

Abstract
The development of inhibitory antibodies to therapeutic factor VIII is the major complication of replacement therapy in patients with hemophilia A. The first step in the initiation of the anti-factor VIII immune response is factor VIII interaction with receptor(s) on antigen-presenting cells, followed by endocytosis and presentation to naïve CD4+ T cells. Recent studies indicate a role for the C1 domain in factor VIII uptake. We investigated whether charged residues in the C2 domain participate in immunogenic factor VIII uptake. Co-incubation of factor VIII with BO2C11, a monoclonal C2-specific immunoglobulin G, reduced factor VIII endocytosis by dendritic cells and presentation to CD4+ T cells, and diminished factor VIII immunogenicity in factor VIII-deficient mice. The mutation of basic residues within the BO2C11 epitope of C2 replicated reduced in vitro immunogenic uptake, but failed to prevent factor VIII immunogenicity in mice. BO2C11 prevents factor VIII binding to von Willebrand factor, thus potentially biasing factor VIII immunogenicity by perturbing its half-life. Interestingly, a factor VIIIY1680C mutant, that does not bind von Willebrand factor, demonstrated unaltered endocytosis by dendritic cells as well as immunogenicity in factor VIII-deficient mice. Co-incubation of factor VIIIY1680C with BO2C11, however, resulted in decreased factor VIII immunogenicity in vivo. In addition, a previously described triple C1 mutant showed decreased uptake in vitro, and reduced immunogenicity in vivo, but only in the absence of endogenous von Willebrand factor. Taken together, the results indicate that residues in the C1 and/or C2 domains of factor VIII are implicated in immunogenic factor VIII uptake, at least in vitro. Conversely, in vivo, the binding to endogenous von Willebrand factor masks the reducing effect of mutations in the C domains on factor VIII immunogenicity.
Introduction
Hemophilia A is a monogenic disorder associated with mutations causing reductions in functional levels of coagulation factor VIII (FVIII). FVIII consists of a heavy chain (A1-a1-A2-a2-B domain) and a light chain (a3-A3-C1-C2) held together by non-covalent interactions.1 It rapidly associates with von Willebrand factor (VWF) in blood, and this interaction is necessary for maintaining its circulatory half-life.2 Current treatment for FVIII deficiency requires prophylactic infusions of plasma-derived or recombinant FVIII. However, up to 30% of patients with severe hemophilia A develop an anti-FVIII immune response, thus rendering treatment ineffective.3 The development of anti-FVIII antibody responses is dependent on T helper cells, requiring antigen uptake and presentation by antigen-presenting cells (APCs).4 Hence, understanding the initial steps involved in FVIII capture by APCs may provide novel strategies to prevent the onset of the immune response.
Several groups, including ours, have investigated the endocytic pathways involved in FVIII uptake. Candidate receptors such as macrophage mannose receptor (MMR, CD206), low-density lipoprotein receptor-related protein (LRP, CD91), or other receptor-associated protein (RAP)-sensitive receptors have been proposed.5–10 Equally, the nature of the residues within FVIII domain(s) that contribute to these interactions is also an area of active investigation. Despite these efforts, the in vivo relevance of these receptors and the nature of the FVIII residues involved in FVIII uptake remain unclear. Recently, Herczenik et al.10 demonstrated that KM33, a human C1 domain-specific monoclonal immunoglobulin G (IgG), inhibits FVIII endocytosis by monocyte-derived dendritic cells (MoDCs) or mouse bone marrow-derived dendritic cells (BMDCs). KM33 engages K2092, F2093 and R2090 residues, involved in the interactions with phospholipid membrane surfaces.11 Additionally, KM33 inhibits interactions of the C1 domain with membrane surfaces, VWF and LRP.12 FVIII uptake by LRP in MoDCs, used as model APCs, has been ruled out,8 while a role for CD206 has been controversially documented.7,9,10 This suggests that FVIII uptake by APCs may involve other endocytic receptor(s). Importantly, FVIII mutants containing alanine substitutions of the K2092, F2093 and R2090 C1 residues, exhibit diminished uptake in vitro and reduced immunogenicity in a mouse model of severe hemophilia A.11 Together, these results point to the significance of membrane-binding residues within the C1 domain for FVIII uptake both in vitro and in vivo. Similar to the C1 domain, the C2 domain of FVIII interacts with phospholipid membrane surface, a binding that involves several basic residues.13
In the study herein, we investigated whether membrane-interacting residues within the C2 domain of FVIII are involved in FVIII uptake. We first show that BO2C11, a well-characterized human monoclonal IgG that engages membrane-binding residues in the C2 domain,14 inhibits FVIII uptake and presentation in vitro, and reduces FVIII immunogenicity in vivo. We also demonstrate that this reduced immunogenicity is independent of the ability of FVIII to interact with endogenous VWF. Additionally, by site-directed mutagenesis, we demonstrate that the R2215 residue, which is part of the BO2C11 epitope, is implicated in the cellular uptake of FVIII by APCs in vitro. Together with the published data, our results suggest a potential synergy between membrane-binding residues in both the C1 and C2 domains of FVIII in mediating FVIII recognition or uptake by APCs in vitro. Furthermore, we also demonstrate that FVIII C domain mutations exhibit diminished immunogenicity in vivo only in the absence of endogenous VWF.
Methods
Reagents
Recombinant human FVIII (Refacto) came from Pfizer (New York, NY, USA). Murine granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4) came from Cellgenix Technology Transfer (Freiburg, Germany). The monoclonal mouse anti-FVIII (mAb6), the human anti-A2 (BO2BII) and anti-C2 (BO2C11) domains antibodies were kind gifts from Drs JM Saint-Remy and M Jacquemin (KUL, Leuven, Belgium). The monoclonal human-derived anti-C1 (KM33) was a gift from Dr J Voorberg (Sanquin, Amsterdam, The Netherlands). The mouse monoclonal anti-A2 domain (GMA-8015) and anti-C2 domain (ESH8) antibodies were purchased from Green Mountain Antibodies (Burlington, VT, USA) and Sekisui Diagnostics (Kings Hill, Kent, UK), respectively. Biotin-labeled GMA-8015 was prepared upon incubation with a 20-fold molar excess of EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, Courtaboeuf, France) for 30 min at room temperature, and the removal of excess biotin by diafiltration was carried out using 30 kDa Amicon Ultra-15 centrifugal filter units (Merck Millipore, Saint-Quentin-en-Yvelines, France). BO2C11 fragment antigen binding (Fab) or F(ab’)2 fragments were digested by papain or pepsin following the manufacturer’s instructions (Thermo Fisher Scientific, Courtaboeuf, France).
Production and purification of recombinant mutated or wild-type FVIII
Complementary DNA (cDNA) encoding human B-domain deleted (BDD) FVIII (FVIIIHSQ), containing the 14-amino acid segment SFSQNPPVLKRHQR in place of the B domain, cloned in the ReNeo mammalian expression plasmid with a geneticin resistance, has been described previously.15 The cDNA encoding FVIIIHSQ was used as a template to generate the R2215A, R2220A, R2215A-R2220A, R2090A-K2092A-F2093A and Y1680C FVIII mutants by splicing by overlap extension mutagenesis as described in the Online Supplementary Methods. The presence of the mutations was confirmed by standard sequencing analysis. The stable expression of wild-type and mutated FVIII by baby hamster kidney-derived cells, and FVIII purification, were performed as described in the Online Supplementary Methods. The concentration of purified wild-type and mutated FVIII was calculated by absorbance at 280 nm using a molar extinction coefficient of 256,300 M−1cm−1 and a molecular weight of 165,300 Da. Specific activities were estimated by a one-stage clotting assay and ranged between 4800–9000 IU/mg. The sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) migration profiles of the different purified recombinant FVIII are shown in the Online Supplementary Figure S1. In parallel, the ability of the different FVIII molecules to generate activated factor X was assessed in a chromogenic assay (Siemens Healthcare Diagnostics, Marburg, Germany).
FVIII binding to VWF and monoclonal antibodies
Ninety-six-well ELISA plates (Maxisorp, Nunc, Roskilde, Denmark) were coated with human plasma-derived VWF (Wilfactin, LFB, Les Ulis, France), BO2C11, BO2BII, ESH8 or KM33 at 1 μg/ml in bicarbonate buffer, pH 9.5, for 1 hr at 37°C. Wells were blocked with 20 mM HEPES, 0.15 M NaCl, 0.05% Tween 20 and 5% bovine serum albumin (BSA), pH 7.4. Wild-type and mutated FVIII were then diluted in blocking buffer and incubated in the coated wells for 1 hr at 37°C. Bound FVIII was revealed using either biotinylated GMA-8015 (1 μg/ml), followed by streptavidin conjugated to horseradish peroxidase (R&D systems, Lille, France) or, in the case of BO2BII-bound FVIII, with ESH8 followed by a horseradish peroxidase conjugated polyclonal goat anti-mouse IgG antibody (Southern Biotech, Anaheim, CA, USA) and the o-phenylenediamine dihydrochloride (OPD) substrate (Sigma-Aldrich, Saint-Louis, MO, USA). Absorbance was read at 492 nm.
Generation of immature human MoDCs and mouse BMDCs
Monocytes were isolated from the blood of healthy donors using anti-CD14+ magnetic microbeads (Miltenyi Biotec, Paris, France). Ethics committee approval was obtained for the use of buffy bags from healthy donors. Monocytes (0.5.106 cells/ml) were cultured in RPMI-1640 (Lonza, Verviers, Belgium) with 10% fetal calf serum (Life Technologies, Saint-Aubin, France), supplemented with GM-CSF (1000 IU/106 cells) and IL-4 (500 IU/106 cells) (Miltenyi Biotec) for 5 days to generate immature MoDCs. After 5 days, the differentiation of MoDCs (> 90% purity) was confirmed by flow cytometry upon loss of CD14 staining (M5E2 clone, BD Pharmingen, San Jose, CA, USA), expression of major histocompatibility complex (MHC) class II and CD1a (HI149 and G46.6 clones, respectively, BD Pharmingen). Acquisition was performed on a LSR II cytometer with FACSDiva software (version 6.1, BD Biosciences, Le Pont au Claix, France). Murine BMDCs were generated as described previously.16 Briefly, bone marrow cells were extracted from FVIII exon 16 knock-out C57BL/6 mice and cultured in Petri dishes (2.106 cells/10 ml/plate) for 10 days in RPMI-1640 supplemented with 10% fetal calf serum, 50 mM 2-mercaptoethanol and 200 U/ml murine recombinant GM-CSF (Cellgenix Technology Transfer, Freiburg, Germany). Supplemented medium was replaced at days 3, 6 and 8. BMDCs purity and phenotype were validated by the expression of CD11c (HL3 clone, BD Pharmingen).
In vitro FVIII uptake by immature MoDCs and BMDCs
B domain-deleted FVIII (20 nM) was pre-incubated with equimolar concentrations of ESH8, BO2C11 or with a 2 molar excess BO2C11 Fab fragments for 30 min at 37°C. Samples were then incubated with 5-day-old immature MoDCs or with 9-day-old immature BMDCs (0.2.106 cells/100 μl) in Iscove’s Modified Dulbecco’s Medium for 30 min at 37°C or 4°C. Cells were washed with ice-cold phosphate buffered saline (PBS) and fixed with BD Cytofix™ Fixation buffer (BD Biosciences) for 20 min at 4°C. Cells were then permeabilized for 30 min at room temperature with permeabilization buffer (eBiosciences), and FVIII was stained using biotinylated GMA-8015 (2 μg/ml), followed by streptavidin-PE (1 μg/ml, BD Biosciences) for 30 min at room temperature. Cells were analyzed by flow cytometry. The uptake was quantified as the difference in median fluorescence intensities between 37°C and 4°C (ΔMFI37°C–4°C), to exclude the signal generated by the binding of FVIII to the cell surface.
In vitro activation of a FVIII-specific HLA-DRB1*0101-restricted mouse CD4+ T cell hybridoma
Activation of the HLA-DRB1*0101-restricted mouse CD4+ T cell hybridoma specific for human FVIII (1G8-A2), was assessed as previously described.17 FVIII (10 nM) pre-incubated or not with 2 molar excess of BO2C11 was incubated with 10,000 MoDCs or 200,000 mitomycin C-treated mouse splenocytes from SUREL1 mice and co-cultured with 100,000 T cells in X-VIVO15 medium (Life Technologies) for 18 hr at 37°C. Controls included T cells incubated alone, or incubated with MoDCs/BMDCs in the presence of concanavalin A (2 μg/ml, Sigma-Aldrich), or in the absence of FVIII. Levels of interleukin-2 (IL-2) secreted in the supernatant by T cells were assessed using the BD OptEIA™ mouse IL-2 ELISA set (BD Biosciences). Of note, MoDCs do not produce IL-2 when incubated with FVIII alone.18
Animals and administration of FVIII
Eight to 12 week-old FVIII exon 16 knock-out C57Bl/6 mice and VWF/FVIII exon 16 double knock-out C57Bl/6 mice (Professor H.H. Kazazian, University of Pennsylvania School of Medicine, Philadelphia, PA, USA, and Drs. C Denis and O Christophe, INSERM U770, Le Kremlin-Bicêtre, France, respectively), were injected intravenously once a week for 4 weeks with either: i) B-domain deleted FVIII (0.2 μg, Refacto, Pfizer) pre-incubated with equimolar amounts of F(ab’)2 fragments of an IgG4 isotype control or BO2C11, ii) B-domain deleted FVIIIY1680C (0.4 μg) alone or pre-incubated with Fab fragments of BO2C11, or iii) B-domain deleted FVIIIHSQ, FVIIIC1 or FVIIIR2215-20A (0.5 or 1 μg). The presence of endotoxins in the different recombinant FVIII was evaluated using the ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit (Genscript, Piscataway, NJ, USA). The measured values were below the accepted threshold (i.e., <0.01 ng endotoxin/20 g mouse weight). Blood was collected on heparinized capillaries by retro-orbital bleeding 4 days after the fourth administration of FVIII. Plasma was collected and kept at −80°C until use. Mice were handled in agreement with French ethical authorities (authorization #23BA53).
Titration of anti-FVIII IgG and FVIII inhibitors
ELISA plates were coated with FVIII (1 μg/ml, Recombinate®, Baxter, Maurepas, France), in bicarbonate buffer pH 9.5, overnight at 4°C. After blocking with PBS, 0.05% Tween 20 and 2% BSA, the mouse plasma was incubated for 1 hr at 37°C. Serial dilutions of the samples were incubated for 1 hr at 37°C, and bound IgG were revealed using a horseradish peroxidase conjugated polyclonal goat anti-mouse IgG antibody (Southern Biotech) and the OPD substrate. Absorbance was read at 492 nm. The monoclonal mouse anti-human FVIII IgG mAb6 was used as a standard. Titers are expressed in arbitrary units (A.U.). Inhibitory titers were estimated by incubating heat-inactivated mouse plasma with human standard plasma (Siemens Healthcare Diagnostics) for 2 hr at 37°C. The residual FVIII procoagulant activity was measured using a chromogenic assay (Siemens Healthcare Diagnostics). Results are expressed in Bethesda units (BU)/ml that correspond to the reciprocal dilution of the mouse plasma that yielded 50% residual FVIII activity.
In vivo clearance of wild-type and mutated FVIII
FVIII exon 16 knock-out mice and VWF/FVIII exon 16 double knock-out C57Bl/6 mice were injected intravenously with wild-type and mutated FVIII (10 nM in 100 μl). Blood was collected in 0.129 M sodium citrate at different time points after FVIII administration. FVIII residual levels in mouse plasma were determined by sandwich ELISA using ESH8 and biotinylated GMA-8015 as capture and detection antibodies, as described above. Data are plotted as a percentage of the initial FVIII level, measured 5 minutes after FVIII infusion, versus time (mean±SEM). Values at 5 min post-injection did not differ between the groups of mice treated with the different FVIII. Experimental data was fitted with a one-phase exponential decay equation using GraphPad Prism software (version 6.01).
Results
FVIII bound to BO2C11 induces diminished immune responses in vivo
We first evaluated whether BO2C11, a human C2-specific anti-FVIII IgG, inhibits FVIII uptake by MoDCs or BMDCs in vitro. We hypothesized that the uptake is restricted within the BO2C11 binding region14 and thus, as a control, employed another C2-specific antibody, ESH8. ESH8 does not bind to the BO2C11 epitope and, unlike BO2C11, does not inhibit FVIII binding to VWF or phosphatidylserine membrane surfaces.19 We incubated 20 nM FVIII or FVIII pre-incubated with equimolar concentrations of anti-C2 antibodies prior to incubation with MoDCs or BMDCs. Additionally, we evaluated the inhibition caused by pre-incubating with a 2-fold molar excess of the Fab fragments of BO2C11. Following the addition of BO2C11 or the corresponding Fab fragments, we observed a more than 70% reduction in FVIII internalization by MoDCs (Figure 1A), and a reduction of about 30% in the case of BMDCs (Online Supplementary Figure 2A). The addition of ESH8 did not reduce FVIII uptake by MoDCs. Similarly, BO2C11 or Fab of BO2C11 inhibited FVIII presentation to a HLA-DR1-restricted CD4+ T-cell hybridoma by more than 80% in the case of both MoDCs (Figure 1B) and splenocytes purified from HLA-DR1 Tg SURE-L1 mice (Online Supplementary Figure S2B), used as sources of APCs. Together, our results implicate a role for C2 membrane-binding residues in FVIII uptake by APCs. We next investigated the effect of BO2C11 on FVIII immunogenicity in vivo. We administered FVIII pre-incubated with F(ab’)2 of an isotype control or of BO2C11 for 4 weeks at weekly intervals. After 4 weeks, BO2C11-bound FVIII exhibited diminished immunogenicity compared to the isotype control-treated mice (Figure 1C).
Figure 1.
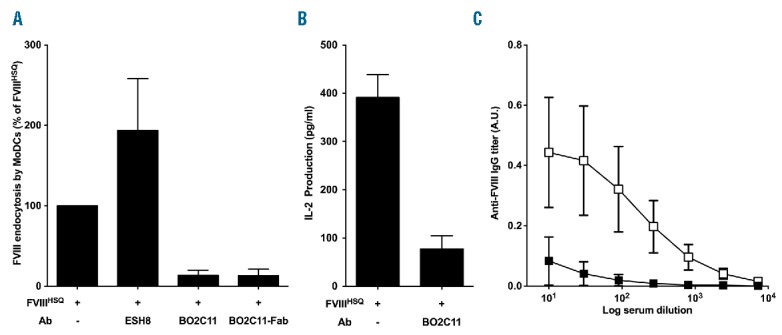
The anti-C2 antibody BO2C11 inhibits FVIII uptake in vitro and modulates FVIII immunogenicity in vivo. Panel A. B domain-deleted FVIII (20 nM) was pre-incubated alone or with equimolar concentrations of ESH8, BO2C11 or with a 2 molar excess BO2C11 Fab fragments. Uptake by human MoDCs was analyzed by fluorescence-activated cell sorting (FACS). Results are expressed as the percentage of median fluorescence intensity (MFI), whereby 100% corresponds to MFI obtained with wild-type FVIII (FVIIIHSQ) incubated alone. The graph is representative of 4 independent donors (mean±SEM). Panel B. Immature MoDCs were cultured for 24 hr in 96 round bottom plates with the FVIII-specific HLA-DRB1*0101-restricted CD4+ T-cell hybridoma (ratio 1:10, clone 1G8-A2) in the presence of B domain-deleted FVIII alone or pre-incubated with 2 molar excess of BO2C11. Activation of T cells was measured by IL-2 secretion in the supernatant by ELISA (BD Biosciences). The graph is representative of 4 experiments (mean±SEM). Panel C. Hemophilic FVIII exon 16 knock-out mice (n=6/group) were injected once a week for 4 weeks with 0.2 μg of B domain-deleted FVIII pre-incubated with 6 μM F(ab’)2 of BO2C11 (closed square) or a human IgG4 isotype control (open square). After 4 weeks, the anti-FVIII antibody response was measured. Anti-FVIII IgG titers are defined as arbitrary units using the mouse monoclonal anti-FVIII IgG mAb6 as a standard. Data are represented as serum dilution versus mean±SEM of absorbance (492 nm). FVIII: factor VIII; MoDCs: monocyte-derived dendritic cells; A.U.: arbitrary unit; IL-2: interleukin-2; IgG: immunoglobulin G; Ab: antibody.
Characterization of FVIII variants mutated in the BO2C11 epitope
We generated FVIII variants, wherein the two arginine residues located at position 2215 and 2220 that belong to the BO2C11 epitope, are mutated to alanine residues. The purified FVIII mutants exhibited specific activities between 4800–8000 IU/mg (Figure 2A) similar to that of non-mutated FVIII (FVIIIHSQ). Substitutions at either R2215 or R2220 did not alter binding to VWF, while substitution of both R2215 and R2220 lead to a marginally reduced binding to VWF (Figure 2B). Substitutions at R2215 or R2220 resulted in diminished binding to BO2C11 (Figure 2C), with substitution at R2220 resulting in a near complete inhibition of FVIII interaction with BO2C11. Double mutation of these residues (FVIIIR2215-20A) provided little additional benefit, confirming that R2220 contributes to most of the binding to BO2C11. Importantly, these FVIII variants retained binding to antibodies targeting other domains of FVIII. In particular, the mutations did not have significant effects on the ability of FVIII to interact with ESH8 (mouse anti-C2 IgG), BO2BII (human anti-A2 IgG) or KM33 (human anti-C1 IgG, Figure 2D–F). Recently, the three residues R2090A/F2092A/K2093A in the FVIII C1 domain that belong to the KM33 epitope, were implicated in FVIII uptake.11 Hence, as a control, we generated the R2090A/F2092A/K2093A FVIII mutant, referred to as “FVIIIC1”. As expected, FVIIIC1 showed drastically reduced binding to KM33 (Figure 2F), and unaltered binding to BO2C11 and ESH8 (Figure 2C–D).
Figure 2.
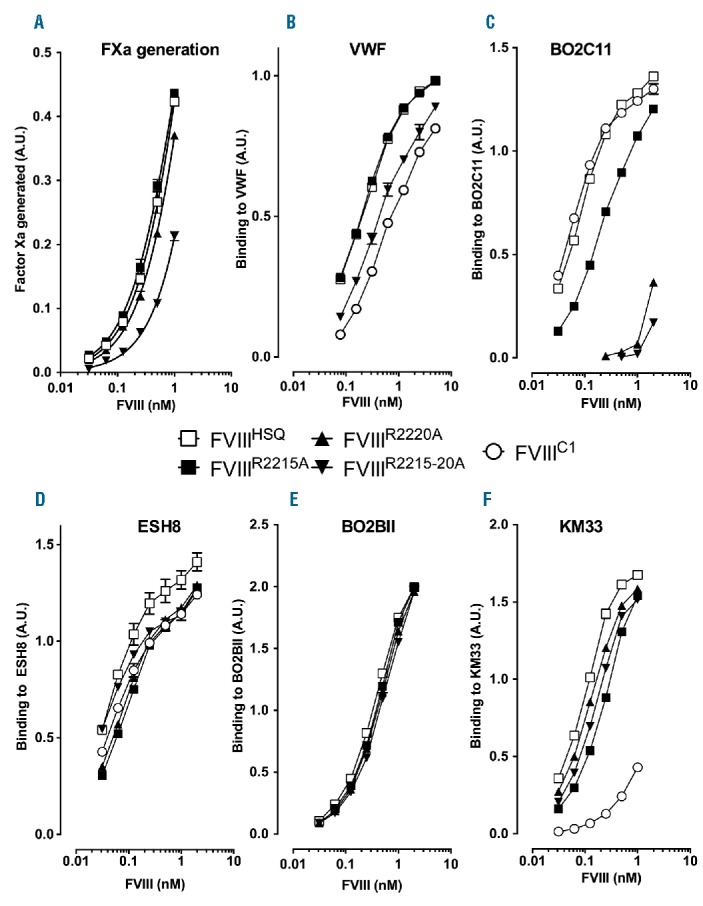
Characterization of FVIII containing alanine substitutions of the residues predicted to interact with BO2C11. Panel A. B domain-deleted wild-type FVIII (FVIIIHSQ), FVIIIR2215A or FVIIIR2220A were serially diluted 2-fold starting at 1 nM. Factor Xa generation was measured using a FVIII chromogenic assay. The reaction was stopped at 10 min using 20% acetic acid and the final absorbance measured at 405 nm. The data are represented as factor Xa generated in arbitrary units and are equivalent to the absorbance at 405 nm. VWF (Panel B), BO2C11 (human anti-C2 domain antibody, Panel C), ESH8 (mouse anti-C2 domain antibody that does not compete with BO2C11, Panel D), BO2BII (human anti-A2 domain antibody, Panel E) and KM33 (human anti-C1 domain antibody, Panel F) were immobilized on microtiter plates. After blocking, FVIIIHSQ, FVIIIR2215A, FVIIIR2220A, FVIIIR2215-20A or FVIIIC1 were serially diluted across the plates, and bound FVIII was revealed using either the biotinylated anti-A2 antibody, GMA-8015, or, in the case of BO2BII-bound FVIII, ESH8, followed by an anti-mouse-HRP antibody. The graphs are represented as absorbance measured at 492 nm (mean±SEM). FXa: factor Xa; VWF: von Willebrand factor; A.U.: arbitrary unit; FVIII: factor VIII.
C2 domain residues are implicated in FVIII uptake by APCs
We next evaluated the role of C2 residues in FVIII uptake and presentation. Human MoDCs and murine BMDCs were incubated with 20 nM FVIII, fixed and permeabilized, and FVIII was revealed with the anti-A2 antibody GMA-8015. FVIII uptake was dose-dependent (Online Supplementary Figure S3) and significantly reduced for FVIIIR2215A and FVIIIR2215-20A (Figure 3A and 3C). We confirmed our observation in an antigen presentation assay to a HLA-DR1-restricted CD4+ T-cell hybridoma using MoDCs derived from a HLA-DR1 donor, and using splenocytes from HLA-DR1 transgenic SURE-L1 mice (Figure 3B and 3D). Importantly, the epitope of CD4+ T-cell hybridoma is located in the 2004–2031 amino acid stretch at the A3-C1 junction of FVIII (data not shown); it is therefore distant from the mutated residues, thus ruling out the fact that the lack of T-cell activation is consecutive to a disruption of the T-cell epitope. We observed a significant decrease in FVIII presentation by human and mouse dendritic cells (DCs), as measured by reduced IL-2 secretion in the case of both FVIIIR2215A and FVIIIR2215A-R20A. Our present data indicate that the C2 domain participates in FVIII uptake and involves at least R2215.
Figure 3.
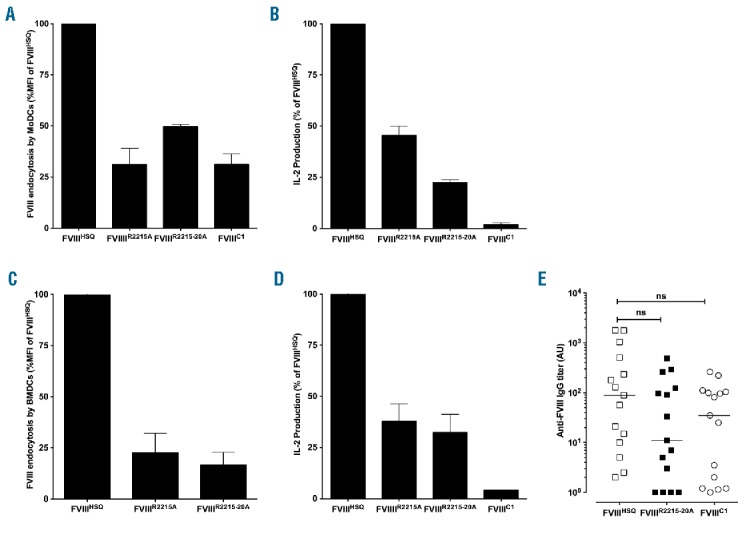
domain mutations alter FVIII endocytosis and presentation by APCs, but do not alter FVIII immunogenicity in vivo. B domain-deleted wild-type FVIII (FVIIIHSQ) or the mutants FVIIIR2215A, FVIIIR2215-20A or FVIIIC1 were added at 20 nM to MoDCs (Panel A) or BMDCs (Panel C) for 30 min. Internalized FVIII was detected as described in Methods. Results are expressed as the percentage of MFI, whereby 100% corresponds to MFI obtained with FVIIIHSQ. Panels B and D represent activation of a FVIII-specific HLA-DRB1*0101-restricted T-cell hybridoma by B domain-deleted FVIII-loaded (10 nM) HLA-matched human MoDCs or splenocytes from SURE-L1 mice, respectively. Supernatant was collected after 24 hr and the IL-2 produced by the activated T cells was measured. Representative of three experiments (mean±SEM). Panel E. FVIII-deficient mice were injected intravenously once weekly for 4 weeks with 1 μg of B domain-deleted FVIIIHSQ, FVIIIC1 or FVIIIR2215-20A. One week after the last injection, blood samples were collected. Anti-FVIII IgG titers are defined as arbitrary units based on standard curves generated using mAb6. Statistical significances were assessed using the two-tailed nonparametric Mann-Whitney U test. ns: not significant; FVIII: factor VIII; MoDCs: monocyte-derived dendritic cells; A.U.: arbitrary unit; IL-2: interleukin-2; IgG: immunoglobulin G; MFI: median fluorescence intensity; BMDCs: bone marrow-derived dendritic cells.
To further evaluate the importance of the C2 mutants in in vivo immunogenic uptake, we compared the immunogenicity of the different FVIII variants in FVIII-deficient mice. Cage-matched siblings were administered intravenously 4 times weekly with 1 μg of FVIIIHSQ, FVIIIR2215-20A or FVIIIC1, used as a control. In contrast to previous reports,11 FVIII-deficient mice generated an antibody response not only to FVIIIHSQ but also to FVIIIC1 (Figure 3E). In addition, FVIIIR2215-20A also presented with the same immunogenicity as FVIIIHSQ. The data suggest that mutating residues in the C1 and C2 domains of FVIII, at least in our hands, has no effect on FVIII immunogenicity in FVIII-deficient mice in vivo.
The immunogenicity of FVIII is independent from its ability to interact with endogenous VWF
Because BO2C11 inhibits FVIII interaction with VWF, we investigated the possibility that FVIII immunogenicity in the presence of BO2C11 (Figure 1C) was diminished due to its faster clearance. To address this potential bias, we generated a FVIII variant, FVIIIY1680C, which cannot associate with VWF. The specific activities of FVIIIHSQ and FVIIIY1680C were similar as assessed by one-stage and chromogenic assay (7300±165 and 6300±185 IU/mg, respectively, mean±SD). FVIIIY1680C exhibited a significant reduction in its ability to interact with VWF by ELISA (Figure 4A). Accordingly, FVIIIY1680C exhibited a rapid clearance in FVIII-deficient mice with a t1/2 of approximately 20 minutes (Figure 4B). We also confirmed that the Y1680C mutation does not alter the in vitro endocytosis of FVIII by MoDCs (Figure 4C). Next, we performed comparative immunogenicity studies of FVIIIHSQ and FVIIIY1680C using FVIII-deficient mice. Remarkably, FVIIIY1680C was as immunogenic as FVIIIHSQ (Figure 4D). The inhibitor titers were also not significantly different (Figure 4E). In addition, the immunogenicity of FVIIIY1680C in FVIII-deficient mice was similar to that of FVIIIHSQ in double FVIII/VWF-deficient mice (data not shown).
Figure 4.
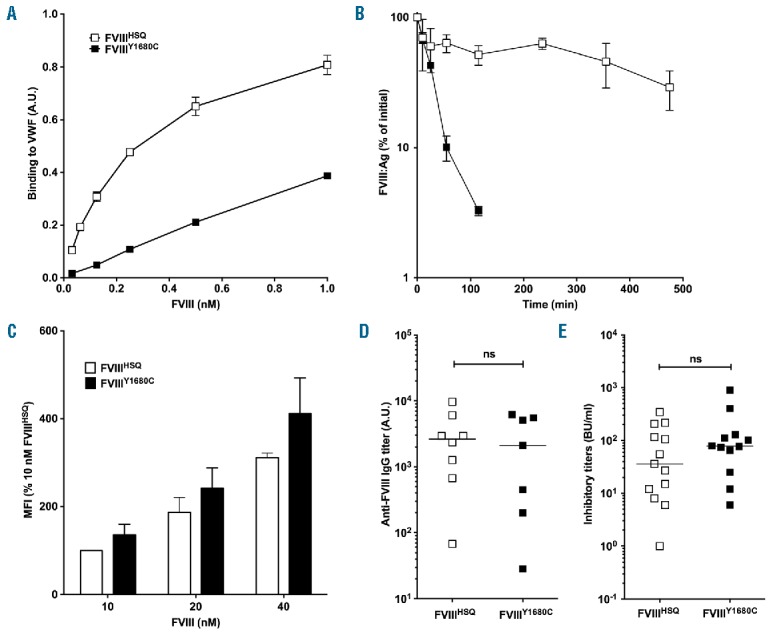
FVIII binding to VWF does not alter endocytosis in vitro or immunogenicity in vivo. Panel A. The binding of B domain-deleted FVIIIHSQ and FVIIIY1680C to VWF was studied using an ELISA as described in the Methods. Results (mean±SEM) are expressed in arbitrary units. Panel B. FVIIIHSQ (10 nM, open squares) or FVIIIY1680C (closed squares) in 100 μl were administered into FVIII-deficient mice and the residual FVIII levels were measured at different time points (n=3 mice per time point) by ELISA (FVIII:Ag). The data is plotted as a percentage of the initial FVIII level (measured 5 minutes after administration, mean±SEM) versus time. Panel C. FVIIIHSQ or mutants FVIIIY1680C were added at increasing concentrations (10, 20 and 40 nM) to immature MoDCs (n=3 different donors) for 30 min at 37°C in serum-free medium and in the absence of VWF. Cells were subsequently washed, fixed, permeabilized and stained for FVIII using a FITC-labeled anti-A2 domain-specific antibody followed by fluorescence-activated cell sorting (FACS). Data are expressed as the percentage of MFI (mean±SEM), whereby 100% corresponds to the MFI observed for 10 nM of FVIIIHSQ. Panels D and E. FVIII-deficient mice were administered with FVIIIHSQ (0.5 μg, open squares) or FVIIIY1680C (0.5 μg, closed squares), intravenously once a week for 4 weeks. Anti-FVIII IgG titers were measured using an ELISA as described above (D). Inhibitory titers towards FVIII were assessed using a modified Bethesda assay (E). Horizontal bars depict medians. The statistical significance was assessed using the two-tailed non-parametric Mann-Whitney U test. ns: not significant; FVIII: factor VIII; VWF: von Willebrand factor; A.U.: arbitrary unit; MFI: median fluorescence intensity; IgG: immunoglobulin G; Ag: antigen.
C domain mutations exhibit reduced immunogenicity only in the absence of VWF
Having demonstrated that the ability of FVIII to interact with endogenous VWF does not modulate the magnitude of the anti-FVIII immune response, we investigated the immune protective effect of BO2C11 using FVIIIY1680C. FVIIIY1680C alone or pre-incubated with a 2 molar excess of BO2C11 Fab fragments was administered to FVIII-deficient mice at weekly intervals for 4 weeks. Five days after the last FVIII administration, the anti-FVIII immune response was measured. We observed a significant reduction both in the anti-FVIII antibody response (Figure 5A), and FVIII-specific T-cell proliferation (Online Supplementary Figure S4), when FVIIIY1680C was pre-incubated with BO2C11 Fab fragments. In contrast, in double FVIII/VWF-deficient mice, the immunogenicity of FVIIIR2215-20A (1300±303 μg/ml, 90±17 BU/ml, mean±SEM) was not statistically different from that of FVIIIHSQ (2258±596 μg/ml, 180±62 BU/ml, Figure 5C,D, P>0.2), despite an increased residence time in the circulation of FVIIIR2215-20A (Figure 5B, 41 min [95% confidence interval (CI): 39–51]) as compared to FVIIIHSQ (17 min [13–23]). Conversely, FVIIIC1 showed a statistically significant reduction in immunogenicity in double FVIII/VWF-deficient mice as compared to FVIIIHSQ, both in terms of levels of anti-FVIII IgG (460±144 and 2258±596 μg/ml, respectively; P<0.001) and inhibitory titers (38±17 and 180±62 BU/ml, respectively; P<0.05). Similar observations were obtained with a FVIII mutant that combined the triple C1 mutation and a replacement of the R2215 residue with a serine (Online Supplementary Figure S5).
Figure 5.
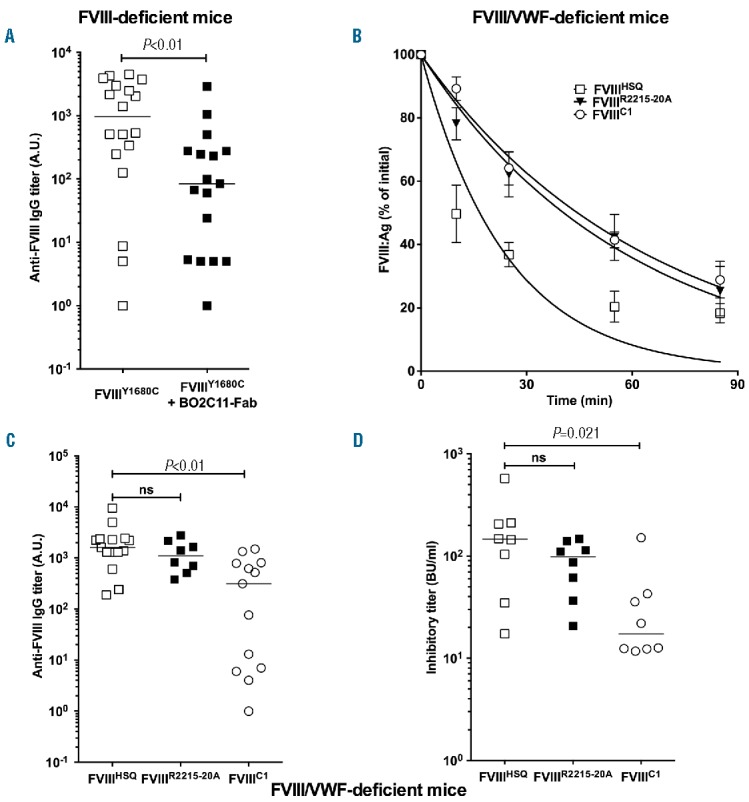
Contribution of the C1 and C2 domains of FVIII to FVIII immunogenicity in the absence of binding to VWF. Panel A. FVIII-deficient mice were administered with B domain-deleted FVIIIY1680C (0.4 μg, open squares) or FVIIIY1680C pre-incubated with a 2-fold molar excess of BO2C11 Fab (closed squares) intravenously once a week for 4 weeks. One week after the last injection, blood samples were collected. Anti-FVIII IgG titers are defined as arbitrary units based on standard curves generated using mAb6. The graph depicts a pool of two independent experiments. Panel B. B domain-deleted FVIIIHSQ, FVIIIC1 or FVIIIR2215-20A (10 nM in 100 μl) were administered to FVIII-deficient mice and the residual FVIII levels were measured at different time points (n=3 mice per time point) by ELISA. The data are plotted as a percentage of the initial FVIII levels (measured 5 minutes after administration) versus time (mean±SEM) and are representative of 2 independent experiments. Experimental data were fitted using a one-phase decay curve to determine the in vivo half-lives. Panels C and D. Double FVIII/VWF-deficient mice were injected intravenously once a week for 4 weeks with 0.5 μg of FVIIIHSQ, FVIIIC1 or FVIIIR2215-20A. One week after the last injection, blood samples were collected. Anti-FVIII IgG titers were measured as described above (C). Inhibitory titers towards FVIII were assessed using a modified Bethesda assay (D). Horizontal bars depict medians. Statistical significances were assessed using the two-tailed non-parametric Mann-Whitney U test. ns: not significant: FVIII: factor VIII; IgG: immunoglobulin G; A.U.: arbitrary unit; Fab: fragment antigen binding; VWF: von Willebrand factor; Ag: antigen.
Discussion
The endocytosis of an antigen by APCs is not sufficient for the induction of a primary CD4+ T-cell-dependent immune response. It is however a prerequisite to its presentation to naïve antigen-specific CD4+ T cells. Over the years, several receptors have been identified for FVIII5,6,20,21 that have mostly been implicated in FVIII catabolism.8,22 To date, the only receptor incriminated in FVIII internalization by APCs is the C-type lectin receptor CD206, that binds mannose-ending glycans linked to Asn239 in the A1 domain of FVIII, and Asn2118 in the C1 domain.7 The importance of the C1 domain of FVIII in binding to phospholipids and participating in FVIII endocytosis has recently been proposed. Thus, Lys 2092 and Phe 2093 in C1 were shown to participate in binding to phosphatidylserine and to platelets.23,24 Interestingly, co-incubation of FVIII with KM33, a monoclonal anti-C1 IgG that targets the R2090, K2092 and F2093 residues, prior to injection into FVIII-deficient mice, was associated with a decrease in FVIII immunogenicity.10 Additionally, a triple R2090A/K2092A/F2093A FVIII mutant demonstrated reduced endocytosis by MoDCs and macrophages, and reduced immunogenicity in FVIII-deficient mice.11 Because the C2 domain of FVIII shares structural homology with the C1 domain,25 and is a major membrane-binding motif26,27 containing several charged/basic residues, we investigated whether it also participates in the endocytic process by APCs, that leads to FVIII processing and presentation to CD4+ T cells. Our results demonstrate that masking part of the C2 domain with BO2C11, a human monoclonal IgG that interacts with solvent-exposed basic and hydrophobic side chains on the C2 membrane-binding loops,14 reduces FVIII endocytosis by MoDCs and presentation to T cells in vitro, as well as immunogenicity in mice. The protective effect of BO2C11 was epitope-specific, since ESH8, a non-overlapping mouse monoclonal anti-C2 IgG, failed to block FVIII uptake in vitro. ESH8 in fact increased the uptake process. Since the binding of ESH8 to FVIII was proposed to prevent a conformational change in the C2 domain of the FVIII light chain,28 our observation may partly relate to a particular conformation of ESH8-bound FVIII, that orients FVIII to facilitate the uptake process. Interestingly, the mutation of key charged residues on C2 that belong to the BO2C11 epitope, resumed the effect of BO2C11 on FVIII endocytosis and presentation in vitro. Our results are thus reminiscent of the aforementioned findings on the role played by the FVIII C1 domain in immunogenic FVIII uptake. Together, the data demonstrate that both the C1 and C2 domains of FVIII play a role in FVIII endocytosis by MoDCs. Since lactadherin, a potent competitor for phosphatidylserine binding, does not inhibit FVIII endocytosis by MoDCs,11 FVIII uptake is probably independent of binding to phospholipids. Rather, we speculate that FVIII uptake in the absence of VWF in vitro involves charged interactions with a putative highly negatively charged receptor, or a potential role for co-receptors, such as heparan sulfate proteoglycans. The respective contribution, either redundant, additive or synergistic, to FVIII internalization of residues in the C1 and C2 domains, and of mannose-ending N-linked glycans in the A1 and C1 domains, as well as the sequence of events that allows FVIII binding at the cell surface and internalization, remain to be deciphered.
While co-incubation of FVIII prior to injection into FVIII-deficient mice, with BO2C11 or with KM3310 reduced the anti-FVIII IgG response, neither the FVIIIR2215-20A C2 mutant nor the triple FVIIIC1 mutant demonstrated reduced immunogenicity in FVIII-deficient mice in our hands. These observations may be explained either by a steric hindrance of the C domains of FVIII by the antigen-binding domains of BO2C11/KM33, or by the implication in FVIII internalization of additional amino acids within the BO2C11 epitope on C2, or within the KM33 epitope on C1. Alternatively, the findings may be accounted for by an interfering effect of endogenous VWF. Indeed, the T2086-S2095 region of the C1 domain was recently shown to be buried upon association with VWF,29 and the triple FVIIIC1 mutant retains the ability to interact with VWF. In support of this, in the absence of endogenous VWF, the triple FVIIIC1 mutant showed reduced immunogenicity as compared to FVIIIHSQ in vivo. In contrast, the FVIIIR2215-20A C2 mutant, that also retains most of VWF binding and demonstrates reduced endocytosis in vitro, was as immunogenic as FVIIIHSQ in mice lacking VWF. Of note, the lack of reduced immunogenicity of the triple FVIIIC1 mutant in FVIII-deficient mice is at odds with the report by Wroblewska et al.;11 it remains unclear whether the observed difference may relate to differences in levels of VWF in different strains of mice.30 Although the C2 domain has been identified as an essential membrane interactive motif, with the C1 domain providing additional membrane binding affinity,23,26,27 the results suggest that C2 residues do not play a predominant role in immunogenic FVIII in the absence of endogenous VWF. Instead, we speculate that the in vivo role in endocytosis of the targeted R2215 and R2220 basic residues in the C2 domain is only secondary to that of other membrane accessible residues within the BO2C11 epitope. This is supported by the observation that the pre-incubation of the FVIIIY1680C mutant, which does not bind VWF, with BO2C11, reduced its immunogenicity in FVIII-deficient mice. Additional mutational analysis of membrane accessible hydrophobic residues within the BO2C11 epitope shall shed light on the specific residues of the C2 domain of FVIII that are implicated in the in vivo immunogenic uptake process.
VWF has controversially been proposed to reduce the immunogenicity of therapeutic FVIII.31 Unexpectedly, the present results indicate that the presence of endogenous VWF, or the capacity of exogenous FVIII to bind endogenous VWF, does not alter the immunogenicity of FVIII in mice. Indeed, FVIII presented with the same degree of immunogenicity in FVIII-deficient mice and in double FVIII/VWF-deficient mice. Likewise, FVIIIY1680C with impaired binding to VWF induced similar anti-FVIII IgG levels as native FVIII in FVIII-deficient mice. The potential immunomodulatory role of VWF towards FVIII has been suggested by a suspected reduced prevalence of FVIII inhibitors in hemophilia A patients treated with exogenous VWF-containing plasma FVIII concentrates, as compared to patients receiving recombinant or highly purified products.32,33 Controversial results have, however, been obtained upon studying large retrospective and prospective patient cohorts.34,35 In parallel, studies performed in pre-clinical mouse models of hemophilia A have indicated that pre-incubation of recombinant FVIII with exogenous VWF leads to a reduction in the levels of inhibitory anti-FVIII antibodies following administration to mice,36–39 although contradictory results have occasionally been generated.40 In vitro, a protective role for VWF on FVIII endocytosis by human MoDCs was clearly shown,10,41 although presentation of processed FVIII-derived peptides could still be detected, at least in vitro.42 In addition, under shear stress, VWF fails to block FVIII internalization by macrophages, and both VWF and FVIII co-localize within the same cells.43 This is in agreement with the observation that exogenous FVIII and exogenous VWF may be co-detected in splenic macrophages after injection into double FVIII/VWF-deficient mice.44 The internalization of FVIII and VWF was, however, inhibited by the LRP antagonist receptor-associated protein (RAP) in the case of macrophages under shear stress, but not in the case of MoDCs in static conditions,8 suggesting that this process is more relevant to FVIII catabolism than immunogenicity. Interestingly, the intricate role played by VWF in FVIII immunogenicity is reminiscent of its complex role towards FVIII catabolism. Indeed, binding of FVIII to VWF dictates FVIII residence time in the circulation, as shown in patients with type 3 von Willebrand disease.45 Conversely, VWF-binding may mediate, at least in part, the catabolism of therapeutic FVIII,20,46 which is further suggested by the fact that the half-life of modified Fc-fused or PEGylated FVIII barely exceeds that of VWF.47,48
Taken together, the results indicate that residues in the C1 and/or C2 domains of FVIII are implicated in immunogenic FVIII uptake, at least in vitro. Conversely, in vivo, the binding to endogenous VWF masks the reducing effect of mutations in the C domains on FVIII immunogenicity.
Supplementary Material
Acknowledgments
KM33 and VWF-deficient mice were kind gifts from Dr Jan Voorberg (Department of Plasma Proteins, Sanquin-AMC Landsteiner Laboratory and Van Creveld Laboratory, Amsterdam, The Netherlands) and Dr Olivier Christophe (Institut National de la Santé et de la Recherche Médicale U770, Le Kremlin-Bicêtre, France), respectively. FVIIIHSQ in the ReNeo plasmid and BHK-M cells were kind gifts from Prof Pete Lollar (Aflac Cancer and Blood Disorders Center, Department of Pediatrics, Emory University, Atlanta, GA, USA). The monoclonal mouse and human anti-FVIII antibodies, mAb6, BO2BII and BO2C11 were kind gifts from Drs JM Saint-Remy and M Jacquemin (KUL, Leuven, Belgium). We would like to extend our thanks to the Centre d’Explorations Fonctionnelles (Centre de Recherche des Cordeliers, Paris, France) for assistance.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/2/271
Funding
This work was supported by Institut national de la santé et de la recherche médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), and Université Pierre et Marie Curie (UPMC) Paris 6 (France), and by an unrestricted grant from Grifols (Investigator-Sponsored Research Program, Barcelona, Spain). MI and IP were the recipients of fellowships from Ministère de l’enseignement supérieur et de la recherche (France).
References
- 1.Fay PJ. Factor VIII structure and function. Int J Hematol. 2006;83(2):103–108. [DOI] [PubMed] [Google Scholar]
- 2.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983–3996. [PubMed] [Google Scholar]
- 3.Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339(8793):594–598. [DOI] [PubMed] [Google Scholar]
- 4.Lacroix-Desmazes S, Navarrete AM, Andre S, Bayry J, Kaveri SV, Dasgupta S. Dynamics of factor VIII interactions determine its immunologic fate in hemophilia A. Blood. 2008;112(2):240–249. [DOI] [PubMed] [Google Scholar]
- 5.Lenting P, Neels J, van den Berg B, et al. The light chain of factor VIII comprises a binding site for low density lipoprotein receptor-related protein. J Biol Chem. 1999; 274(34):23734–23739. [DOI] [PubMed] [Google Scholar]
- 6.Saenko E, Yakhyaev A, Mikhailenko I, Strickland D, Sarafanov A. Role of the low density lipoprotein-related protein receptor in mediation of factor VIII catabolism. J Biol Chem. 1999;274:37685–37692. [DOI] [PubMed] [Google Scholar]
- 7.Dasgupta S, Navarrete AM, Bayry J, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci U S A. 2007;104(21):8965–8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dasgupta S, Navarrete AM, Andre S, et al. Factor VIII bypasses CD91/LRP for endocytosis by dendritic cells leading to T-cell activation. Haematologica. 2008;93(1):83–89. [DOI] [PubMed] [Google Scholar]
- 9.Repesse Y, Dasgupta S, Navarrete AM, Delignat S, Kaveri SV, Lacroix-Desmazes S. Mannose-sensitive receptors mediate the uptake of factor VIII therapeutics by human dendritic cells. J Allergy Clin Immunol. 2012;129(4):1172–1173. [DOI] [PubMed] [Google Scholar]
- 10.Herczenik E, van Haren SD, Wroblewska A, et al. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J Allergy Clin Immunol. 2012; 129(2):501–509, 9 e1–5. [DOI] [PubMed] [Google Scholar]
- 11.Wroblewska A, van Haren SD, Herczenik E, et al. Modification of an exposed loop in the C1 domain reduces immune responses to factor VIII in hemophilia A mice. Blood. 2012;119(22):5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meems H, van den Biggelaar M, Rondaij M, van der Zwaan C, Mertens K, Meijer AB. C1 domain residues Lys 2092 and Phe 2093 are of major importance for the endocytic uptake of coagulation factor VIII. Int J Biochem Cell Biol. 2011;43(8):1114–1121. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, Pipe SW, Miao H, Jacquemin M, Gilbert GE. A membrane-interactive surface on the factor VIII C1 domain cooperates with the C2 domain for cofactor function. Blood. 2011;117(11):3181–3189. [DOI] [PubMed] [Google Scholar]
- 14.Spiegel PC, Jr, Jacquemin M, Saint-Remy JM, Stoddard BL, Pratt KP. Structure of a factor VIII C2 domain-immunoglobulin G4kappa Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood. 2001;98(1):13–19. [DOI] [PubMed] [Google Scholar]
- 15.Healey JF, Barrow RT, Tamim HM, et al. Residues Glu2181-Val2243 contain a major determinant of the inhibitory epitope in the C2 domain of human factor VIII. Blood. 1998;92(10):3701–3709. [PubMed] [Google Scholar]
- 16.Lutz MB, Kukutsch N, Ogilvie AL, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. [DOI] [PubMed] [Google Scholar]
- 17.Delignat S, Repesse Y, Gilardin L, et al. Predictive immunogenicity of Refacto AF. Haemophilia. 2014;20(4):486–492. [DOI] [PubMed] [Google Scholar]
- 18.Pfistershammer K, Stockl J, Siekmann J, Turecek PL, Schwarz HP, Reipert BM. Recombinant factor VIII and factor VIII-von Willebrand factor complex do not present danger signals for human dendritic cells. Thromb Haemost. 2006;96(3):309–316. [DOI] [PubMed] [Google Scholar]
- 19.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110(13):4234–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarafanov A, Ananyeva N, Shima M, Saenko E. Cell surface heparan sulfate proteoglycans participate in factor viii catabolism mediated by low density lipoprotein receptor-related protein. J Biol Chem. 2001;276(15):11970–11979. [DOI] [PubMed] [Google Scholar]
- 21.Bovenschen N, Rijken DC, Havekes LM, Vlijmen BJ, Mertens K. The B domain of coagulation factor VIII interacts with the asialoglycoprotein receptor. J Thromb Haemost. 2005;3(6):1257–1265. [DOI] [PubMed] [Google Scholar]
- 22.Navarrete AM, Dasgupta S, Teyssandier M, et al. Endocytic receptor for pro-coagulant factor VIII: relevance to inhibitor formation. Thromb Haemost. 2010;104(6):1093–1098. [DOI] [PubMed] [Google Scholar]
- 23.Meems H, Meijer AB, Cullinan DB, Mertens K, Gilbert GE. Factor VIII C1 domain residues Lys 2092 and Phe 2093 contribute to membrane binding and cofactor activity. Blood. 2009;114(18):3938–3946. [DOI] [PubMed] [Google Scholar]
- 24.Bloem E, van den Biggelaar M, Wroblewska A, et al. Factor VIII C1 domain spikes 2092–2093 and 2158–2159 comprise regions that modulate cofactor function and cellular uptake. J Biol Chem. 2013;288(41):29670–29679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16(4):597–606. [DOI] [PubMed] [Google Scholar]
- 26.Scandella D, Gilbert GE, Shima M, et al. Some Factor VIII inhibitor antibodies recognize a common epitope corresponding to C2 domain amino acids 2248 through 2312, which overlap a phospholipid-binding site. Blood. 1995;86(5):1811–1819. [PubMed] [Google Scholar]
- 27.Gilbert GE, Kaufman RJ, Arena AA, Miao H, Pipe SW. Four hydrophobic amino acids of the factor VIII C2 domain are constituents of both the membrane-binding and von Willebrand factor-binding motifs. J Biol Chem. 2002;277(8):6374–6381. [DOI] [PubMed] [Google Scholar]
- 28.Saenko EL, Shima M, Gilbert GE, Scandella D. Slowed release of thrombin-cleaved factor VIII from von Willebrand factor by a monoclonal and a human antibody is a novel mechanism for FVIII inhibition. J Biol Chem. 1996;271(44):27424–27431. [DOI] [PubMed] [Google Scholar]
- 29.Chiu PL, Bou-Assaf GM, Chhabra ES, et al. Mapping the interaction between factor VIII and von Willebrand factor by electron microscopy and mass spectrometry. Blood. 2015;126(8):935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shavit JA, Manichaikul A, Lemmerhirt HL, Broman KW, Ginsburg D. Modifiers of von Willebrand factor identified by natural variation in inbred strains of mice. Blood. 2009; 114(26):5368–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oldenburg J, Lacroix-Desmazes S, Lillicrap D. Alloantibodies to therapeutic factor VIII in hemophilia A: the role of von Willebrand factor in regulating factor VIII immunogenicity. Haematologica. 2015;100(2):149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood. 2006; 107(1):46–51. [DOI] [PubMed] [Google Scholar]
- 33.Calvez T, Laurian Y, Goudemand J. Inhibitor incidence with recombinant vs. plasma-derived FVIII in previously untreated patients with severe hemophilia A: homogeneous results from four published observational studies. J Thromb Haemost. 2008;6(2):390–392. [DOI] [PubMed] [Google Scholar]
- 34.Gouw SC, van der Bom JG, Auerswald G, Escuriola Ettinghausen C, Tedgard U, van den Berg HM. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007; 109(11):4693–4697. [DOI] [PubMed] [Google Scholar]
- 35.Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368(3):231–239. [DOI] [PubMed] [Google Scholar]
- 36.Behrmann M, Pasi J, Saint-Remy JM, Kotitschke R, Kloft M. Von Willebrand factor modulates factor VIII immunogenicity: comparative study of different factor VIII concentrates in a haemophilia A mouse model. Thromb Haemost. 2002;88(2):221–229. [PubMed] [Google Scholar]
- 37.Delignat S, Dasgupta S, Andre S, et al. Comparison of the immunogenicity of different therapeutic preparations of human factor VIII in the murine model of hemophilia A. Haematologica. 2007;92(10):1423–1426. [DOI] [PubMed] [Google Scholar]
- 38.Kallas A, Kuuse S, Maimets T, Pooga M. von Willebrand factor and transforming growth factor-beta modulate immune response against coagulation factor VIII in FVIII-deficient mice. Thromb Res. 2007; 120(6):911–919. [DOI] [PubMed] [Google Scholar]
- 39.Delignat S, Repesse Y, Navarrete AM, et al. Immunoprotective effect of von Willebrand factor towards therapeutic factor VIII in experimental haemophilia A. Haemophilia. 2012;18(2):248–254. [DOI] [PubMed] [Google Scholar]
- 40.Qadura M, Waters B, Burnett E, et al. Recombinant and plasma-derived factor VIII products induce distinct splenic cytokine microenvironments in hemophilia A mice. Blood. 2009;114(4):871–880. [DOI] [PubMed] [Google Scholar]
- 41.Dasgupta S, Repesse Y, Bayry J, et al. VWF protects FVIII from endocytosis by dendritic cells and subsequent presentation to immune effectors. Blood. 2007;109(2):610–612. [DOI] [PubMed] [Google Scholar]
- 42.Sorvillo N, Hartholt RB, Bloem E, et al. von Willebrand factor binds to the surface of dendritic cells and modulates peptide presentation of factor VIII. Haematologica. 2016;101(3):309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro-Nunez L, Dienava-Verdoold I, Herczenik E, Mertens K, Meijer AB. Shear stress is required for the endocytic uptake of the factor VIII-von Willebrand factor complex by macrophages. J Thromb Haemost. 2012;10(9):1929–1937. [DOI] [PubMed] [Google Scholar]
- 44.van Schooten CJ, Shahbazi S, Groot E, et al. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. 2008;112(5):1704–1712. [DOI] [PubMed] [Google Scholar]
- 45.Goodeve AC, Eikenboom JC, Ginsburg D, et al. A standard nomenclature for von Willebrand factor gene mutations and polymorphisms. On behalf of the ISTH SSC Subcommittee on von Willebrand factor. Thromb Haemost. 2001;85(5):929–931. [PubMed] [Google Scholar]
- 46.Pegon JN, Kurdi M, Casari C, et al. Factor VIII and von Willebrand factor are ligands for the carbohydrate-receptor Siglec-5. Haematologica. 2012;97(12):1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123(3):317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126(9):1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.