

Chronic benign lymphoproliferation and autoimmune cytopenias are the main features requiring treatment in FAS mutant patients with autoimmune lymphoproliferative syndrome (ALPS).1,2 Successful use of the mTOR inhibitor rapamycin was initially reported in the treatment of refractory cytopenia in 3 ALPS-FAS patients.3 The remarkable efficacy as a second-line agent for this indication was confirmed in a recent prospective study including a further 9 ALPS-FAS patients.4 Here, we analyze aspects of rapamycin therapy that have so far not been addressed including first- versus second-line therapy, comprehensive biomarker responses, and the consequences of stopping rapamycin by reporting our experience in 28 ALPS-FAS patients.
We performed a retrospective survey of ALPS patients enrolled into research protocols in Paris, France (DC2011-1338) and Freiburg, Germany (DRKS00000298). Patients were included if they fulfilled NIH diagnostic criteria of ALPS5 with genetical confirmation, and had received rapamycin for more than 6 months. Lymphoproliferation was defined as enlarged spleen or lymphadenopathy (≥ 2 lymph nodes in ≥ 2 sites enlarged for ≥ 3 months). Autoimmune cytopenia required autoantibodies or documented response to immunosuppression. In patients not previously treated with steroids or immunosuppressive drugs, rapamycin was regarded as first-line therapy. IVIG was not considered immunosuppression. Rapamycin was initiated at 1–2.8 mg/m2/day (d) aiming for plasma levels of 2–10 ng/mL. Treatment responses were evaluated at 6–9 months and at last follow up. Complete remission (CR) was defined as normal blood counts with platelets over 100×109/L, absent splenomegaly (palpable <2 cm) and lymphadenopathy and cessation of immunosuppression including steroids. Partial remission (PR) was defined as persistent symptoms but a 50% or greater decrease in spleen size and/or in a reference lymph node and cessation of steroids without relapse of cytopenia.
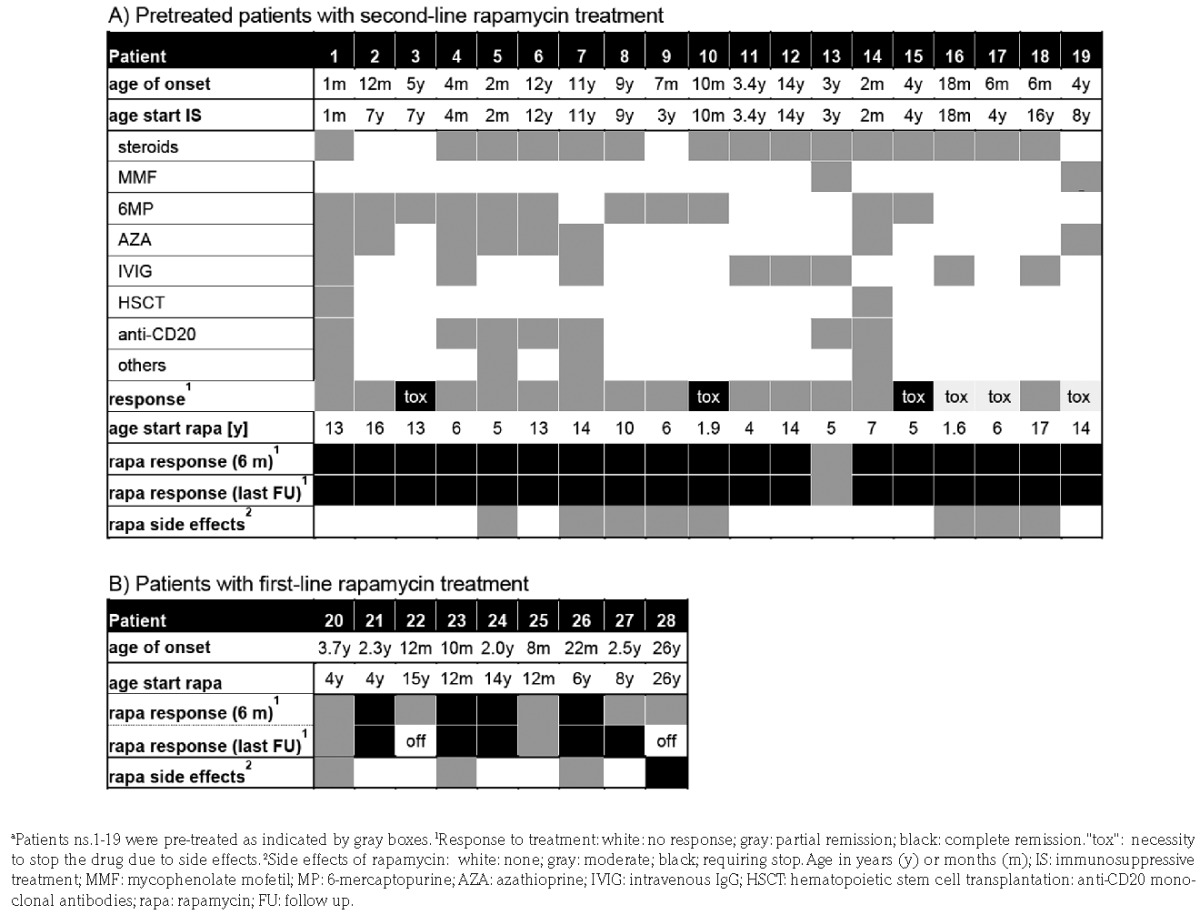
Of 28 patients, 19 had heterozygous germline TNFRSF6 mutations, one had a homozygous mutation; 8 had somatic mutations (Online Supplementary Table S1). Median age at disease onset was 4 (0–26) years (Table 1). The indication for rapamycin was lymphoproliferation in 8, lymphoproliferation and autoimmunity in 18, and autoimmunity in 2 patients. Overall, 19 patients had autoimmune cytopenia [16 in 1 lineage (mostly AIHA), 2 in 2 lineages, 1 in 3 lineages]. Before rapamycin, 7 patients had no previous therapy, 2 had received IVIG. In these 9 patients, rapamycin was considered first-line therapy (Table 1). It was initiated as monotherapy in 8 patients and in P21 together with steroids and IVIG. The remaining 19 patients had received up to 6 lines of previous therapies for a median of 3.7 (0.3–13) years. Two patients had undergone hematopoietic stem cell transplantation. Both had disease relapses due to graft rejection (P1) and mixed chimerism (P14). In these patients, rapamycin was considered second-line therapy (Table 1). At initiation of rapamycin, 3 of 19 had no response to previous treatment, 13 were in PR and 3 had responded, but had side effects (Table 1). Except for 2 cases, ongoing therapy was maintained (steroids n=9, steroids+6MP n=2, 6MP n=4, AZA n=3, MMF n=1). Rapamycin was introduced at a median dose of 2 (1–2.8) mg/m2. After 6–9 months of treatment, 22 patients (79%) were in CR and 6 (21%) in PR. Rapamycin add-on therapy led to CR in 94% (17 of 18). The other immunosuppressive drugs were discontinued in all patients within six months, except for one with PR (P13). In that patient, who had presented with severe AIHA, rapamycin resolved the splenomegaly and stabilized, but did not fully control AIHA. Nevertheless, corticosteroids could be terminated. First-line rapamycin induced CR in 4 and PR in 5 patients (Table 1 and Online Supplementary Figure S1). The incomplete treatment response was associated with documented poor compliance in 2 patients (P22 and P28). The other 3 PR patients were satisfied with the response achieved and no dose escalation or alternative therapies were attempted. Importantly, rapid stabilization of cytopenias was achieved among all 5 patients who received rapamycin as first-line therapy for AIHA or ITP. At last follow up, 26 patients were still on rapamycin. P28 developed IgG4-related disease while on rapamycin and it was decided to stop treatment, P22 stopped because of feeling “unwell” on the medication. Median treatment duration was 2.8 (0.5–5.6) years. All 23 patients with initial CR maintained CR (Table 1). Rapamycin dose was reduced in 14 of 22 patients where this information was available. At last follow up, serum levels were available in 10 patients and CR was maintained in all 6 patients at levels between 2–5 ng/mL.
Table 1.
Treatment history and rapamycin response of individual patients.
As described,3,6 DNT cells rapidly decreased upon rapamycin treatment, but only 7 of 25 patients (33%) reached normal percentages (Figure 1A). IL-10 levels decreased, but remained more than 20 pg/mL in 14 of 23 patients (61%) (Figure 1C). sFASL decreased, but only 2 of 28 patients reached normal values (Figure 1D). Significant biomarker decreases were observed in both first- and second-line treated patients. VitB12 levels remained elevated in 16 of 23 (69%) patients. While 6 of 19 patients with second-line treatment showed a drop in VitB12 levels, 7 of 19 showed an increase (Figure 1B). All patients with rapamycin first-line treatment showed a decrease (Figure 1B). Among 22 patients in CR, 2 had complete normalization and 5 had close to normal values for DNT cells, IL-10 and sFASL, while 15 patients still had elevated biomarkers. There was no statistical difference in biomarker responses between PR and CR patients. Thirteen patients experienced mild adverse effects, including mouth ulcers (n=4), mild proteinuria (n=2), increased blood pressure (n=2), and transient skin rash, Helicobacter Pylori associated gastritis, transient liver enzyme elevation concomitant to herpes zoster and EBV infection in one patient each. In 6 patients, rapamycin was temporarily stopped due to incompliance (n=3), complete remission (n=2) or elevated blood pressure (n=1). All 6 had rapid relapse of lymphoproliferation and 2 relapsed with autoimmune cytopenias, accompanied by rapid re-augmentation of biomarkers (Figure 2A and B). Five patients restarted treatment. All had the same response as during the first treatment episode (Figure 2A).
Figure 1.
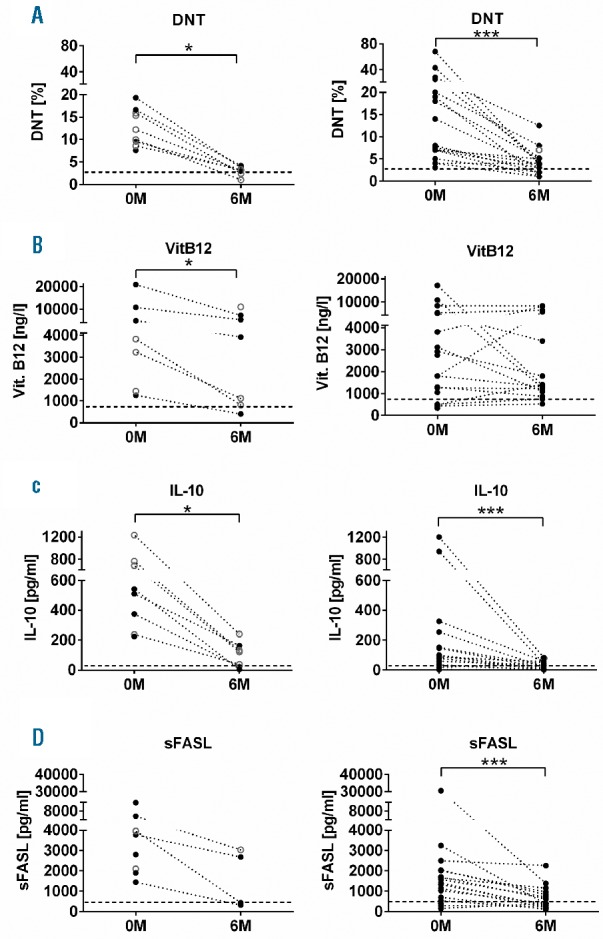
Biomarker responses at six months in patients receiving rapamycin as first-line or second-line treatment. The percentage of DNT cells among CD3+TCRαβ+ lymphocytes (A), the serum levels of VitB12 (B), IL-10 (C) and sFASL (D) were determined before rapamycin therapy and 6–8 months after initiation of treatment in patients who received rapamycin as first-line therapy (left panels) or after prior immunosuppressive treatment (right panels). Patients with complete remission are depicted in black, partial remissions are indicated by open gray circles. Analyses were performed using PRISM-software (GraphPad software, San Diego, USA). Populations were compared using the Wilcoxon matched-pairs signed rank t-test. P<0.05 was considered significant. *P<0.05; ***P<0.001.
Figure 2.
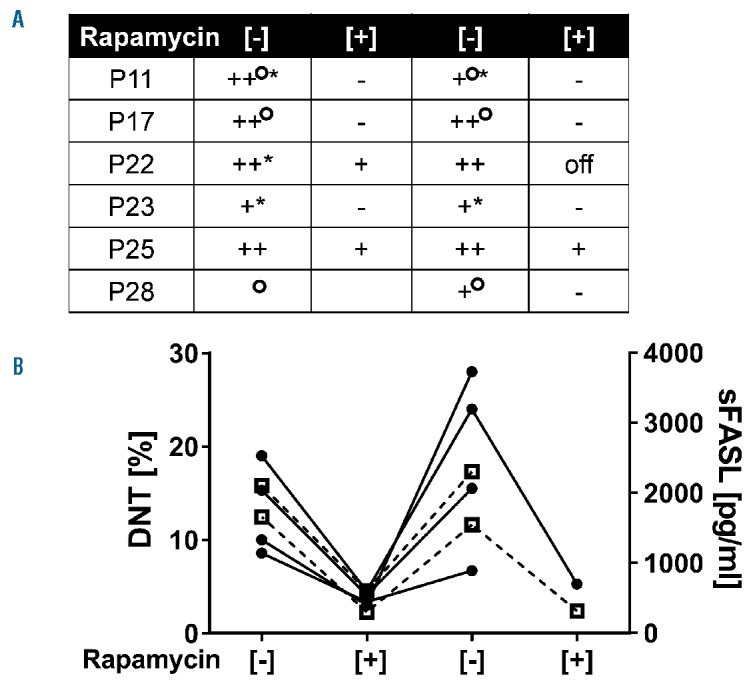
Effect of treatment discontinuation. Summary of observations in 6 patients who discontinued rapamycin treatment. (A) Status of rapamycin treatment is indicated by [−]/[+]. The spleen size is indicated as follows: − not palpable; + palpable but <5 cm; ++ >10 cm or up to umbilicus. °Additional lymphadenopathy. *Cytopenia. (B) Percentages of DNT cells (left y-axis, black circles) and sFASL levels (right y-axis, open boxes/broken lines) at different stages of therapy.
Our results provide novel, practically relevant insights on rapamycin therapy in ALPS-FAS based on the pioneering original observations by Teachey et al.3,7 First, we confirm the efficacy and safety of rapamycin by reporting 28 additional ALPS-FAS patients, all of whom responded to treatment. Our results also confirm the good tolerability within the treatment period of up to six years. Nevertheless, long-term side effects of rapamycin can be relevant and careful monitoring is required. Second, we present the first data on ALPS-FAS patients receiving rapamycin as first-line therapy. We were not only able to stop all other treatments in patients who had received several lines of prior therapy, but we also achieved excellent responses using rapamycin as the first and single agent. This included rapid control of cytopenia and lymphoproliferative manifestations. Nevertheless, immediate stabilization of autoimmune cytopenia cannot be expected and may require initial concomitant treatment. We propose to consider rapamycin as a first-line treatment in ALPS-FAS patients.
In genetically proven cases, there is a clear biological rationale for rapamycin. Lymphoproliferation in ALPS is not just due to an accumulation of DNT cells that cannot die, but these cells and their single positive precursor cells8 are highly proliferative in vivo.6,9 This proliferative activity is associated with hyperactive mTOR signaling.6 Blocking proliferative activity and induction of apoptosis in DNT cells and their precursors are two non-mutually exclusive explanations for the impressive effect of rapamycin on lymphoproliferation.6,7 The rapid relapse of disease after stopping treatment indicates that the cells giving rise to DNT cells are either not fully eliminated or are rapidly regenerated once targeted mTOR inhibition is stopped. How rapamycin abrogates autoimmune manifestations remains unclear. Fas deficient B cells escape germinal center selection and undergo enhanced somatic hypermutation.10 Rapamycin could either directly affect B-cell signaling or survival and/or it could indirectly influence germinal center function by decreasing DNT and IL-10. While mTOR inhibition represents targeted molecular treatment for ALPS, this is less clear in patients with autoimmunity and lymphoproliferation in the context of other diseases clinically resembling ALPS-FAS. Although there are other immunodeficiencies characterized by mTOR activation (eg. activated PI3K delta syndrome11), the rapamycin response in these diseases is much more variable.4 At present, we therefore advocate first-line rapamycin only in ALPS-FAS patients. Third, we present new observations relevant for the guidance of long-term therapy. Rapamycin levels less than 5 ng/mL were sufficient to maintain disease control in most patients. However, stopping rapamycin was associated with rapid relapse in all cases. Based on these observations, we currently start with 2 mg/m2/d and adapt this up to 10 ng/mL. Once full remission has been achieved, we titrate down to 2–5 ng/mL. Because at least the lymphoproliferative manifestations tend to attenuate with age, the necessity of life-long treatment remains unclear. Another open issue of long-term treatment is whether rapamycin reduces or increases the risk of lymphoma. While biomarkers have proven useful for establishing a diagnosis of ALPS-FAS,12–14 their value in guiding therapy appears limited. Although a significant decrease was observed, CR was accompanied by biomarker normalization in no more than one-third of the patients. In summary, our results further establish rapamycin as an excellent targeted therapy for ALPS-FAS patients and provide support for its use as a first-line agent in this disease.
Supplementary Material
Acknowledgments
We thank the patients, their families and referring physicians who made this study possible.
Footnotes
Funding: this work was supported by the German Federal Ministry of Education and Research (BMBF 01 EO 0803 grant to the Center of Chronic Immunodeficiency and BMBF 01GM1111B grant to the PID-Net Initiative).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81(6):935–946. [DOI] [PubMed] [Google Scholar]
- 2.Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–1349. [DOI] [PubMed] [Google Scholar]
- 3.Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145(1):101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127(1):17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116(14):e35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volkl S, Rensing-Ehl A, Allgauer A, et al. Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood. 2016; 128(2):227–238. [DOI] [PubMed] [Google Scholar]
- 7.Teachey DT, Obzut DA, Axsom K, et al. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood. 2006;108(6):1965–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rensing-Ehl A, Volkl S, Speckmann C, et al. Abnormally differentiated CD4+ or CD8+ T cells with phenotypic and genetic features of double negative T cells in human Fas deficiency. Blood. 2014; 124(6):851–860. [DOI] [PubMed] [Google Scholar]
- 9.Sneller MC, Straus SE, Jaffe ES, et al. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90(2):334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janda A, Schwarz K, van der Burg M, et al. Disturbed B-lymphocyte selection in autoimmune lymphoproliferative syndrome. Blood. 2016;127(18):2193–2202. [DOI] [PubMed] [Google Scholar]
- 11.Angulo I, Vadas O, Garcon F, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caminha I, Fleisher TA, Hornung RL, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125(4):946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, et al. FAS-L, IL-10, and double-negative CD4− CD8− TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113(13):3027–3030. [DOI] [PubMed] [Google Scholar]
- 14.Rensing-Ehl A, Janda A, Lorenz MR, et al. Sequential decisions on FAS sequencing guided by biomarkers in patients with lymphoproliferation and autoimmune cytopenia. Haematologica. 2013; 98(12):1948–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.