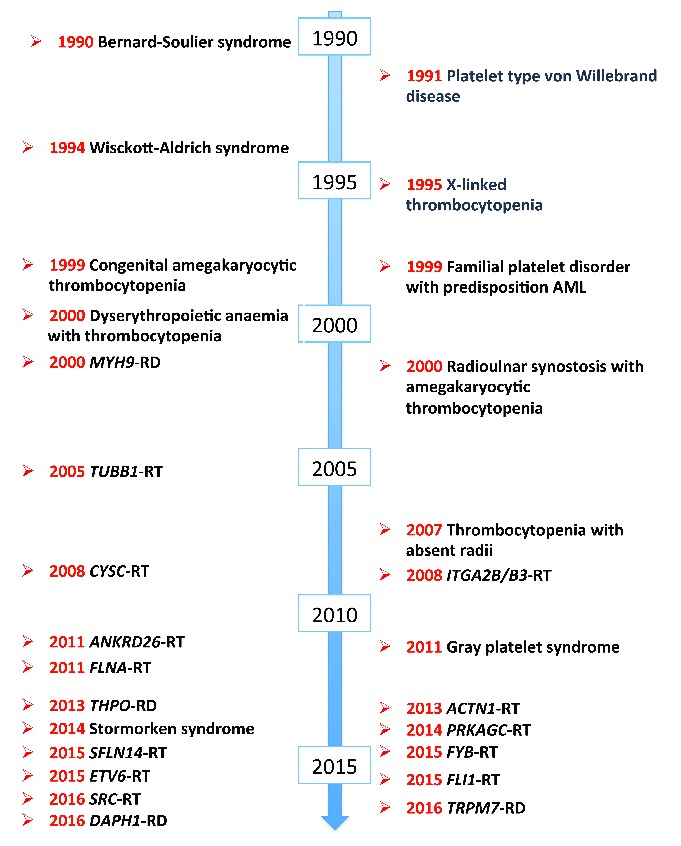
Identification of the etiology of inherited thrombocytopenias. Until the 1980s, only a very few forms of inherited thrombocytopenia had been identified on the basis of their peculiar clinical pictures. Subsequently, the availability of more and more effective gene sequencing techniques resulted in the unveiling of the etiology of the already known forms and the identification of an increasing number of new diseases. Of note, 14 new causative genes have been identified since 2010. We presently know more than 30 forms of inherited thrombocytopenias deriving from mutations in different genes, but despite this explosion of knowledge, almost 50% of patients still remain without a diagnosis because their illnesses have not yet been identified. AML: acute myeloid leukemia; related disorder (RD) and related thrombocytopenias (RT) suffixes refer to syndromic and non-syndromic inherited thrombocytopenias (respectively) deriving from mutations in the genes indicated in italics.
Huge improvements have been obtained in recent years also for other forms of thrombocytopenia, but, as for ITP and ITs, further research is required to respond to the many unanswered questions and improve patient care. We refer interested readers to the “EHA Roadmap for European Hematology Research”, where this matter is discussed in detail by leading experts in different thrombocytopenic disorders.11
At variance with thrombocytopenias, past and recent achievements in the field of defects of platelet function have been limited and knowledge of these conditions is still very unsatisfactory. This is surprising, since both acquired and inherited platelet dysfunctions are more prevalent than the corresponding forms of thrombocytopenia. For instance, it has been shown that many common liver and kidney disorders affect platelet function and may result in bleeding tendency,15 but we have little information on this matter, and both diagnosis and treatment of these conditions are poorly defined. Concerning inherited forms, most patients still receive diagnoses, as ‘primary secretion defect’ or ‘granule disorder’, that are merely descriptive, because the underlying genetic defects, as well as the pathogenic mechanisms, are unknown.16
A major problem that hindered progress in the field is that the definition itself of platelet dysfunction has remained vague. This has been because, despite recent and important efforts to reach a consensus among experts,16,17 we do not have validated criteria for recognizing and classifying these conditions. Moreover, our ability to predict the risk of bleeding in the individual patient remains limited and, therefore, we are often in doubt as to whether or not to administer prophylactic treatments in situations of hemostatic challenges, such as giving birth or surgery. Finally, also the therapeutic armamentarium for these conditions is still very limited and, in many cases, there is no clear evidence of efficacy. Knowledge of functional platelet defects, therefore, requires a big leap forward to be made. A major advance would be the identification of a comprehensive laboratory approach for diagnosing these disorders. Acquiring the ability to identify the bleeding risk of each patient by in vitro techniques would be even better. These are very ambitious goals, the achievement of which requires the joint evaluation of the clinical and laboratory features of large series of patients. Organizing these kind of studies is challenging, especially for the inherited forms that need international collaboration for achieving a sufficient sample size. However, reaching these objectives would represent the starting point for further studies designed to identify both the pathogenic mechanisms of these conditions and new effective treatments. Moreover, the application of next generation sequencing techniques to large series of well characterized patients is expected to identify a large number of inherited platelet dysfunctions that are still waiting to be recognized. Initial evidence of the effectiveness of this approach has been recently published.18,19
In conclusion, as in many other fields of medicine, also in the area of thrombocytopenias and platelet function disorders we are observing the apparent paradox that the advance of knowledge increases the number of questions to be answered instead of reducing it. So, theoretically, it is to be expected that medical research and the consequent improvement of human health is a never-ending story.
Supplementary Material
References
- 1.Oeppen J, Vaupel JW. Demography. Broken limits to life expectancy. Science. 2002;296(5570):1029–1031. [DOI] [PubMed] [Google Scholar]
- 2.Werlhof PG. Disquisitio medica et philologica de variolis et anthracibus, signis differentiis, medelis disserit etc. Nicolai Foersteri; Hannover, 1735. [Google Scholar]
- 3.Bizzozero G. [Ueber einen neuen Forrnbestandteil des Blutes und dessen Rolle bei der Thrombose und Blutgerinnung. Virchows Archiv fur Pathologische Anatomie und Physiologie und fur Klinische Medizin]. 1882;90:261–332. [Google Scholar]
- 4.Brohm F. Quoted by Krauss, E. Über purpura. Inaugural dissertation. Heidelberg, 1883. [Google Scholar]
- 5.Kaznelson P. [Verschwinden der hämorrhagischen Diathese bei einem Falle von “essentieller Thrombopenie” (Frank) nach Milzexstirpation: splenogene thrombolytische Purpura]. Wiener Klinische Wochenschrift. 1916;29:1451–1454. [Google Scholar]
- 6.Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med. 1951;38:1–10. [PubMed] [Google Scholar]
- 7.Evans RS, Takahashi K, Duane RT, Payne R, Liu C. Primary thrombocytopenic purpura and acquired hemolytic anemia; evidence for a common etiology. AMA Arch Intern Med. 1951;87(1):48–65. [DOI] [PubMed] [Google Scholar]
- 8.Wintrobe MM, Cartwright GE, Palmer JG, Kuhns WJ, Samuels LT. Effect of corticotrophin and cortisone on the blood in various disorders in man. AMA Arch Intern Med. 1951;88(3):310–336. [DOI] [PubMed] [Google Scholar]
- 9.Schick BP. Hope for treatment of thrombocytopenia. N Engl J Med. 1994;331(13):875–876. [DOI] [PubMed] [Google Scholar]
- 10.Balduini CL, Savoia A, Seri M. Inherited thrombocytopenias frequently diagnosed in adults. J Thromb Haemost. 2013;11(6):1006–1019. [DOI] [PubMed] [Google Scholar]
- 11.Engert A, Balduini C, Brand A, et al. The European Hematology Association Roadmap for European Hematology Research: a consensus document. Haematologica. 2016;101(2):115–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balduini CL, Noris P. Innovation in the field of thrombocytopenias: achievements since the beginning of the century and promises for the future. Haematologica. 2016;101(1):2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerrits AJ, Leven EA, Frelinger AL, 3rd, et al. Effects of eltrombopag on platelet count and platelet activation in Wiskott-Aldrich syndrome/X-linked thrombocytopenia. Blood. 2015;126(11):1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pecci A, Gresele P, Klersy C, et al. Eltrombopag for the treatment of the inherited thrombocytopenia deriving from MYH9 mutations. Blood. 2010;116(26):5832–5837. [DOI] [PubMed] [Google Scholar]
- 15.Jalal DI, Chonchol M, Targher G. Disorders of hemostasis associated with chronic kidney disease. Semin Thromb Hemost. 2010;36:34–40. [DOI] [PubMed] [Google Scholar]
- 16.Gresele P; Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis. Diagnosis of inherited platelet function disorders: guidance from the SSC of the ISTH. J Thromb Haemost. 2015;13(2):314–322. [DOI] [PubMed] [Google Scholar]
- 17.Cattaneo M, Cerletti C, Harrison P, et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J Thromb Haemost. 2013;11:1183–1189. [DOI] [PubMed] [Google Scholar]
- 18.Simeoni I, Stephens JC, Hu F, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016;127(23):2791–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson B, Lowe GC, Futterer J, et al. ; UK GAPP Study Group. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica. 2016;101(10):1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.