

Abstract
Variants in ETV6, which encodes a transcription repressor of the E26 transformation-specific family, have recently been reported to be responsible for inherited thrombocytopenia and hematologic malignancy. We sequenced the DNA from cases with unexplained dominant thrombocytopenia and identified six likely pathogenic variants in ETV6, of which five are novel. We observed low repressive activity of all tested ETV6 variants, and variants located in the E26 transformation-specific binding domain (encoding p.A377T, p.Y401N) led to reduced binding to corepressors. We also observed a large expansion of megakaryocyte colony-forming units derived from variant carriers and reduced proplatelet formation with abnormal cytoskeletal organization. The defect in proplatelet formation was also observed in control CD34+ cell-derived megakaryocytes transduced with lentiviral particles encoding mutant ETV6. Reduced expression levels of key regulators of the actin cytoskeleton CDC42 and RHOA were measured. Moreover, changes in the actin structures are typically accompanied by a rounder platelet shape with a highly heterogeneous size, decreased platelet arachidonic response, and spreading and retarded clot retraction in ETV6 deficient platelets. Elevated numbers of circulating CD34+ cells were found in p.P214L and p.Y401N carriers, and two patients from different families suffered from refractory anemia with excess blasts, while one patient from a third family was successfully treated for acute myeloid leukemia. Overall, our study provides novel insights into the role of ETV6 as a driver of cytoskeletal regulatory gene expression during platelet production, and the impact of variants resulting in platelets with altered size, shape and function and potentially also in changes in circulating progenitor levels.
Introduction
The genetic determinants of non syndromic autosomal dominant (AD) thrombocytopenia with normal platelet size remain largely unknown, yet it is important to identify such variants because they may predispose carriers to hematological malignancy. Germline variants in RUNX1 cause a familial platelet disorder with an increased risk of acute myeloid leukemia (FPD/AML), while variants in the 5′ untranslated region (UTR) of ANKRD26 have also been shown to predispose individuals to hematologic malignancies. Recently, germline variants in ETV6 (TEL) have been reported to underlie AD thrombocytopenia with predisposition to leukemia.1–3 ETV6, which was initially identified as encoding a tumor suppressor in humans, is often found fused with partner genes in samples from human leukemia of myeloid and lymphoid origin.4 Somatic ETV6 variants have also been found in solid tumors, T-cell leukemias and myelodysplastic syndromes, hence the widespread interest in this gene.5,6 ETV6 encodes an E26 transformation-specific (Ets) family transcriptional repressor. It can bind DNA via a highly conserved Ets DNA-binding consensus site located at the C-terminus. The N-terminal domain (pointed domain) is necessary for homotypic dimerization and interaction with the Ets family protein FLI.7,8 The central region is involved in repressive complex recruitment (including SMRT, Sin3A and NCOR)9 and autoinhibitory activity.10
ETV6 plays an important role in hematopoiesis. In mice, ETV6 is essential for hematopoietic transition from the fetal liver to the bone marrow (BM).11 Conditional disruption of the ETV6 gene has shown that ETV6 plays a unique, non redundant role in megakaryocytopoiesis. Data concerning ETV6 involvement in megakaryocytopoiesis in humans remains scarce, however, a recent study has shown that patients expressing a mutated form of ETV6 displayed abnormal megakaryocyte (MK) development with a likely impact on platelet production.1
We have assessed the biological impact of six likely pathogenic variants in ETV6, of which five are novel. We describe in detail how variants in ETV6 lead to increased megakaryocyte proliferation and various cytoskeleton-related platelet defects that include altered platelet shape, reduced Rho GTPase expression in platelets, decreased proplatelet (PPT) formation and reduced platelet spreading. Additionally, we show that patients exhibit elevated levels of circulating CD34+ progenitors and a predisposition to myelodysplastic syndrome and leukemia.
Methods
Platelets and circulating CD34+-cells analysis
Blood samples were collected after informed written consent, in accordance with our local Institutional Review Boards and the Declaration of Helsinki. Platelet-rich plasma (PRP), washed platelets and circulating CD34+ cells were prepared according to standard procedures. For electron microscopy (EM), platelets were fixed in glutaraldehyde and processed as previously described.12 For platelet spreading, fibronectin-adherent platelets were stained with Alexa Fluor 488 phalloidin (filamentous (F)-actin) and Alexa Fluor 594 DNAse I (globular (G)-actin). Filopodia and lamellipodia were manually quantified. For clot retraction, coagulation of PRP was triggered using thrombin, and clots were allowed to retract. Images were recorded using a CoolSNAP CCD camera and analyzed to evaluate the reduction of the initial clot surface (ImageJ). The platelet survival assay was based on the method of Thakur and colleagues.13
High-throughput and Sanger sequencing
DNA samples from 957 patients enrolled in the BRIDGE-BPD project were subjected to whole-genome or whole-exome sequencing, and the results were used for variant calling as described previously.14,15 DNA samples from eight patients in the French cohort were subjected to whole-exome sequencing or Sanger sequencing at the ETV6 lotus. Sequence analysis was carried out using Chromas X software, and aligned using Multalin.16
Site-directed mutagenesis and luciferase assays
ETV6 cDNA was ligated into a pcDNA3 expression vector, and mutagenesis was performed using the GENEART® Site-directed Mutagenesis System kit (Life technologies).17 Transcriptional regulatory properties of wild-type (WT) and mutant ETV6 (mutETV6) as well as ETV6 corepressor binding,18 were determined by using the luciferase reporter systems in transfected GripTite™ 293 macrophage scavenger receptor (MSR) cells.
Immunoassays
Immunoblots were performed with antibodies directed against human ETV6; SMRT, RHOA (Santa Cruz Biotechnology), CDC42, RAC1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore) and MYH10 (Cell Signaling Technology) antibodies. Chemiluminescence signals were detected and quantified (CCD camera-based ImageQuant LAS 4000, GE Healthcare). Levels of thrombopoietin (TPO) and stromal cell-derived factor 1α (SDF1α) were quantified via ELISA (Abcam). For the co-immunoprecipitation assays, whole cell extracts were prepared in NP-40 buffer and pre-cleared with protein A/G magnetic beads (Millipore). Immunoprecipitation of the cell extracts with anti-ETV6-coated beads was carried out overnight.
Megakaryocyte differentiation and quantification of proplatelet-bearing megakaryocytes
CD34+ cells were grown in serum-free medium supplemented with TPO and stem cell factor (SCF) (Life Technologies).19 At culture day 10, we assessed ploidy in the Hoechst+CD41+CD42a+ cell population20 (Navios, BD Biosciences). Proplatelets were quantified between day 11 and 15. Microtubule and F-actin organization was determined in megakaryocytes adhering to fibrinogen with fluorescently labeled polyclonal rabbit anti-tubulin antibody (Sigma-Aldrich) and phalloidin (Life Technologies).
Lentiviral particle production and CD34+ cell transduction
Lentiviral particles were prepared as previously described.21,22 CD34+ cells were infected twice. After 8 hours, the cells were washed and cultured in serum-free medium.
Clonogenic progenitor assays
CD34+ cells were plated in human methylcellulose medium H4434 (STEMCELL Technologies), supplemented with erythropoietin (EPO), interleukin-3 (IL-3), SCF, granulocyte colony-stimulating factor (G-CSF), interleukin-6 (IL-6) and TPO to quantify erythroid (burst forming unit-erythroid (BFU-E), colony-forming unit-erythroid (CFU-E), granulocytic/macrophage (CFU-GM), mixed (CFU-GEMM) and megakaryocyte (CFU-MK)) progenitors at day 12.23
Statistical analyses
Analyses were performed using GraphPad Prism software. Statistical significance was determined via a two-tailed Mann-Whitney test. P<0.05 was considered statistically significant.
Results
Identification of affected families
Screening of patients with thrombocytopenia for rare non-synonymous variants in ETV6 revealed six families with patients carrying one of six possibly pathogenic variants. The variants encode p.P214L, which has been previously reported,3 and the novel substitutions p.I358M, p.A377T, p.R396G, p.Y401N and p.Y401H (Figure 1A). Family studies by Sanger sequencing showed segregation between the ETV6 variant and thrombocytopenia in all cases for which DNA samples were available (Figure 1B). Henceforth, we refer to the likely pathogenic variants described above as mutETV6.
Figure 1.
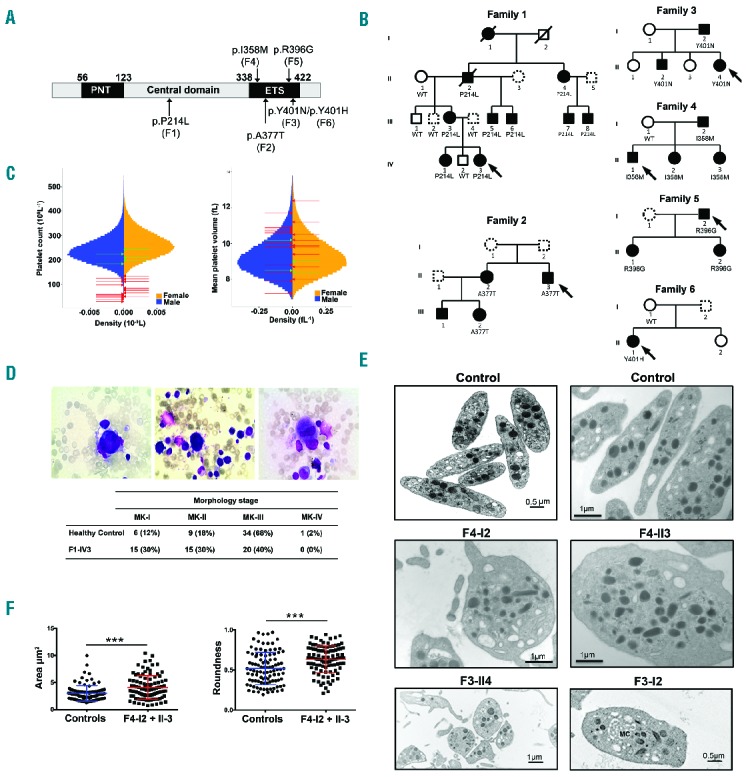
Identification of variants in ETV6 underlying AD thrombocytopenia, megakaryocyte and platelet characteristics. (A) Schematic representation of the different domains of the ETV6 protein. The N-terminal domain (PNT), central domain and C-terminal domain containing a DNA-binding domain (ETS) are depicted. Arrows indicate the location of the ETV6 variants and the corresponding family is mentioned in brackets. (B) Pedigrees for the affected families. Squares denote males, circles denote females and slashes represent deceased family members. Black filled symbols represent thrombocytopenic family members and dotted line symbols represent non-tested members. The families F1, F2, F3, F4, F5 and F6 carried the ETV6 p.P214L, p.A377T, p.Y401N, p.I358M, p.R396G and p.Y401H variants, respectively, which segregated with thrombocytopenia. See Table 1 for blood cell count values. (C) Sex-stratified histograms of platelet count and mean platelet volume measurements, obtained using a Coulter hematology analyzer, from 480,001 UK Biobank volunteers, after adjustment for technical artifacts. The red arrows superimposed upon the histograms indicate the sex and values for patients with a deleterious variant in ETV6. The green arrows indicate the sex and values for relatives homozygous for the corresponding wild-type allele. (D) BM smears (May-Grünwald-Giemsa staining) from family F1 propositus (F1-IV3). Left: a relatively immature MK with reduced cytoplasm. Middle: a micromegakaryocyte without granules, with immature cytoplasm (basophilic) and nucleus. Signs of impaired proplatelet formation can be observed. Right: a mature MK of reduced size with a hypolobulated nucleus. Table 1 indicates the % of MKs at each stage of maturation in the BM samples from family F1 proposita (F1-IVI3) and a healthy control. (E) Ultrastructural aspects of platelets from patients F3-I2 and F3-II4, F4-I2 and F4-II3 and unrelated healthy controls. Upper panel: aspect of healthy platelets; middle panel: series of mostly rounder platelets from patients F4-I2 and F4-II3, lower panel: a series of platelets emphasizing anisocytosis in patient F3-114 and a platelet from patient F3-I2 with abnormal membrane complex (MC). Note the heterogeneous presence of α-granules with an occasional granule of increased size. (F) The platelet area and roundness was quantified. Perfect round platelets would have a value of 1. Values are the means and SD as quantified for 50 randomly selected platelets per subject using two-tailed unpaired t-test with Welch’s correction. ***P<0.0001. WT: wild-type; MK: megakaryocyte; PNT: pointed; ETS: ET6 transformation-specific.
Description of the families
Our study included six families with mutETV6 variants. The proband of the first family (F1-IV3) was a 7-year-old girl who was admitted for emergency care due to the suspicion of acute leukemia with asthenia, weakness, paleness, severe thrombocytopenia (44 × 109/L) and anemia (hemoglobin: 50 g/L). BM examination unequivocally dismissed a diagnosis of leukemia, and the anemia was attributed to an iron deficiency subsequent to repeated episodes of severe epistaxis. The patient underwent a red blood cell transfusion, and the anemia was progressively corrected via iron supplementation. However, the platelet count remained low (50 × 109/L). Clinical examination of the parents and two siblings did not reveal any particular bleeding tendency. However, the patient’s mother (F1-III3) had undergone a splenectomy at the age of 17 because of chronic thrombocytopenia, and she exhibited subnormal platelet counts (116–210 × 109/L) at the time of examination. To gain further insight into the possibility of inherited thrombocytopenia, we screened the extended family for platelet counts. An AD form of thrombocytopenia was evidenced (Figure 1B), with normal mean platelet volume (MPV) compared with a large population of blood donors (Figure 1C). Of note, fourteen out of twenty-three carriers exhibit MPV >9 fL (Table 1). Plasma TPO levels were decreased in affected F1 members (n=4): 160 ± 9 pg/mL vs. controls (n=8): 296 ± 39 pg/mL, P=0.02. May-Grünwald-Giemsa staining of BM smears of patient F1-IV3 showed that megakaryocytes were present, although a high proportion were of medium size, in the early stages of maturation and tended to be hypolobulated (Figure 1D). The 7-year-old patient’s grandfather (F1-II2) was diagnosed with refractory anemia with excess blasts type 2 (RAEB-2) at the age of 70. Individuals from the second and third families had platelet counts between 60 and 125 × 109/L (Table 1). BM from F2-II3 displayed a delay in granulocyte maturation and dyserythropoiesis (data not shown). Peripheral blood smears revealed platelet anisocytosis (data not shown), as confirmed by EM (Figure 1E), which further highlighted the presence of occasional hypogranular platelets with a poorly organized open canalicular system. Patient F2-I1 presented with refractory anemia with excess blasts (RAEB) and required BM transplantation. The propositus (F4-II1) from pedigree 4 was referred with AML type M0 at the age of 8 years, and suffered from epistaxis, ecchymosis and infections. After 2 years of chemotherapy treatment, the BM showed no blasts and the peripheral blood counts normalized, except for a persistent low platelet count (Table 1). BM studies showed the presence of many hypolobulated small megakaryocytes (data not shown). Thrombocytopenia was also present in his father and two sisters without any bleeding problems (Table 1). EM investigation of platelets from affected members F4-I2 and F4-II3 showed the presence of both larger and smaller platelets that, significantly, were of round shape rather than of discoid shape (Figure 1E,F; P<0.0001). These platelets had normal dense and α-granules numbers, but some α-granules were elongated (data not shown). Pedigree 5 was referred for genetic testing of AD thrombocytopenia in a father with very mild bleeding problems (propositus F5-I2), and his 2 asymptomatic daughters (Figure 1B). BM investigation in F5-II2 showed the presence of dysmegakaryopoiesis with almost no mature megakaryocytes (data not shown). A mother (F6-I1) and daughter (propositus F6-II1) from pedigree 6 (Figure 1B) were diagnosed with platelet dense storage pool deficiency (SPD), with platelet aggregation defects and abnormal dense granules. Thrombocytopenia was only recorded for the daughter, who suffered from severe menorrhagia and had an increased bleeding tendency with bruising and nosebleeds. The mother had a normal platelet count and did not carry the ETV6 variant. No clinical information or DNA was available from the father. Therefore, the ETV6 variant in F6-II1 could be present as a de novo or somatic variant. SPD in the mother and daughter was likely to be caused by another additional genetic factor. Indeed, in contrast to the obvious platelet aggregation and secretion defects for these two patients, such abnormalities were not present in the other five families, except for a decreased aggregation response to arachidonic acid as the only consistent finding in every family (Online Supplementary Table S1). Consistent with normal dense granules found by EM (Figure 1E), adenosine triphosphate (ATP) secretion and mepacrine uptake and release were normal (Online Supplementary Table S1). Flow cytometry analysis of key platelet surface receptors (αIIbβ3, glycoprotein (GP) Ibα, GPIa, GPIV, CD63 and CD62P) was also normal (Online Supplementary Table S2).
Table 1.
Hematological parameters in the six studied family members.
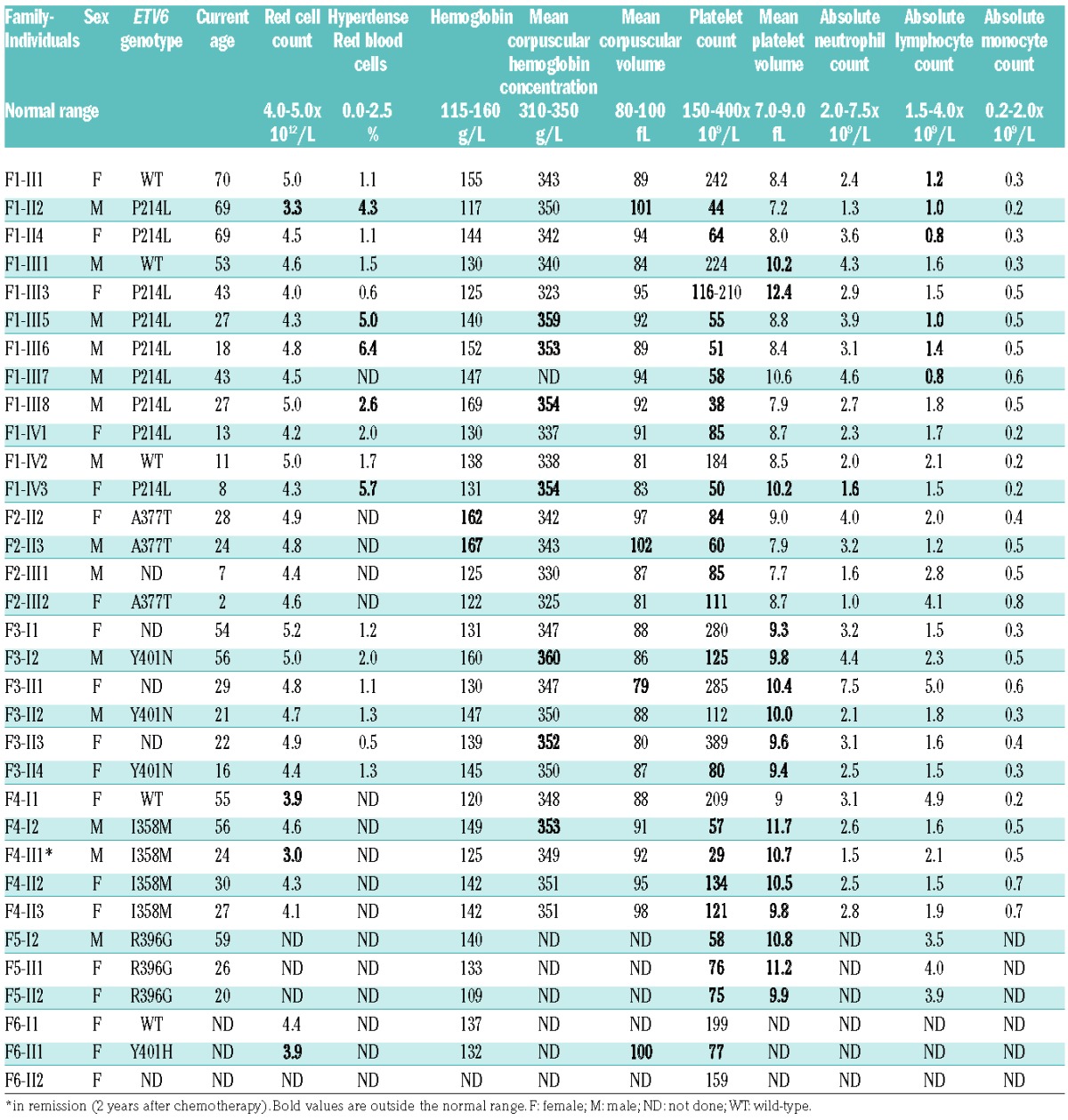
A platelet survival assay was performed on patient F3-II4 (Online Supplementary Table S3), and revealed decreased platelet lifespan (4.6 days) without significant splenic or hepatic sequestration. Notably, this patient had not undergone platelet transfusion. Patient F1-III3 underwent a 111In-oxine platelet survival assessment (autologous transfusion) in 1981 prior to a splenectomy, which revealed short platelet half-life (24 h vs. 3.5 days in the control) and hepatic and splenic platelet sequestration, with predominant sequestration in the liver (data not shown). Patient F1-III3 was assessed for anti-human leukocyte antigen (HLA) antibodies on several occasions (National Center of Blood Transfusion, Marseille, France), but all results were negative (data not shown).
Variants in ETV6 lead to a functional defect in transcriptional activity
Western blot analysis showed that ETV6 protein expression was not reduced in platelets from the patients, nor in GripTite™ 293 MSR cells transfected with the ETV6 variants (Figure 2A,B). To investigate the transcriptional regulatory properties of mutETV6 compared with WT ETV6, we analyzed repressive activity. Cotransfection of the reporter plasmid along with expression of a plasmid encoding WT ETV6 resulted in an almost 90% inhibition of luciferase activity. The substitution of WT ETV6 with any of the mutETV6 variants led to a significant reduction in repressive activity (85% to 100%) (Figure 2C).
Figure 2.
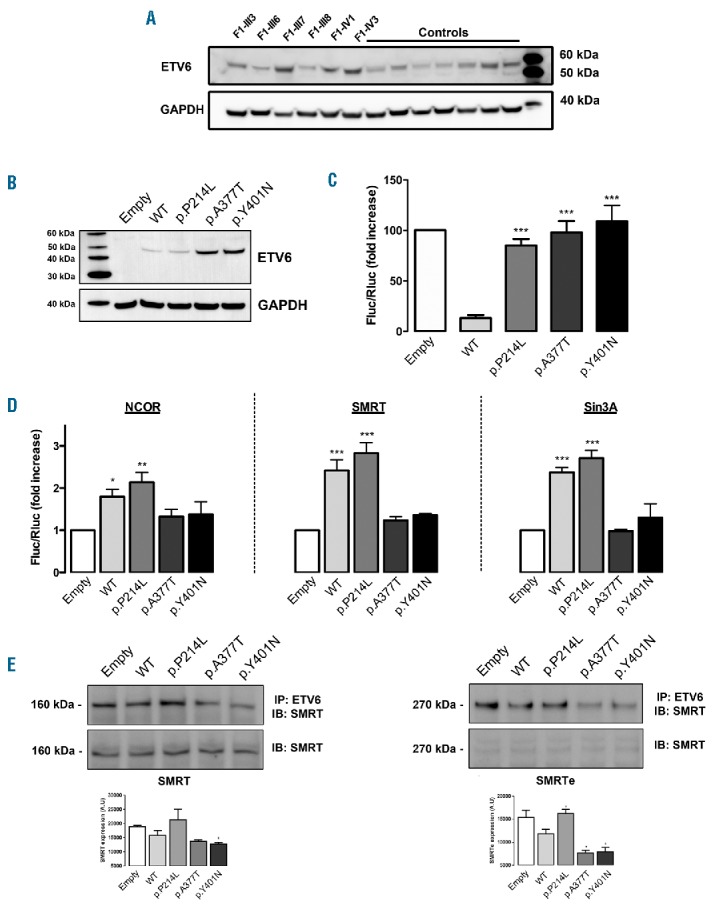
Effect of the variants on repressive activity and corepressor recruitment. (A) Western blot analysis of ETV6 expression in platelets of 6 affected F1 members and 7 external controls. GAPDH was used as a protein loading control. (B–C) GripTite™ 293 MSR cells were co-transfected with the luciferase reporter plasmid containing 3 tandem copies of the Ets Binding Site (EBS) upstream of HSV-Tk (E743tk80Luc), pCDNA3.1 expression vector (empty, WT or mutETV6) or pGL473 Renilla luciferase control vector. (B) Western blot analysis of ETV6 expression in whole cell lysates of GripTite™ 293 MSR transfected with WT ETV6 or mutETV6 expression vectors. GAPDH was used as a protein loading control. The data are representative of 4 to 8 independent experiments. (C) The firefly to renilla luminescence ratios (Fluc/Rluc) were calculated to compensate for transfection efficiency. The data represent the mean ± SEM of 4 to 8 independent experiments, student’s t-test ***P<0.001 (each condition was compared with WT). (D) Effects of the ETV6 variants on corepressor recruitment. Mammalian two-hybrid analysis of the protein interactions between WT NCOR, SMRT or Sin3A (expressed using the GAL4 DNA-binding domain (DBD) plasmid) and WT ETV6 or mutETV6 (expressed using the GAL4-VP16 activation domain vector). The results are expressed as mean ± SEM of 3 to 8 independent experiments, student’s t-test *P<0.05, **P<0.01, ***P<0.001. (E) Immunoprecipitation of endogenous corepressor SMRT and ETV6 from GripTite™ 293 MSR cells transfected with WT and mutETV6. Immunoprecipitation was performed on cell lysates with ETV6 antibody. The total cell lysates (lower panel) and immunoprecipitates (upper panel) were analyzed via immunoblotting with anti-SMRT antibody. Quantification of band intensity for SMRT and SMRT-extended (SMRTe) is shown below the western blot. The results are expressed as mean ± SEM, student’s t-test, *P<0.05 vs. WT. GAPDH: glyceraldehyde-3-phosphate dehydrogenase; WT: wild-type; A.U: arbitrary unit; IP: immunoprecipitation; IB: immunoblot.
To evaluate whether this reduction in repressive activity perhaps resulted from variations in nuclear corepressor complex recruitment, we investigated the interaction of ETV6 with NCOR, SMRT and Sin3A using a mammalian two-hybrid assay. p.P214L ETV6 interacted with NCOR, SMRT and Sin3A, whereas p.A377T and p.Y401N ETV6 did not (Figure 2D). Immunoprecipitation assays showed that the p.A377T and p.Y401N variants reduced ETV6 binding to SMRT and SMRTe (Figure 2E)
Increased numbers of circulating CD34 positive cells in affected family members
F1 carriers (F1-III3, F1-III7, F1-III8, F1-IV1 and F1-IV3) exhibited a 4- to 6-fold increase in circulating CD34+/CD38+ cells compared with healthy donors (Figure 3A,B). Similarly, F3-I2 and F3-II4 exhibited a 5- and 3-fold increase in circulating CD34+ cells compared with controls (0.16% and 0.09%, respectively, vs. 0.035%). The expression levels of immature cell markers CD133 and CD117 did not differ between F1 members and controls. Expression of the myeloid lineage marker CD33 contrasted with the absence of megakaryocyte lineage markers CD123, CD41, CD61 and CD42b (data not shown). Additionally, plasma levels of SDF1α did not vary between patients (F1: 1966 ± 95 pg/mL; n=8) and controls (2068 ± 75 pg/mL; n=9).
Figure 3.
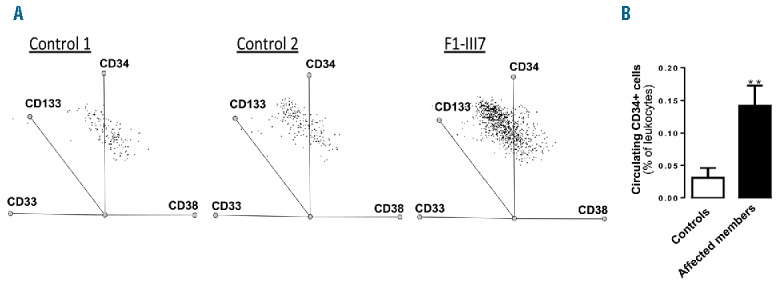
Increased numbers of circulating CD34 positive cells in variant carriers. Flow cytometry analysis of CD34+ cells. (A) Representative CD34+/CD38+ dot plot of cells from 2 controls and 1 patient (F1-III7). (B) Histograms show the percentage of CD34+ cells in 8 controls and 5 affected family members (F1-III3, F1-III7, F1-III8, F1-IV1, F1-IV3) (mean ± SEM, student’s t-test, **P<0.01).
Variants in ETV6 cause megakaryocyte hyperplasia but reduced proplatelet formation in vitro
The percentage of CD41+CD42a+ megakaryocytes derived from CD34+ (gated on Hoechst+ cells) was significantly higher in mutETV6 carriers (Figure 4A,B). No significant difference in mean ploidy was detected between patients and healthy donors (Figure 4C). Accordingly, the number of CD34+-derived CFU-GM/G/M colonies was higher in patients compared with controls (Figure 4D). The number of megakaryocyte progenitors (CFU-MK) from patients F3-I2 and F3-II4 did not differ from controls, although the size of CFU-MKs was significantly increased in the two patients (Figure 4E,F), thereby suggesting an increased proliferation of megakaryocyte precursors in the presence of mutETV6.
Figure 4.
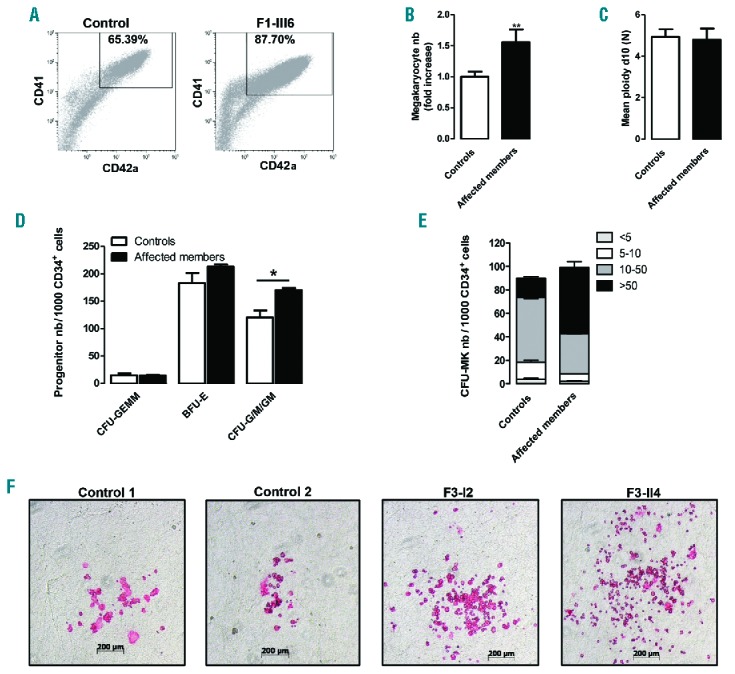
Megakaryocyte differentiation and colony-forming cell potential. (A–C) In vitro megakaryocyte (MK) differentiation in control or patient peripheral blood CD34+ cells, the cells were analyzed at culture day 10. (A) The data show a representative dot plot of CD41 and CD42a expression in Hoechst+ cells from a control individual and F1-III6. The gate represents mature MKs. (B) The histogram represents the MK (CD41+CD42a+Hoechst+) numbers (nb) in the affected family members (n=9) expressed as fold increase over healthy controls (n=10), student’s t-test, **P<0.01. (C) The ploidy level (N) was analyzed for CD41+CD42a+ MKs, and mean ploidy was calculated using the percentage of cells with 2N, 4N, 8N, 16N and 32N. (D) Methylcellulose assay. The histograms present the number of erythroid (BFU-E), granulo-monocyte (CFU-G/M/GM) and mixed (CFU-GEMM) progenitors from two patients of family F3 with the p.Y401N variant (F3-I2 and F3-II4) and two independent controls. Mean ± SEM, student’s t-test, *P<0.05. (E) Fibrin clot culture. The histograms present the number of MK progenitors (CFU-MK) from two independent controls and two patients (F3-I2 and F3-II4). The CFU-MKs are divided into four categories: <5 MKs per colony, 5–10 MKs per colony, 10–50 MKs per colony or >50 MKs per colony. Error bars represent ± SD of triplicate experiments. (F) Representative pictures of CFU-MKs after CD41 immunostaining. Control 1 and Control 2 represent 2 independent controls, and F3-I2 and F3-II4 are two affected patients. CFU-GEMM: colony-forming unit–granulocyte, erythrocyte, monocyte, megakaryocyte; BFU-E: burst forming unit-erythroid; CFU-G/M/GM: colony-forming unit-granulocytes, macrophages, granulocyte-macrophages; CFU-MK: colony-forming unit-megakaryocyte.
Proplatelet-bearing megakaryocytes derived from controls showed multiple branched thin extensions, swellings and tips. In contrast, megakaryocytes from patients formed very few proplatelets with a reduced number of thicker extensions. Although there was no swelling, tips were of increased size (Figure 5A). A 2- to 15-fold decrease in the percentage of proplatelet-bearing megakaryocytes was observed in carriers of the p.P214L (F1-III3, F1-III7 and F1-IV3) and p.Y401N (F3-II2 and F3-II4) variants (Figure 5B). β-tubulin and F-actin staining confirmed the megakaryocyte-proplatelet extension defect together with a reduced concentration of both actin filaments and microtubules in the residual larger megakaryocyte cell body (Figure 5C). Additionally, β-tubulin failed to accumulate normally in the few extension tips observed in mutETV6 megakaryocytes compared with controls.
Figure 5.
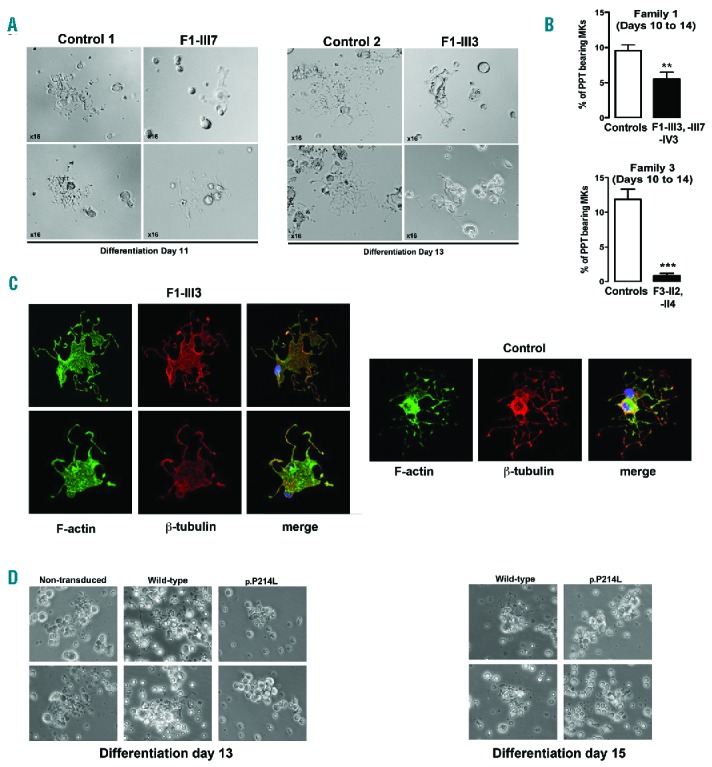
ETV6 variants lead to defective proplatelet formation. (A-B) In vitro MK differentiation induced from control or patient peripheral blood CD34+ progenitors in the presence of TPO and SCF. (A) Representative microscopic images of PPT formation in control (n=2) and patient (F1-III7, F1-III3) MKs after 11 or 13 days of culture. (B) The histograms show the percentage of PPT-bearing MKs from members of 2 families (F1-III3, F1-IV3, F1-III7, F3-I2, F3-II4) and 5 independent controls evaluated (3 to 5 evaluations) between culture days 10 to 15. The percentage of PPT-forming MKs was estimated by counting MKs exhibiting ≥1 cytoplasmic processes with areas of constriction. Double-blinded researchers quantified a total of 300–500 cells. The results are expressed as mean ± SEM, student’s t-test **P<0.01 and ***P<0.001. (C) F-actin and β-tubulin staining on PPT-forming MKs from F1-III3 and a control individual, adhering to fibrinogen. Confocal images were acquired at day 12 of culture (x60). (D) In vitro MK differentiation was induced from control peripheral blood CD34+ progenitors transduced with WT or mutETV6 (family F1, c.641C>T, p.P214L) lentiviral particles in the presence of TPO and SCF. Microscopic images of PPT formation were acquired at days 13 and 15 of culture. PPT: proplatelet; MKs: megakaryocytes; F-actin: filamentous actin.
To confirm that mutETV6 leads to a defect in proplatelet formation, CD34+ cells from healthy donors were transduced with lentivirus containing ETV6 sequences encoding the WT or the p.P214L mutant. Non-transduced cells were also included as control. After 13 and 15 days of culture in the presence of TPO and SCF, cells transduced with mutETV6 lentivirus did not form proplatelets, in contrast to non-transduced cells or those transduced with WT ETV6 (Figure 5D).
mutETV6 does not alter MYH10 expression but was associated with decreased expression and activity of the key regulators of the actin cytoskeleton CDC42 and RHOA
To assess potential cooperation between the ETV6, RUNX1 and FLI1 pathways we examined MYH10 protein expression levels in patient platelets. We did not detect increased MYH10 levels in platelets from ETV6 patients (F1-III6 and F1-III8), which contrasted with RUNX1 and FLI1 defects24 (Online Supplementary Figures S1A and S1B).
Proplatelet formation is dependent on massive reorganization of the actin cytoskeleton. Rho GTPase family members (e.g., CDC42, RAC1 and RHOA) are key regulators of actin cytoskeleton dynamics in platelets25 and megakaryocytes. p.P214L (n=5) and p.Y401N (n=2) variants led to significantly reduced platelet expression levels of CDC42 and RHOA, without affecting RAC1 expression (Figure 6A). Likewise, CDC42 and RHOA messenger ribonucleic acid (mRNA) levels were decreased in megakaryocytes from F1 and F3, while RAC1 mRNA levels remained unaffected (Figure 6B). Notably, patient F3-I2, with 112 × 109 platelets/L, exhibited only slightly decreased levels of CDC42 and RHOA in platelets (Figure 6C) compared with other affected members. CDC42 and RHOA levels significantly correlated with platelet count (n=6 from F1 and F3) (P=0.03 and r=0.84 for CDC42; P=0.008 and r=0.92 for RHOA). To confirm the specificity of this effect, we quantified CDC42 protein levels in FLI1 deficient patients with thrombocytopenia (n=2) (122 and 131 × 109/L). None of these patients exhibited reduced levels of CDC42 (Online Supplementary Figure S1C).
Figure 6.
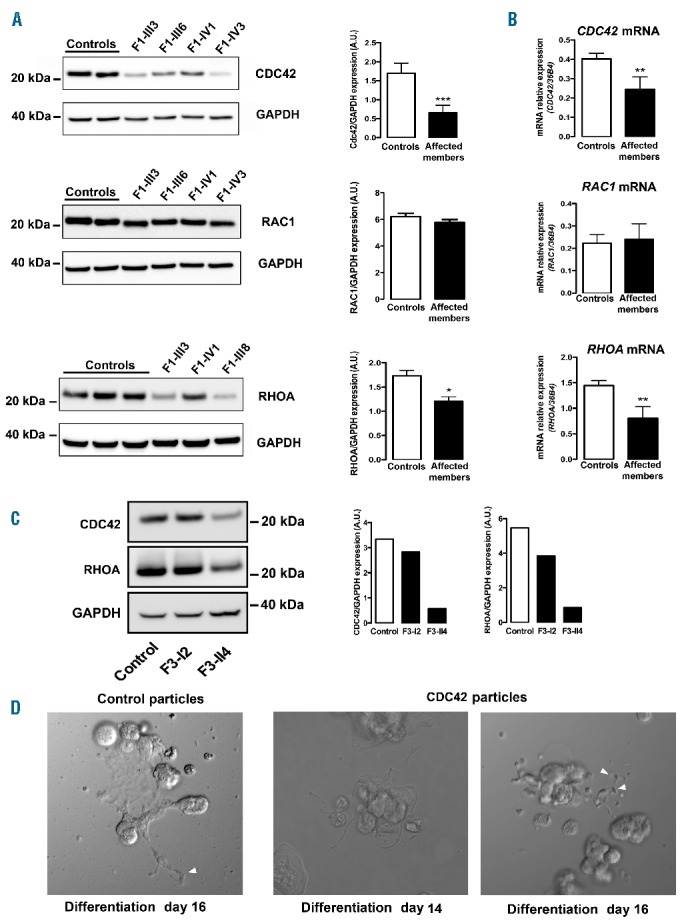
Rho GTPase expression analysis. (A) Western blot analysis and quantification of CDC42, RAC1 and RHOA expression in platelet lysates from healthy controls (n=7 for CDC42, n=4 for RAC1, n=4 for RHOA) and affected members from F1 (n=6 for CDC42, n=4 for RAC1 and n=5 for RHOA). GAPDH was used as a protein loading control. The results are expressed as mean ± SEM, student’s t-test *P<0.05 and ***P<0.001. (B) Quantification of CDC42, RAC1 and RHOA mRNA levels in CD34+-derived megakaryocytes (MKs) from healthy controls (n=10) and affected family members from F1 (n=6) and F3 (n=2). mRNA expression levels were measured via reverse transcription polymerase chain reaction (RT-PCR), and expression levels were normalized to housekeeping 36b4 RNA. The results are expressed as mean ± SEM, student’s t-test, **P<0.005. (C) Western blot analysis and quantification of CDC42, RHOA and GAPDH expression in platelets from two affected members of the F3 family (F3-I2 and F3-II4) and a healthy control. (D) In vitro MK differentiation was induced from F1-III7 CD34+ progenitors transduced with control or CDC42 lentiviral particles in the presence of TPO and SCF. Microscopic images of proplatelet (PPT) formation were acquired at days 14 and 16 of culture. The arrows indicate thinner PPT extensions and swellings in the presence of CDC42. Extensions were enlarged in the control. mRNA: messenger RNA; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; A.U: arbitrary unit.
Overexpression of CDC42 in CD34+-derived megakaryocytes from patients (F1-III3, F1-III7, F1-III8) did not fully reverse the phenotype, although it did improve the proplatelet-bearing megakaryocyte phenotype. Transduced cells produced thinner extensions and swellings, which were not observed in control transduced cells (Figure 6D).
mutETV6 alters platelet spreading
The reduced expression of CDC42 and RHOA suggests that ETV6 is involved in cytoskeletal reorganization, and thus does not only have an important role in platelet shape (EM showed more round platelets) and proplatelet formation, but also in regulating platelet spreading. We assessed whether the p.P214L transition in ETV6 affects the spreading of platelets over immobilized fibronectin and clot retraction. Platelets showed a reduced capacity to form filopodia and lamellipodia, under unstimulated and adenosine diphosphate (ADP)-stimulated conditions, respectively (Figure 7A). The G/F actin ratio was significantly higher in the rare patient platelets that spread (Figure 7B). Furthermore, reduced clot retraction velocity was noticeable in the mutETV6 (F1-lll7) (Figure 7C).
Figure 7.
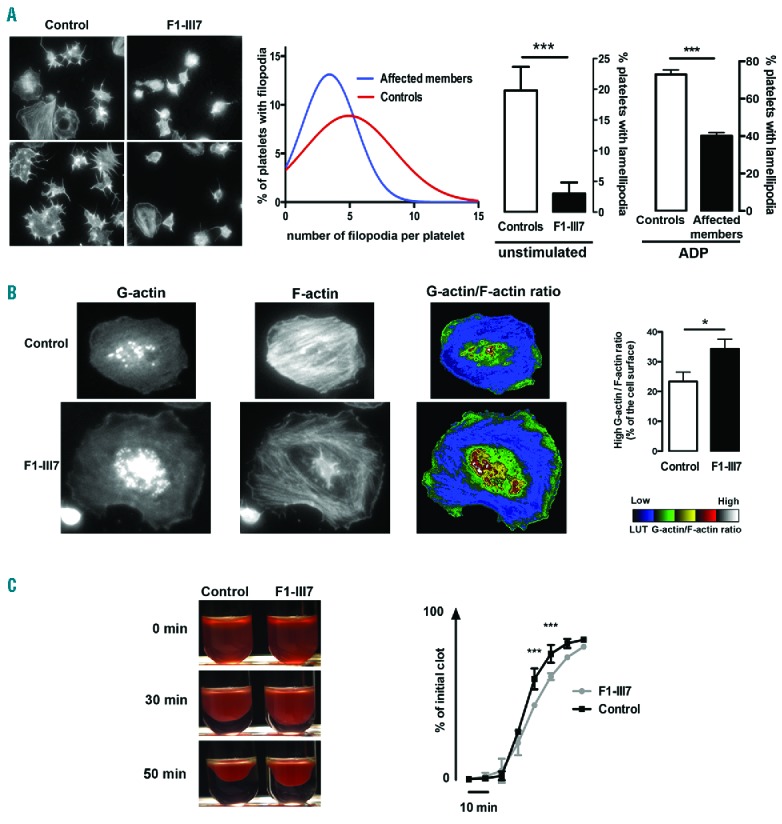
Platelet spreading and clot retraction. (A) Left: Representative images of unstimulated platelets spread over immobilized fibronectin. Middle: filopodia formation was quantified according to the number of extensions per unstimulated platelet derived from affected individuals (F1-III3, F1-III7) and healthy controls (n=2). Right: Quantification of lamellipodia-forming cells, at resting and ADP-stimulated conditions, from affected members (F1-III7, F1-III8) and healthy controls (n=2). The data are expressed as mean ± SEM of 5 different view fields. Student’s t-test, ***P<0.001. (B) Actin polymerization quantification in spread unstimulated platelets. Left: representative images of G-actin, F-actin and the G-actin/F-actin ratio in control platelets and the rare spread platelets detected in F1-III7. Platelets were spread over fibronectin and stimulated with ADP. Right: quantification of the area with the high G-actin/F-actin ratio. Quantification of the ratio was performed according to the look-up table as the percentage of the platelet surface (n=20 different cells; mean ± SEM. Student’s t-test *P<0.05). (C) Clot retraction. Left: representative images at 0, 30 and 50 minutes. Right: quantification of the extent of clot retraction expressed as percentage of the initial clot (mean ± SEM, n=2 for F1-III7 and n=4 for controls. Two-way ANOVA, ***P<0.001). F-actin: filamentous actin; G-actin: globular-actin; ADP: adenosine diphosphate; LUT: look-up table.
Discussion
Herein, we presented six families with AD thrombocytopenia associated with germline variants in ETV6. CD34+-derived megakaryocytes from mutETV6 carriers showed a reduced ability to form proplatelets. The variants in the Ets domain impaired interaction with the corepressors NCOR, SMRT and Sin3A. Patient platelets were more round and had a reduced capacity to form filopodia and lamellipodia, which was associated with reduced expression levels of cytoskeletal regulators CDC42 and RHOA. Additionally, mutETV6 carriers displayed increased numbers of circulating CD34+ progenitor cells, which may contribute to a predisposition to hematologic malignancy.
Loss of ETV6 function has been reported to contribute to leukemia, predominantly due to somatic variants and fusion transcripts.3 Four amino acid substitutions described in this study are listed in the Catalog Of Somatic Mutations In Cancer (COSMIC). The p.P214L, p.R396G and p.A377T variants were present in digestive tract tumors,26 while p.Y401C has been associated with AML.27 More recently, germline ETV6 variants have also been found to predispose to cancer. Eleven patients carrying mutETV6 (p.P214L, p.R399C, p.R369Q, p.L349P, p.N385fs or p.W380R) developed acute lymphocytic leukemia or myelodysplastic syndrome.1–3,28 Two affected members of the families F1 and F2 had myelodysplasia with RAEB and one member of the F4 family was successfully treated with chemotherapy for AML-M0.
Variants reduced the repressive activity of ETV6 without altering ETV6 protein expression levels in platelets. The alteration of ETV6 repressive activity can be explained by the modification of ETV6 cellular localization, as p.P214L and four other variants affecting the ETS domain lead to ETV6 sequestration in the cytoplasm in both HeLa transfected cells and cultured megakaryocytes.1–3 However, three of these variants only partially prevented nuclear localization, thereby indicating other possible mechanisms. The p.A377T and p.Y401N variants prevented corepressor complex recruitment. These substitutions are located in the ETV6 Ets second and third α-helix, contiguous to amino acids involved in key hydrophobic contacts with the H5 helix of the C-terminal inhibitory domain (aa 426–436),29 thus possibly affecting ETV6 DNA-binding ability. In immunoprecipitation assays, overexpression of WT or p.P214L ETV6 did not modify the interaction between SMRT and ETV6, while p.A377T and p.Y401N ETV6 significantly reduced this interaction. Overall, this suggests that variants in the Ets DNA-binding domain exert a dominant negative effect.
ETV6 has been shown to drive megakaryocyte differentiation of hematopoietic stem cells.30 From the literature, and supported by BM studies in F4-II1 and F5-II2, ETV6 defects seem to result in an increased percentage of small megakaryocytes.1 Our megakaryocyte colony assays confirmed an increased proliferation of early megakaryocyte progenitors, characterized by an increased production of CD41+CD42a+ megakaryocytes compared to control conditions. This may explain the reduced TPO levels observed in the affected members of family F1. Accordingly, the loss of ETV6 in the erythro-megakaryocytic lineage in mice also results in large, highly proliferative early megakaryocytes and mild thrombocytopenia. We cannot exclude that ETV6-driven deregulation of megakaryocyte proliferation may take place in hematopoietic progenitors, thereby promoting oncogenic transformation. Altogether, these data do not support the concept that signaling between the ETV6 and RUNX1/FLI1/ANKRD26 pathways is involved in the underlying mechanism, as variants in these genes were associated with a decreased or normal megakaryocyte colony formation.19,31,32 Furthermore, MYH10 expression levels remained low in patients with mutETV6, which indicates unaltered RUNX1 and FLI1 function.24
Despite the increased early megakaryocyte proliferation potential, CD34+-derived megakaryocytes from patients with mutETV6 showed a reduced capacity to form proplatelets. These altered megakaryocyte features suggest that a defect in cytoskeletal reorganization during proplatelet formation likely causes thrombocytopenia in patients. Sequencing of platelet RNA from patients with p.P214L ETV6 revealed a considerable reduction in the levels of several cytoskeletal transcripts.1 Furthermore, Palmi et al.33 showed that the ETV6-RUNX1 fusion protein, which is associated with a loss of ETV6 repressive activity,34,35 alters the expression of genes regulating cytoskeletal organization. In particular, the ETV6-RUNX1 fusion protein led to reduced expression of CDC42. The mechanism by which the loss of ETV6 repressive activity results in reduced CDC42 and RHOA expression remains to be resolved.
CDC42 is an important mediator of platelet and megakaryocyte cytoskeleton reorganization.36 Therefore, we hypothesize that ETV6 repressive activity is a key regulator of megakaryocyte cytoskeleton remodeling, driven via Rho GTPases in mutETV6 carriers. mutETV6 was associated with a decrease in CDC42 and RHOA expression levels in platelets without affecting RAC1 expression. Additionally, mutETV6 platelets showed defects in functions classically associated with CDC42 (i.e., filopodia formation) and RHOA (i.e., lamellipodia formation and clot retraction).36 EM also showed platelets of variable sizes and having a more circular instead of discoid shape. RNA sequencing previously performed on mutETV6 transfected cells, patient platelets and leukemia cells did not reveal any modification in Rho GTPase mRNA levels,1,3 which may be due to variations in the models applied. Indeed, Rho GTPase mRNA levels were evaluated in CD34+-derived megakaryocytes, and the reduced mRNA levels were confirmed at the protein level in patient platelets. Moreover, we observed a correlation between platelet count and CDC42 and RHOA expression levels, thereby suggesting a relationship between thrombocytopenia severity and Rho GTPase levels. Key regulators of the actin cytoskeleton CDC42 and RHOA have already been shown to be associated with thrombocytopenia, due to defects in cytoskeleton organization.37–41 In affected individuals, we found abnormal tubulin organization in proplatelet-forming megakaryocytes and altered actin polymerization in platelets. Rescue experiments with CDC42 lentiviral particles were not able to fully reverse the phenotype, although the cells produced thinner extensions and swellings, which were barely observed in control cells.
In mice, Cdc42 or RhoA deficiency causes increased platelet clearance.37,38 Such an observation was noted in two patients: one young girl who never received platelets (F3-II4), and a patient for whom splenectomy improved the platelet count (F1-III3). This suggests that ETV6 mutations are linked to several defects with reduced platelet formation and survival, although this latter mechanism requires further confirmation.
Individuals carrying a germline ETV6 variant showed increased numbers of circulating CD34+/CD133+ cells. The phenotype of these stem cells did not differ between patients and controls. This increase has to be considered as a helpful marker of the ETV6-related thrombocytopenia. It may not be attributed to excessive proliferation, as Zhang et al. showed reduced proliferation of CD34+ cells expressing WT or mutETV6.3 Interestingly, the defect in CDC42 expression may also account for increased hematopoietic progenitor mobilization, as chemical inhibition of CDC42 in mice efficiently improved progenitor recruitment in the peripheral blood. Altered interaction between mutated progenitors and the BM microenvironment, as reported in the case of the ETV6-RUNX1 fusion protein, may also be involved.33 Further investigations are required to more precisely delineate the role that ETV6 plays in stem cell progenitor mobilization.
In conclusion, we identified six variants in ETV6, of which five are novel, associated with dominant thrombocytopenia. Our study provides novel insights into the role that ETV6 plays in platelet function, morphology and formation that seem to be driven by changes in the cytoskeleton and potentially also in circulating CD34+ progenitor levels.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/2/282
Funding
Bioinformatics analyzes benefit from the C2BIG computing centre funded by the Région Ile de France and UPMC. This work was partially supported by the ICAN Institute of Cardiometabolism and Nutrition (ANR-10-IAHU-05), the Ligue nationale contre le cancer (Labeled team H Raslova), and the “Fondation pour la Recherche Médicale FRM” (grant to PS FDM20150633607). We thank Dr. J. Ghysdael and Dr. F. Guidez for providing the plasmid constructs, Laboratory of Pr. D. Raoult (URMITE), microscopy unit (P. Weber), Dr Paola Ballerini (Hopital Trousseau) for genotyping; Dr. JC. Bordet for transmission electron microscopy; Dr C Chomiene and C Dosquet for the platelet survival assay; M Crest for experimental help, the French Reference Center on Hereditary Platelet Disorders (CRPP) for patients’ clinical exploration. For the F2-F6 families, study makes use of whole genome sequencing data and analysis approaches generated by the NIHR BioResource - Rare Disease BRIDGE Consortium. The NIHR BioResource - Rare Diseases is funded by the National Institute for Health Research of England (NIHR; award number RG65966)). KF is supported by the Fund for Scientific Research-Flanders (FWO-Vlaanderen, Belgium, G.0B17.13N] and by the Research Council of the University of Leuven (BOF KU Leuven‚ Belgium, OT/14/098].
References
- 1.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47(5):535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topka S, Vijai J, Walsh MF, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. 2015; 11(6):e1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998;32:495–519. [DOI] [PubMed] [Google Scholar]
- 5.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208(13):2571–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwiatkowski BA, Bastian LS, Bauer TR, Jr, Tsai S, Zielinska-Kwiatkowska AG, Hickstein DD. The ets family member Tel binds to the Fli-1 oncoprotein and inhibits its transcriptional activity. J Biol Chem. 1998;273(28):17525–17530. [DOI] [PubMed] [Google Scholar]
- 8.Chakrabarti SR, Sood R, Ganguly S, Bohlander S, Shen Z, Nucifora G. Modulation of TEL transcription activity by interaction with the ubiquitin-conjugating enzyme UBC9. Proc Natl Acad Sci U S A. 1999;96(13):7467–7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Hiebert SW. TEL contacts multiple co-repressors and specifically associates with histone deacetylase-3. Oncogene. 2001;20(28):3716–3725. [DOI] [PubMed] [Google Scholar]
- 10.Green SM, Coyne HJ, 3rd, McIntosh LP, Graves BJ. DNA binding by the ETS protein TEL (ETV6) is regulated by autoinhibition and self-association. J Biol Chem. 2010;285(24):18496–18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang LC, Swat W, Fujiwara Y, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12(15):2392–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nurden P, Debili N, Vainchenker W, et al. Impaired megakaryocytopoiesis in type 2B von Willebrand disease with severe thrombocytopenia. Blood. 2006;108(8):2587–2595. [DOI] [PubMed] [Google Scholar]
- 13.Thakur ML, Walsh L, Malech HL, Gottschalk A. Indium-111-labeled human platelets: improved method, efficacy, and evaluation. J Nucl Med. 1981;22(4):381–385. [PubMed] [Google Scholar]
- 14.Westbury SK, Turro E, Greene D, et al. Human phenotype ontology annotation and cluster analysis to unravel genetic defects in 707 cases with unexplained bleeding and platelet disorders. Genome Med. 2015;7(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turro E, Greene D, Wijgaerts A, et al. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci Transl Med. 2016;8(328):328ra330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barton GJ, Sternberg MJ. A strategy for the rapid multiple alignment of protein sequences. Confidence levels from tertiary structure comparisons. J Mol Biol. 1987;198(2):327–337. [DOI] [PubMed] [Google Scholar]
- 17.Lopez RG, Carron C, Oury C, Gardellin P, Bernard O, Ghysdael J. TEL is a sequence-specific transcriptional repressor. J Biol Chem. 1999;274(42):30132–30138. [DOI] [PubMed] [Google Scholar]
- 18.Guidez F, Petrie K, Ford AM, et al. Recruitment of the nuclear receptor corepressor N-CoR by the TEL moiety of the childhood leukemia-associated TEL-AML1 oncoprotein. Blood. 2000;96(7):2557–2561. [PubMed] [Google Scholar]
- 19.Bluteau D, Balduini A, Balayn N, et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest. 2014;124(2):580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lordier L, Bluteau D, Jalil A, et al. RUNX1-induced silencing of non-muscle myosin heavy chain IIB contributes to megakaryocyte polyploidization. Nat Commun. 2012;3:717. [DOI] [PubMed] [Google Scholar]
- 21.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–267. [DOI] [PubMed] [Google Scholar]
- 22.Raslova H, Komura E, Le Couedic JP, et al. FLI1 monoallelic expression combined with its hemizygous loss underlies Paris-Trousseau/Jacobsen thrombopenia. J Clin Invest. 2004;114(1):77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klimchenko O, Mori M, Distefano A, et al. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood. 2009;114(8):1506–1517. [DOI] [PubMed] [Google Scholar]
- 24.Antony-Debre I, Bluteau D, Itzykson R, et al. MYH10 protein expression in platelets as a biomarker of RUNX1 and FLI1 alterations. Blood. 2012;120(13):2719–2722. [DOI] [PubMed] [Google Scholar]
- 25.Goggs R, Williams CM, Mellor H, Poole AW. Platelet Rho GTPases-a focus on novel players, roles and relationships. Biochem J. 2015;466(3):431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120(18):e83–92. [DOI] [PubMed] [Google Scholar]
- 28.Melazzini F, Palombo F, Balduini A, et al. Clinical and pathogenetic features of ETV6 related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coyne HJ, 3rd, De S, Okon M, et al. Autoinhibition of ETV6 (TEL) DNA binding: appended helices sterically block the ETS domain. J Mol Biol. 2012;421(1):67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakurai T, Yamada T, Kihara-Negishi F, et al. Effects of overexpression of the Ets family transcription factor TEL on cell growth and differentiation of K562 cells. Int J Oncol. 2003;22(6):1327–1333. [PubMed] [Google Scholar]
- 31.Bluteau D, Glembotsky AC, Raimbault A, et al. Dysmegakaryopoiesis of FPD/AML pedigrees with constitutional RUNX1 mutations is linked to myosin II deregulated expression. Blood. 2012;120(13):2708–2718. [DOI] [PubMed] [Google Scholar]
- 32.Breton-Gorius J, Favier R, Guichard J, et al. A new congenital dysmegakaryopoietic thrombocytopenia (Paris-Trousseau) associated with giant platelet alpha-granules and chromosome 11 deletion at 11q23. Blood. 1995;85(7):1805–1814. [PubMed] [Google Scholar]
- 33.Palmi C, Fazio G, Savino AM, et al. Cytoskeletal regulatory gene expression and migratory properties of B cell progenitors are affected by the ETV6-RUNX1 rearrangement. Mol Cancer Res. 2014; 12:1796–1806. [DOI] [PubMed] [Google Scholar]
- 34.Song H, Kim JH, Rho JK, Park SY, Kim CG, Choe SY. Functional characterization of TEL/AML1 fusion protein in the regulation of human CR1 gene promoter. Mol Cells. 1999;9(5):560–563. [PubMed] [Google Scholar]
- 35.Rho JK, Kim JH, Yu J, Choe SY. Correlation between cellular localization of TEL/AML1 fusion protein and repression of AML1-mediated transactivation of CR1 gene. Biochem Biophys Res Commun. 2002;297(1):91–95. [DOI] [PubMed] [Google Scholar]
- 36.Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11(1):35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pleines I, Eckly A, Elvers M, et al. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. 2010;115(16): 3364–3373. [DOI] [PubMed] [Google Scholar]
- 38.Pleines I, Hagedorn I, Gupta S, et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood. 2012;119(4):1054–1063. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki A, Shin JW, Wang Y, et al. RhoA is essential for maintaining normal megakaryocyte ploidy and platelet generation. PLoS One. 2013;8(7):e69315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pleines I, Dutting S, Cherpokova D, et al. Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood. 2013;122(18):3178–3187. [DOI] [PubMed] [Google Scholar]
- 41.Chen C, Song X, Ma S, et al. Cdc42 inhibitor ML141 enhances G-CSF-induced hematopoietic stem and progenitor cell mobilization. Int J Hematol. 2015;101(1):5–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.