

Abstract
Neutrophils, early mediators of the innate immune defense, are recruited to developing thrombi in different types of thrombosis. They amplify intravascular coagulation by stimulating the tissue factor-dependent extrinsic pathway via inactivation of endogenous anticoagulants, enhancing factor XII activation or decreasing plasmin generation. Neutrophil-dependent prothrombotic mechanisms are supported by the externalization of decondensed nucleosomes and granule proteins that together form neutrophil extracellular traps. These traps, either in intact or fragmented form, are causally involved in various forms of experimental thrombosis as first indicated by their role in the enhancement of both microvascular thrombosis during bacterial infection and carotid artery thrombosis. Neutrophil extracellular traps can be induced by interactions of neutrophils with activated platelets; vice versa, these traps enhance adhesion of platelets via von Willebrand factor. Neutrophil-induced microvascular thrombus formation can restrict the dissemination and survival of blood-borne bacteria and thereby sustain intravascular immunity. Dysregulation of this innate immune pathway may support sepsis-associated coagulopathies. Notably, neutrophils and extracellular nucleosomes, together with platelets, critically promote fibrin formation during flow restriction-induced deep vein thrombosis. Neutrophil extracellular traps/extracellular nucleosomes are increased in thrombi and in the blood of patients with different vaso-occlusive pathologies and could be therapeutically targeted for the prevention of thrombosis. Thus, during infections and in response to blood vessel damage, neutrophils and externalized nucleosomes are major promoters of intravascular blood coagulation and thrombosis.
Introduction
Blood neutrophils are among the first immune cells that are recruited to pathogen-infected tissues and sterile injuries.1 After extravasation, and with the assistance of complement, neutrophils ingest pathogens and kill them inside intracellular phagolysosomal granules or, as in the case of sterile injuries, engulf the cellular debris to degrade it. Besides their well-documented functions in combating tissue-based infections and injuries, neutrophils are also involved in restricting blood-based infections. Recent evidence suggests that neutrophils can be central components of intravascular immunity in response to circulating pathogens. Indeed, neutrophils help to prevent circulating pathogens from spreading and are also involved in intravascular microbicidal activities.2,3 To protect the host against pathogens, neutrophils expel neutrophil extracellular traps (NET).4 NET are mainly formed by decondensed nucleosomes and proteins derived from intracellular granules, such as neutrophil elastase (NE) and myeloperoxidase. Thus, apart from their known capacity to restrict infections by intracellular mechanisms, neutrophils also use extracellular tools to protect the host from infection-induced damage.
Activation of intravascular blood coagulation and the formation of thrombi in microvessels (microvascular thrombosis) under certain conditions support a distinct mechanism of intravascular immunity named immunothrombosis.2 This biological form of “protective” thrombosis immobilizes circulating bacteria, restricts tissue invasion, and limits the survival of circulating bacteria in organs such as the liver and spleen. NET/extracellular nucleosomes were identified as major effectors of intravascular immunity supported by microvascular thrombosis in mice in vivo.5 In parallel, it was shown that NET derived from neutrophils are present at sites of pathological thrombus formation in large arteries in experimental mouse models as well as in coronary thrombi of patients.6 Notably, a substantial fraction of neutrophil-derived extracellular nucleosomes did not exhibit the typical morphology of NET and were present in fragmented forms.
Apart from being causally involved in microvascular thrombosis as part of the physiological host response to bacterial infection, extracellular nucleosomes, the major constituents of NET, were shown to critically promote the development of arterial thrombosis in mouse models in vivo.5 Besides this, NET were also detected in animal models of deep vein thrombosis and were shown to enhance thrombus formation in vivo.7,8 Thus, neutrophils and NET/extracellular nucleosomes are crucial promoters of thrombosis under physiological and pathological conditions, in different vascular beds, and under diverse conditions of vessel injury and infection. This review summarizes the mechanisms supporting propagation of thrombosis by neutrophils. In particular, we discuss how NET/nucleosomes are generated/externalized at the cellular level, demonstrate the molecular events supporting their procoagulant functions, as well as emphasize the critical role of neutrophils in the activation of various types of thrombosis in vivo.
Mechanisms of chromatin release from activated/dead cells and host defense functions of neutrophil extracellular traps
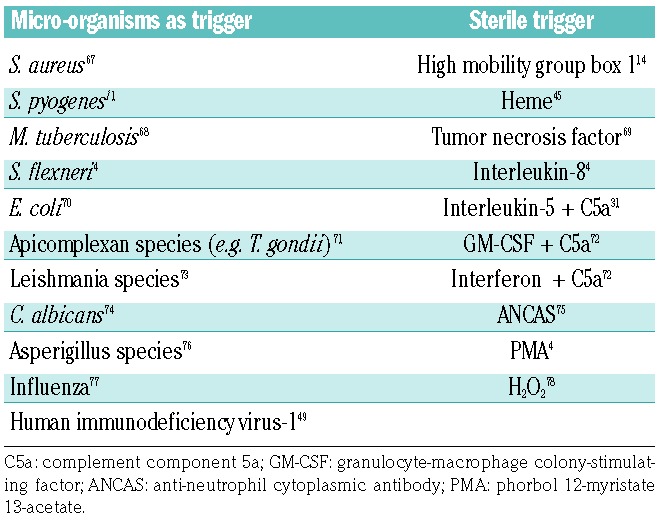
Various triggers such as cytokines, bacterial components, the experimental agonist phorbol-12-myristate-13-acetate, or activated platelets can stimulate the activation of neutrophils (Table 1), which in turn leads to degranulation and the concomitant release of decondensed chromatin fibers (designated as NET) into extracellular compartments.9 Such fibers, which can also be formed by eosinophils and mast cells and greatly exceed the size of the neutrophils themselves, enable trapping, and eventually, killing of micro-organisms. The process of NET formation, also called NETosis, is dependent on the enzyme peptidylarginine deiminase-4 (PAD4).10 PAD4 catalyzes the conversion of histone-associated arginine residues into the non-canonical amino acid citrulline. This is mediated by the conversion of the imino group of arginine into a keto-group. Replacement of arginine residues by citrullines in turn causes dissociation of histones from the tightly packed DNA backbone that is wrapped around unmodified histones, and thereby induces chromatin decondensation.
Table 1.
Triggers of NETosis.
In parallel, nuclear membranes begin to vesiculate and neutrophil granules disintegrate. Thereby, granule proteins, including myeloperoxidase, come into contact with nuclear components such as chromatin. Myeloperoxidase is a major trigger for NET generation in addition to PAD4.9 Further molecules involved in NET formation remain to be identified. Disintegration of the nuclei and granules allows the fusion of different intracellular membranes. The cells round up and in an abrupt event, the decondensed chromatin, together with various granule components, is expelled to form NET. NET were found to capture Gram-positive and Gram-negative bacteria and to be responsible for microbicidal activities in vitro.4 In line with this, neutrophils isolated from PAD4-deficient mice (PAD4−/−) showed reduced NET formation and microbicidal activities. In addition, these mice were found to be more susceptible to infection with S. pyogenes in vivo.11 Formation of NET does not necessarily result in neutrophil death. Indeed, following NET formation the generated neutrophil fragments have been described to be present in abscesses in vivo.12
Apart from activated cells, also dying or dead cells, in particular apoptotic cells, can release nucleosomes.13 Extracellular nucleosomes, especially when complexed with high mobility group box protein 1, can activate antigen-presenting cells, including dendritic cells, and thereby disrupt tolerance against nucleosomes/double-stranded DNA which might favor autoimmune diseases such as systemic lupus erythematosus.14 Stimulation of NETosis in lupus, which might be supported by reactive oxygen species originating from mitochondria, could well contribute to the increased risk of both arterial and venous thrombosis in this disease.15 In the case of tumor cells, nucleosome release has been shown to occur from both apoptotic and necrotic cells.16 The molecular mechanisms supporting release of nucleosomes from apoptotic and late apoptotic cells are largely unknown. It has been suggested that the serine protease factor seven activating protease (FSAP or hyaluronic acid binding protein-2) plays a major role in the release reaction,17 whereby FSAP is found associated with the released nucleosomes. Similarly to RNA, the DNA components of nucleosomes could also promote auto-activation of FSAP, which in turn might contribute to some of the prothrombotic actions of extracellular nucleosomes, given that FSAP supports arterial thrombosis.18, 19
Extracellular nucleosomes enhance blood coagulation and platelet activation
Platelets are increasingly recognized as critical players in immune responses.20–22 Their immunoregulatory effects are in part related to platelet interactions with innate immune cells such as neutrophils and monocytes. In line with this, activated platelets are substantially involved in the formation of NET by neutrophils.5,23 This can be mediated by high mobility group box protein 1 exposed on the surface of activated platelets and by adhesive interactions of platelet P-selectin with neutrophil P-selectin glycoprotein ligand-1.24,25 The expelled NET in turn allow the adhesion of additional platelets and promote their activation, mediated in particular by histones.7,26 NET likely also bind von Willebrand factor as well as fibrinogen.
Interactions between activated platelets and activated neutrophils can result in an activation of the extrinsic pathway of coagulation both in human and mouse systems.5,27 Fibrin formation via the extrinsic pathway is centrally initiated by the cell membrane protein tissue factor (TF). P2Y12-mediated platelet activation by the platelet agonist ADP plays a critical role in induction of TF activity by platelet-neutrophil interactions.28 Such procoagulant activity can originate in principle from neutrophil TF, platelet TF or both.27,29 Nonetheless, so far no consensus has been reached regarding the functional relevance of neutrophil and platelet TF. Neutrophils and platelets release microparticles that have been suggested to express TF and to be trapped by NET.30 In line with this, TF has been detected in association with NET inside venous thrombi in vivo.8 Eosinophils, which expel DNA-histone complexes upon activation as well, serve as a major intravascular pool of TF.31,32 Thus, under certain conditions, neutrophils and eosinophils might co-expose TF and extracellular DNA.
To prevent pathological vessel occlusion due to intravascular blood coagulation, it is essential that the potent procoagulant role of TF can be silenced under physiological conditions. Suppression of TF activation is mediated by different mechanisms, including proteins that prevent the translocation of phosphatidylserine to the outer leaflet of the plasma membrane, given that phosphatidylserine exposure activates TF.33 Protein disulfide isomerase serves as another mechanism to promote the extrinsic pathway by oxidation (internal disulfide bond formation) of TF.34 Protein disulfide isomerase, which can be released from activated platelets and from endothelial cells at sites of vascular injury, has been shown to be associated with NET in deep vein thrombosis.8
Activation of the extrinsic pathway of blood coagulation by TF is controlled by the natural anticoagulant protein tissue factor pathway inhibitor (TFPI), which directly inhibits coagulation factors VIIa and Xa involving the Kunitz 1 and 2 domains of TFPI. To disinhibit TF-driven blood coagulation, neutrophil serine proteases such as NE and cathepsin G, locally degrade and thereby inactivate TFPI.5 Since these proteases and TFPI both bind to NET, they greatly enhance inactivation of TFPI.5 Isolated nucleosomes (derived from insects and mammalian cells) mimic this effect, and can thus also act as a stimulus of TF-dependent fibrin formation. While the role of NET in promoting intravascular fibrin formation and thrombosis in vivo has been relatively well documented (see below), the question as to whether nucleosomes released from normal apoptotic cells and/or tumor cells also act as prothrombotic triggers requires future investigation.
Apart from activating the extrinsic pathway of blood coagulation, NET can also promote factor XII activation, and thereby stimulate the contact pathway of blood coagulation.8 Moreover, NET might enhance fibrin formation by inhibiting tissue plasminogen activator-mediated fibrinolysis.35 The procoagulant mechanisms induced by NET, in particular their ability to increase blood coagulation via TFPI degradation, are likely mediated in part by nucleic acids, which provide a polyanionic surface that, like other polyanions (including, for example, RNA and polyphosphates), allows the proteolytic activation of coagulation factors such as factor XII.36–38 NE-dependent degradation of endogenous anticoagulants, activation of the contact pathway, and inhibition of fibrinolysis, support propagation of blood coagulation, rather than triggering its initiation. Overall, neutrophils and NET thus operate through multiple pathways that propagate fibrin formation and enhance recruitment and activation of platelets.
Immunothrombosis: intravascular fibrin formation as part of the innate immune defense
Systemic bacterial infections can be a lethal threat to an organism. The mechanisms of host defense against infections by circulating bacteria are still not fully understood. Recently, it was shown that neutrophil serine proteases (NE, cathepsin G) and NET/extracellular nucleosomes trigger the formation of immunothrombosis in liver and spleen sinusoids during systemic bacterial infection in vivo.5 The ability of NET to promote fibrin formation enables microvascular thrombi to limit the dissemination, tissue invasion as well as the survival of circulating E. coli. The procoagulant role of neutrophils and their released NET during immunothrombosis critically depends on neutrophil serine proteases such as NE. NET can thus serve as a platform for NE-mediated activation of intravascular coagulation in vivo. Consistent with the role of NE-induced TFPI cleavage for the antimicrobial activity of intravascular blood coagulation, infusion of a TFPI mutant specifically resistant to cleavage by NE and cathepsin G (T87F/L89A), which almost completely suppressed microvascular fibrin formation, markedly increased tissue invasion of E.coli and enhanced bacterial survival.5
However, apart from its beneficial role in combating circulating pathogens, NET-induced microvascular thrombosis under certain conditions can become detrimental to the host.39,40 This is particularly true for disseminated intravascular coagulation, a serious complication of sepsis, which is likely a direct pathological consequence of immunothrombosis. In line with this, NET have been shown to foster the development of sepsis.41 In particular, NET have been detected in several organs during sepsis, including lungs, or even circulating in the systemic blood stream.42 Thus, in severe sepsis, the prothrombotic actions of NET may have deleterious side effects on the blood supply and functions of multiple organs. In line with a role of NET in pathological microvascular thrombosis in humans, patients with acute thrombotic microangiopathies show impaired DNase-mediated NET degradation.43 Moreover, NET, predominantly via their histone components, can directly induce endothelial (and epithelial) cell death.44
In addition to their role in microbial infections, NET are also main regulators of microvascular thrombosis in sterile inflammatory processes and tumor cell metastasis. Indeed, NET are involved in veno-occlusive crises of sickle cell disease and contribute significantly to the mortality associated with this disease.45 Interestingly, heme released from lysed erythrocytes was identified as a new trigger for NETosis under these conditions. In sickle cell crises, NET do not only cause microvascular thrombosis, but also generate excessive damage to pulmonary tissue, the main cause of mortality in this setting, which could be reversed by DNase I treatment. Similarly, NET have been detected within the pulmonary microcirculation during transfusion-related acute lung injury, and contribute significantly to morbidity and mortality by increasing endothelial permeability.46 Another pathological side-effect of NET formation in the microcirculation may be promotion of tumor metastasis, whereby NET formed in the liver sinusoids in response to infection have been shown to support the adhesion and trapping of circulating tumor cells.47
NETosis can also be detected in viral infections and various viruses (such as influenza and human immunodeficiency virus-1) are able to induce the formation of NET, which may bind and thereby neutralize viruses.48,49 A new host-protective effect of NET has been described in fungal infections: neutrophils that are exposed to a micro-organism such as C. albicans, which cannot be phagocytosed because of its large size, initiate NETosis to capture the pathogen. Vice versa, if the microbe can be phagocytosed by the neutrophil, NET formation is inhibited.50 Interestingly, NET may not only stimulate, but may also restrict inflammatory reactions. Once they have formed densely packed aggregates, NET can degrade neutrophil-derived inflammatory mediators by means of their own serine proteases, thereby limiting inflammatory reactions during gout.51 However, it is still not clear whether this mechanism is also relevant for resolution of microbial infections and microvascular thrombosis. Hence, neutrophils and the procoagulant mechanisms supported by them can be seen to be as efficient tools in fighting bacterial and viral infections, but can also cause substantial collateral damage to host tissues.
Detection and functional role of neutrophil extracellular traps in arterial thrombosis
While the development of microvascular thrombosis in general is relatively slow in nature and triggered by diverse stimuli, arterial thrombosis is a fast process with a uniform trigger, especially disruption of the endothelial cell layer and exposure of the subendothelial matrix, following rupture at sites of atherosclerotic plaques. The sudden loss of blood supply induced by thrombosis in coronary arteries results in myocardial infarction and stroke. TF plays a central role in inducing arterial thrombosis. TF is highly concentrated in atherosclerotic plaques in both cellular and acellular regions. Plaque rupture leads to the exposure of TF to the blood and together with platelet adhesion, activation and aggregation at sites of turbulent flow these interactions lead to the rapid development of arterial occlusions.52
Notably, it could be demonstrated that neutrophils and NET/extracellular nucleosomes are also of major relevance for the development of arterial thrombosis. Using a model of ligation-induced thrombosis of the carotid artery, NET/extracellular nucleosomes were detected in association with neutrophils adhering to the damaged endothelium in vivo.5 The NET detected in arterial thrombi were intact in some cases; in other cases, they were fragmented. These fragmented NET were clearly derived from neutrophils, as they stained positive for myeloperoxidase. Blocking NET with anti-H2A/H2B-DNA antibody decreased fibrin formation at the site of injury. Moreover, this treatment delayed the time to vessel occlusion and strongly reduced the duration of occlusion without affecting firm adhesion of platelets.5 These findings established for the first time a causal role for NET in large vessel thrombosis in mouse models.
The procoagulant mechanisms induced by neutrophils in arterial thrombosis partially overlapped with the mechanisms supporting the development of microvascular thrombosis. Accordingly, fibrin formation and arterial vessel occlusion were strongly reduced in mice deficient for neutrophil serine proteases.5 As mentioned, TFPI inactivation by NET-associated neutrophilic proteases participates in arterial thrombosis since TFPI was degraded at the site of vessel injury and thrombus formation in wild-type mice, but not in neutrophil serine protease-deficient animals. Moreover, the TFPI mutant T87F/L89A, which is resistant to cleavage by neutrophil serine proteases, decreased arterial thrombosis more efficiently than did native TFPI.
The importance of these findings is underscored by the detection of NET in association with neutrophils in specimens of human coronary thrombi (Figure 1) and at lesion sites of patients with acute myocardial infarction and stent thrombosis.6,53,54 NET could only be detected in early stages of coronary thrombosis, but not in organized thrombi, which could be attributed to digestion of NET by plasma DNases or disruption of NET by heparin treatment applied during cardiac catheterization.53 Increased NET burden in coronary thrombi correlated with infarct size, and DNase treatment (in particular together with tissue plasminogen activator) promoted resolution of coronary thrombi ex vivo, suggesting that a combination of established antithrombotic and NET-disrupting therapy might be beneficial in the therapy of acute coronary syndromes.6 In addition, neutrophils are by far the most important innate leukocyte subtype in stent thrombosis.54 Apart from neutrophils, eosinophils were present in all types of stent thrombosis, indicating a highly sensitive determinant that could induce thrombosis via eosinophil-associated TF.32
Figure 1.

Neutrophil extracellular traps (blue; arrows) and neutrophil elastase (red) in thrombi recovered from human coronary arteries. Left and right images show thrombi from two different patients.
Neutrophils propagate venous thrombosis
Deep vein thrombosis and its complication pulmonary embolism are frequent disorders that contribute considerably to the mortality associated with cardiovascular diseases. One of the main triggers of venous thrombosis is decreased or stagnant blood flow as in the setting of immobilization. Using a mouse model of reduced blood flow in the inferior vena cava it was demonstrated that the processes leading to occlusion of venous vessels are reminiscent of microvascular thrombosis.2,8,55 The development of deep vein thrombosis was found to be driven by a tight co-operation between platelets, monocytes and neutrophils resulting in both the initiation and propagation of fibrin formation. In particular, neutrophil-derived extracellular nucleosomes were detected in venous thrombi in mice in vivo.7,8 Overall, NET profoundly enhanced deep vein thrombosis by a yet to be analyzed mechanism.8 Potentially, NET might play different roles in the development of venous thrombosis. For example, NET could damage the endothelium in large veins through the cytotoxic potential of histones.
Platelet-neutrophil interactions at the site of deep vein thrombosis formation were found to induce NETosis and to be of substantial relevance for thrombogenesis in the context of deep vein thrombosis in general.8 NET in turn support propagation of blood coagulation as indicated by the inhibition of fibrin formation following infusion of anti-H2A/H2B-DNA antibody and DNase I. Notably, NE-dependent TFPI degradation, which contributes to arterial thrombosis, is not an essential element for deep vein thrombosis, suggesting that alternate, yet unknown pathways support the NET-induced thrombotic process in veins. (K. Stark, B. Engelmann, S. Massberg, unpublished data, 2017;56) Fibrin formation during development of deep vein thrombosis was dependent both on the extrinsic and on the contact pathways of blood coagulation. Correspondingly, intravascular TF most likely expressed by monocytes, but to a lesser extent exposed on NET, as well as factor XIIa critically mediated venous thrombosis. This was suggested by experiments performed with mice lacking hematopoietic TF or myeloid TF and in FXII-deficient mice.8 FXII can bind to extracellular chromatin and thus be activated, which contributes to the propagation of intravascular clot formation. NET, apart from activating the coagulation system during venous thrombosis also bind platelets, a process mediated by von Willebrand factor.7,8 In addition, NET can bind erythrocytes, and histones can induce a procoagulant phenotype in these anucleated cells by inducing the exposure of phosphatidylserine; nevertheless, the relevance of this observation for deep vein thrombosis in vivo is unclear.8,57
Recently, it has been described that NET and circulating nucleosomes are present in human thromboembolism, suggesting that extracellular nucleosomes may be of relevance to deep vein thrombosis in patients.58,59 Apart from immobilization, cancer is another important risk factor for venous thrombosis and is associated with hypercoagulability, which could in part be explained by an increased activation of neutrophils and their enhanced ability to form NET in tumor-bearing mice.60
Neutrophil extracellular traps/extracellular chromatin as a marker and therapeutic target of thrombosis
In line with the central role of neutrophils and extracellular chromatin in different types of experimental thrombosis, extracellular nucleosomes and distinct components of them such as citrullinated histones have been shown to be increased in plasma of patients with sepsis, arterial thrombosis, atherosclerosis, and in deep vein thrombosis.5–8,41,53,58,59,61–63 Since nucleosomes are not only externalized by neutrophils but also by apoptotic and necrotic cells, and since the plasma levels of nucleosomes have been shown to be increased under various pathological conditions (e.g. ischemia/reperfusion, cancer), the diagnostic evaluation of nucleosome-driven thrombosis requires the use of additional markers.64
Additional markers might include, for example, plasma markers of neutrophil activation such as NE as well as D-dimer levels.6,58 Inhibition of the prothrombotic functions of NET/extracellular nucleosomes by specific antibodies, such as anti-H2A/H2B-DNA antibody, or their degradation by DNase I robustly inhibits thrombosis in different vascular beds in animal models.5,7,8 Inhibition of NET formation in PAD4-deficient mice does not change bleeding times.65 Future studies will need to address in more detail whether blocking/degrading NET is associated with bleeding complications. Overall, neutralizing antibodies targeting nucleosomes and their histone components, DNases as well as PAD4 inhibitors, are interesting candidate molecules for the prevention of various types of thrombosis in humans.66
Conclusions
During infections and inflammatory responses, neutrophils promote intravascular blood coagulation and thrombosis. A major mechanism allowing neutrophils to shape platelet activation and fibrin formation is via extrusion of NET/extracellular nucleosomes (Figure 2). Platelets are critically involved in neutrophil-regulated thrombosis since they promote NET formation and are themselves activated by extracellular chromatin. NET-induced blood coagulation is probably a key mediator of intravascular immunity which, supported by microvascular thrombosis, restricts the dissemination and survival of circulating bacteria. However, the defense against circulating pathogens by neutrophil-induced prothrombotic mechanisms most likely comes at a high cost. Indeed, NET markedly promote vessel occlusion in experimental models of arterial and deep vein thrombosis. Moreover, high numbers of neutrophils and extracellular nucleosomes have been detected in thrombi and blood of patients with arterial and deep vein thrombosis. Overall, this suggests that neutrophils and their NET may contribute to cardiovascular diseases induced by thrombosis, such as myocardial infarction, stroke, and venous thromboembolism.
Figure 2.

Mechanisms of neutrophil extracellular trap(NET)-mediated thrombosis (model). PSGL-1: P-selectin glycoprote in ligand-1; HMGB1: high mobility group box 1; RAGE: receptor for advanced glycation end-products; PAD4: peptidylarginine deiminases 4; NE: neutrophil; MPO: myeloperoxidase; TF: tissue factor; MP: microparticle; vWF: von Willebrand factor.
As illustrated in Figure 2, NETosis can be induced by interactions of activated platelets (red) with neutrophils (blue), which result in the formation of intact and fragmented NET in different vascular beds in vivo. The externalized nucleosomes promote the propagation of intravascular blood coagulation, von Willebrand factor binding, and platelet adhesion/activation, which fosters thrombosis both in the microcirculation and in large vessels. During infection and inflammation, TF may be exposed on NET-embedded microparticles, whereby platelet-neutrophil conjugates could also participate in the initiation of intravascular fibrin formation.
Supplementary Material
Acknowledgment
Work performed in the laboratories of Bernd Engelmann and Steffen Massberg mentioned in this review has been supported by grants from the DFG, including SFB 1123, and from Deutsche Krebshilfe.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/2/206
References
- 1.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–531. [DOI] [PubMed] [Google Scholar]
- 2.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45. [DOI] [PubMed] [Google Scholar]
- 3.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. [DOI] [PubMed] [Google Scholar]
- 5.Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16(8):887–896. [DOI] [PubMed] [Google Scholar]
- 6.Mangold A, Alias S, Scherz T, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015;116(7):1182–1192. [DOI] [PubMed] [Google Scholar]
- 7.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107(36): 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012; 198(5):773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yipp BG, Petri B, Salina D, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18(9):1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisetsky DS. The origin and properties of extracellular DNA: from PAMP to DAMP. Clin Immunol. 2012;144(1):32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urbonaviciute V, Furnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205(13):3007–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61(4):1659–1665. [PubMed] [Google Scholar]
- 17.Zeerleder S, Zwart B, te Velthuis H, et al. Nucleosome-releasing factor: a new role for factor VII-activating protease (FSAP). FASEB J. 2008;22(12):4077–4084. [DOI] [PubMed] [Google Scholar]
- 18.Nakazawa F, Kannemeier C, Shibamiya A, et al. Extracellular RNA is a natural cofactor for the (auto-)activation of factor VII-activating protease (FSAP). Biochem J. 2005;385(Pt 3):831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramaniam S, Thielmann I, Morowski M, et al. Defective thrombus formation in mice lacking endogenous factor VII activating protease (FSAP). Thromb Haemost. 2015;113(4):870–880. [DOI] [PubMed] [Google Scholar]
- 20.Duerschmied D, Bode C, Ahrens I. Immune functions of platelets. Thromb Haemost. 2014;112(4):678–691. [DOI] [PubMed] [Google Scholar]
- 21.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. [DOI] [PubMed] [Google Scholar]
- 22.Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. 2014;12(6):426–437. [DOI] [PubMed] [Google Scholar]
- 23.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. [DOI] [PubMed] [Google Scholar]
- 24.Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074–2088. [DOI] [PubMed] [Google Scholar]
- 25.Sreeramkumar V, Adrover JM, Ballesteros I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller I, Klocke A, Alex M, et al. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17(3):476–478. [DOI] [PubMed] [Google Scholar]
- 28.Leon C, Alex M, Klocke A, et al. Platelet ADP receptors contribute to the initiation of intravascular coagulation. Blood. 2004;103(2):594–600. [DOI] [PubMed] [Google Scholar]
- 29.Maugeri N, Brambilla M, Camera M, et al. Human polymorphonuclear leukocytes produce and express functional tissue factor upon stimulation. J Thromb Haemost. 2006;4(6):1323–1330. [DOI] [PubMed] [Google Scholar]
- 30.Kambas K, Mitroulis I, Apostolidou E, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One. 2012;7(9):e45427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yousefi S, Gold JA, Andina N, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14(9):949–953. [DOI] [PubMed] [Google Scholar]
- 32.Moosbauer C, Morgenstern E, Cuvelier SL, et al. Eosinophils are a major intravascular location for tissue factor storage and exposure. Blood. 2007;109(3):995–1002. [DOI] [PubMed] [Google Scholar]
- 33.Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide iso-merase in tissue factor activation. Thromb Haemost. 2014;111(4):590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reinhardt C, von Bruhl ML, Manukyan D, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118(3):1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varju I, Longstaff C, Szabo L, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost. 2015; 113(6):1289–1298. [DOI] [PubMed] [Google Scholar]
- 36.Kannemeier C, Shibamiya A, Nakazawa F, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA. 2007;104(15):6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller F, Mutch NJ, Schenk WA, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139(6):1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. 2006;103(4):903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfeiler S, Massberg S, Engelmann B. Biological basis and pathological relevance of microvascular thrombosis. Thromb Res. 2014;133 (Suppl 1)S35–37. [DOI] [PubMed] [Google Scholar]
- 40.Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost. 2015;13 (Suppl 1)S82–91. [DOI] [PubMed] [Google Scholar]
- 41.Camicia G, Pozner R, de Larranaga G. Neutrophil extracellular traps in sepsis. Shock. 2014;42(4):286–294. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka K, Koike Y, Shimura T, et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One. 2014;9(11):e111888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jimenez-Alcazar M, Napirei M, Panda R, et al. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J Thromb Haemost. 2015;13(5):732–742. [DOI] [PubMed] [Google Scholar]
- 44.Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7(2):e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123(24):3818–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cools-Lartigue J, Spicer J, McDonald B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123(8):3446–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narasaraju T, Yang E, Samy RP, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saitoh T, Komano J, Saitoh Y, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12(1):109–116. [DOI] [PubMed] [Google Scholar]
- 50.Branzk N, Lubojemska A, Hardison SE, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15(11):1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20(5):511–517. [DOI] [PubMed] [Google Scholar]
- 52.Jackson SP. Arterial thrombosis–insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. [DOI] [PubMed] [Google Scholar]
- 53.de Boer OJ, Li X, Teeling P, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost. 2013;109(2):290–297. [DOI] [PubMed] [Google Scholar]
- 54.Riegger J, Byrne RA, Joner M, et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: a report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur Heart J. 2016;37(19):1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. J Thromb Haemost. 2013;11 (Suppl 1)233–241. [DOI] [PubMed] [Google Scholar]
- 56.Martinod K, Witsch T, Farley K, Gallant M, Remold-O’Donnell E, Wagner DD. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost. 2016;14(3):551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semeraro F, Ammollo CT, Esmon NL, Esmon CT. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J Thromb Haemost. 2014;12(10):1697–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Montfoort ML, Stephan F, Lauw MN, et al. Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2013;33(1):147–151. [DOI] [PubMed] [Google Scholar]
- 59.Savchenko AS, Martinod K, Seidman MA, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost. 2014;12(6):860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Demers M, Krause DS, Schatzberg D, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA. 2012;109(32):13076–13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeerleder S, Zwart B, Wuillemin WA, et al. Elevated nucleosome levels in systemic inflammation and sepsis. Crit Care Med. 2003;31(7):1947–1951. [DOI] [PubMed] [Google Scholar]
- 62.Megens RT, Vijayan S, Lievens D, et al. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost. 2012;107(3):597–598. [DOI] [PubMed] [Google Scholar]
- 63.Borissoff JI, Joosen IA, Versteylen MO, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33(8):2032–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holdenrieder S, Stieber P. Clinical use of circulating nucleosomes. Crit Rev Clin Lab Sci. 2009;46(1):1–24. [DOI] [PubMed] [Google Scholar]
- 65.Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110(21):8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta. 2013;1829(10):1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pilsczek FH, Salina D, Poon KK, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185(12):7413–7425. [DOI] [PubMed] [Google Scholar]
- 68.Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb). 2009;89(1):29–37. [DOI] [PubMed] [Google Scholar]
- 69.Gupta AK, Joshi MB, Philippova M, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584(14):3193–3197. [DOI] [PubMed] [Google Scholar]
- 70.Grinberg N, Elazar S, Rosenshine I, Shpigel NY. Beta-hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infect Immun. 2008;76(6):2802–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abi Abdallah DS, Lin C, Ball CJ, King MR, Duhamel GE, Denkers EY. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect Immun. 2012;80(2):768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martinelli S, Urosevic M, Daryadel A, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem. 2004;279(42):44123–44132. [DOI] [PubMed] [Google Scholar]
- 73.Guimaraes-Costa AB, Nascimento MT, Froment GS, et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci USA. 2009;106(16):6748–6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006;8(4):668–676. [DOI] [PubMed] [Google Scholar]
- 75.Kessenbrock K, Krumbholz M, Schonermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15(6):623–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bruns S, Kniemeyer O, Hasenberg M, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6(4):e1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One. 2011;6(7):e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.