

Exposure to mutagenic sources such as ionizing radiation or chemical agents leads to damage of the genome, however, DNA double-strand breaks (DSB) and the subsequent repair machinery pose a basic necessity for a functional immune response.1
Two pathways restore genomic integrity. Non-homologous end-joining (NHEJ) is error-prone and leads to T-cell receptor (TCR) and immunoglobulin diversity; defects in the NHEJ process result in severe combined immunodeficiency, radiosensitivity and developmental errors.2
On the other hand, homologous recombination (HR) conveys a high-fidelity process. It is restricted to the S and Gap 2 (G2) phase as a homologous “sister chromatid” template is required. HR is vital in providing repair to DSB and DNA interstrand crosslinks (ICL).3 ICL are toxic DNA lesions preventing the separation of the two complementary double helix strands. Chemotherapeutic agents, such as mitomycin C (MMC), nitrogen mustards and platinum compounds, can induce irreversible covalent linkage.1 Fanconi anemia (FA) is an autosomal recessive disorder, caused due to a mutation in one of at least 18 genes, leading to bone marrow failure, developmental delay and an increased incidence of malignancies.4 The inability to repair ICL is a key feature of all FA genes. BRCA2 (FANCD1) and other genes involved in breast and ovarian cancer also show regulation of ICL repair.5 We report on an 18-year-old man with profound combined immunodeficiency, Epstein-Barr virus (EBV) lymphoproliferative disease and chemosensitivity similar to FA patients, and a missense mutation in the RAD52 gene. While RAD51 and its paralogs RAD51C, BRCA1, BRCA2 and PALB2 have been associated with FA and DNA damage repair; the precise function of RAD52 in the recombination process is still unclear. We suspect a modification in the single-stranded (ss)DNA annealing function of the protein and hence a disturbed homologous recombination, leading to cancer and lymphoproliferative disease in the patient and his family. The patient presented with leukopenia with predominantly reduced lymphocytes (B, CD4 and CD8 T cells, and the expansion of TCRγδ cells). Candidate primary immunodeficiencies (PIDs) were ruled out by various immunological investigations. Immunohistochemistry of an extracted lymph node revealed polymorphic monoclonal EBV-associated B-cell lymphoproliferative disease of type III latency (Online Supplementary Figure S1). The patient’s mother died early from breast carcinoma (age 39), and further cases of colon carcinoma were reported in the maternal lineage. We performed whole exome sequencing on the peripheral blood of the patient and the patient’s sister and father, and stored samples of the deceased patient’s mother, which revealed 122 single nucleotide variants (SNVs) common in both the patient and the mother. Three were in PID genes (BCL10, CHD7 and PRF1), which were excluded due to the clinical presentation. Only one SNV came to our attention as we saw a connection to a known DNA repair pathway. Interestingly, our in-house tool to detect SNVs near known PID genes also highlighted the same SNV to be related to a known PID with DNA repair disorders (Online Supplementary Figure S2). Both the patient and his mother showed a heterozygous missense mutation in RAD52 (c.547C>T, P183S) in the ssDNA-annealing domain of the protein (Online Supplementary Figure S3), confirmed by capillary sequencing. BRCA1/2 analysis in the mother did not show any abnormalities. We did not find any de novo mutations in a known or likely PID-causing gene.
Molecular dynamic (MD) simulations were performed to compare the average structure of wild-type (WT) RAD52 with the mutant one. We illustrated the influence of the P183S mutation on dynamics and structure (Figure 1). As expected for a proline-to-serine substitution, we demonstrated a change in configuration and an increase in the distance between the Cα atom of residue 183 and the close α4’ helix. The average structures of both protein variants are highly structurally similar, indicating only a local influence of the P183S mutation. However, structural changes triggered by the P183S mutation caused a subsequent reorientation of a K169 side chain, which was predicted to disturb ssDNA binding and, hence, the ssDNA annealing function of RAD52 (for detailed analysis see the Online Supplementary Material). Primary human fibroblasts were exposed to MMC, a known DNA intercalating agent, and cultured for 48h as part of our diagnostic strategy in patients with cytopenia of unknown origin. Cells exposed to MMC showed an increased number of G2 phase cells, indicating that these cells were in a cycle arrest following damage by MMC. A G2 increase was not seen in healthy controls (Figure 2A, second left chart), while the increase was similar in an internal comparison with fibroblasts from a MMC sensitive disease control patient (Figure 2A, right chart). Lymphoblastoid cell lines (LCLs) from the patient’s bone marrow were analyzed in a routine flow cytometric cellular DNA content analysis, showing repeatedly abnormal aneuploid DNA content (Figure 2B).
Figure 1.
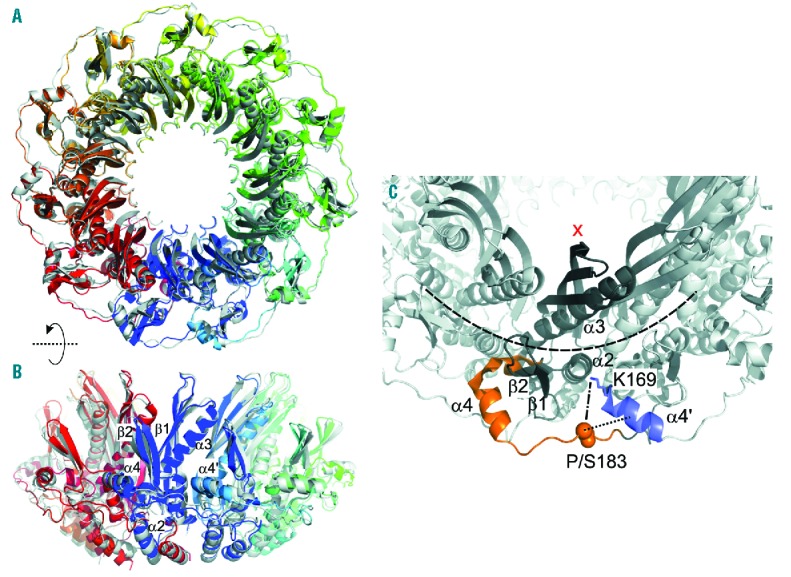
Overall structural similarity of wild-type and mutant RAD52. (A) Overlay of average structures of wild-type RAD52 (colored by a gradient from blue to red) and the S183 mutant (white). The average structures were obtained from MD simulations of 80 ns length; the proteins are depicted in cartoon representation. The average structures differ by a Cα atom root-mean-square deviation (rmsd) of 1.0 Å (B) As in panel (A), but rotated by 90° (C) Local influence of the P183S mutation on dynamics and structure. Cutaway of the RAD52 11-mer (white), with one protomer colored in dark grey. The orange region depicts helix α4, the preceding region of 310 helices, and part of the loop between α4 and α5 where the largest changes in root mean square fluctuation (rmsf) are observed between wild-type and mutant RAD52. The backbone atoms of residue 183 are depicted as spheres. Helix α4’ of the neighboring protomer is colored blue. The distance measured between the Cα atom of residue 183 and the center of α4’ is shown as a dotted black line. The side chain of K169 is shown in stick representation. The distance measured between atom Cγ of P183 or Oγ of S183 and Nz of K169, respectively, is indicated by a dotted-dashed line. The orientation of the side chain of K169 was determined with respect to the center of the 11-mer (red cross). The dashed arc indicates the position of ssDNA according to suggestions.6
Figure 2.
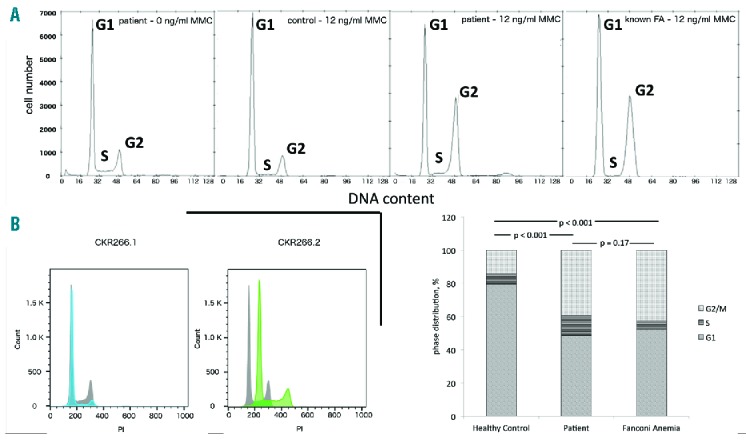
Disturbance of DNA repair in patient samples. (A) Mitomycin C (MMC) sensitivity assay - patient fibroblasts (without MMC, left chart) were exposed to MMC (12 ng/ml) and cultured for 48h. Staining with 4′,6-diamidino-2-phenylindole (DAPI) shows an increased number of G2 phase cells, indicating that cells are in a cycle arrest after induced damage by MMC, a known DNA intercalating agent (second right chart). A G2 increase is not seen in healthy controls (second left chart), but detected in a similar quantity in an internal control with fibroblasts from a MMC sensitive patient control (right chart). Cell cycle distribution among mentioned samples – a χ2 test shows a significant difference between healthy control and patient sample and MMC sensitive patient control, while there is no difference between patient sample and MMC sensitive patient control (bottom right chart) (B) Flow cytometric DNA content measurement of two different LCLs generated from patient’s bone marrow stained with PI following ethanol fixation. Both cell lines were cultured and processed under the same standard conditions from the same specimen. Left: Patient LCL#1 cells (blue) show a slightly increased DNA amount than normal control LCLs (grey). Right: Patient LCL#2 cells (green) show aneuploidy (polyploidy in another experiment), compared to healthy human samples. PI: propidium iodide; FA: fanconi anemia; MMC: mitomycin C.
SsDNA binding of purified mutant P183S and WT protein was assessed in an electrophoretic mobility shift assay (EMSA). The P183S-ssDNA complexes showed an increased mobility shift in repeated experiments, although densitometric analyses of EMSA bands revealed a lower amount of complex formation in the P183S-ssDNA bands despite an increasing protein concentration (Figure 3A,B). Similar changes, such as the Y171A variant, have been seen in other publications.7
Figure 3.
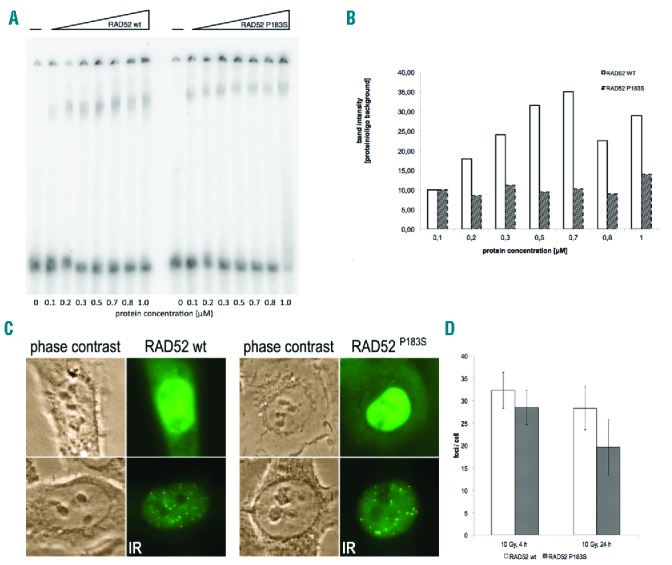
Functional impairment of RAD52P183S. (A) Electrophoretic mobility shift assay (EMSA) of wild-type (WT) and mutant synthetic RAD52 protein (0,1–1 mM) complexes which were formed with a 50bp ssDNA. The purified mutant protein forms faster migrating complexes compared to WT, despite (B) densitometric analyses of EMSA bands for WT and mutant RAD52/ssDNA complexes revealing a lower amount of complex formation irrespective of applied protein concentration in the mutant. Data are representative of three independent experiments. (C) Representative examples of HT-1080 cells stably expressing GFP-RAD52 or GFP-RAD52P183S after irradiation with 10 Gy and a further 24 hours of culture. Phase contrast and fluorescence microscopy. (D) Foci counting per cell in both cell lines after 10 Gy and 4h or 24h of subsequent culture. Irradiation with 1 Gy did not show any difference (data not shown).
We transfected HT-1080 cells with WT EGFP-RAD52 and EGFP-RAD52P183S via bicistronic expression vectors, previously used for stable expression in human cells. We assayed the ability of the P183S variant and WT to produce DNA breaks detectable as RAD52 foci by immunofluorescence in transfected HT-1080 cells. The cells were irradiated with 1 Gy or 10 Gy, and cultured for 4h or 24 hours, respectively. While all conditions showed a numerically decreased foci formation in the P183S cell line, the higher (10 Gy) irradiation and longer (24 hours) culture period showed a further, but nonsignificant, decrease in the mutant (Figure 3C,D). Previously, RAD51 foci after MMC induction were counted in the patient’s primary fibroblasts, and showed no abnormalities (data not shown), thus we did not repeat that assay in EGFP-RAD52P183S transfected cells.
In conclusion, we describe a patient with T-cell depletion associated comorbidities, such as EBV lymphoproliferative disease and severe varicella infection. The patient’s past medical history suggests a combined immunodeficiency. The patient presented to us with hypersplenism, hence their white blood cell (WBC) count was low and the patient’s immunological phenotype could not be elucidated completely. The most striking abnormality was the detection of FA-like chemosensitivity in the patient’s fibroblasts. The overlap of genes involved in breast cancer and FA8 and increased mortality due to adenocarcinoma in the maternal lineage led us to the assumption of an inherited disease, or at least of a variant of the latter conferring a higher risk of disease.
The discovery of FA genes has revealed the molecular pathway (FA/BRCA) of DNA maintenance. The core element is E3 ligase-mediated monoubiquitination of the protein complex D2-I, which is carried out by an upstream complex consisting of classical FA proteins. Downstream of this step other proteins associate with the D2-I complex and apply HR to the induced DSB. The proteins involved in this part of the FA/BRCA pathway have a major role in HR and vice versa.9 RAD52, which was not associated with this pathway in humans, exists in an undecameric ring structure and consists of an N-terminal ssDNA annealing, central replication protein A (RPA) interacting and C-terminal RAD51 mediating domain.10 Initial findings suggested an indispensable role of RAD52 in yeast, but increasing evidence attributes RAD52 to HR in mammalian cells. During DSB repair, DNA strand exchange takes place in which a template strand invades a homologous DNA strand. RAD51 recombinase binds to RPA-coated ssDNA to form a nucleoprotein filament able to invade duplex DNA. The assembly of this filament is mediated by RAD52. After capture of the second end of the DSB and strand extension, RAD52 anneals RPA-coated ssDNA.7,10–12 The heterozygous P183S mutation lies in the ssDNA annealing domain, and our assays suggest that ssDNA annealing of mutant P183S is impaired. A second DNA binding site was identified in RAD52 for double-stranded (ds)DNA, and showed that impaired ability to form RAD52-ssDNA leads to a less stable complex with dsDNA, which additionally leads to decreased HR.7 We show evidence of some reduced formation of RAD52 foci in the P183S-transfected HT-1080 cell line after irradiation, suggesting a reduced capacity of HR. Furthermore the MMC assay indicates a disturbance of ICL repair, in which HR plays a major role. Synthetic lethality of RAD52 inactivation in BRCA1, BRCA2 and PALB2 deficient cells has been described, suggesting RAD52 as part of an alternative repair pathway parallel to RAD51-mediated HR.13,14 We and others propose the alternative RAD52 pathway as being required for DNA stability, and attribute a tumor suppressor function. Additive germline mutations in other DNA repair genes might confer an increased tumor risk. We are aware of the possible interaction of RAD52 with NHEJ genes (LIG4 or MRN complex genes), as also depicted in our gene association network (see Online Supplementary Material). However, due to full aplasia when we investigated the patient, we could not check for NHEJ related disturbances (e.g., the TCR repertoire). We observed a heterozygous mutation in the patient and his mother, with reduced messenger RNA (mRNA) expression and suggestive assays for reduced HR capacity. In line with emerging data in genome-wide association studies in other patients with malignancies, results from targeted synthetic lethality studies and our own findings in reduced RAD52 foci and ssDNA annealing, we strongly suggest that RAD52 plays a major role in DNA repair and cancer predisposition.
Supplementary Material
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol. 2010;45(1):23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair. 2014;16:84–96. [DOI] [PubMed] [Google Scholar]
- 3.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. [DOI] [PubMed] [Google Scholar]
- 4.Ameziane N, May P, Haitjema A, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hussain S, Wilson JB, Medhurst AL, et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004;13(12):1241–1248. [DOI] [PubMed] [Google Scholar]
- 6.Kagawa W, Kurumizaka H, Ishitani R, et al. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol Cell. 2002;10:359–371 [DOI] [PubMed] [Google Scholar]
- 7.Kagawa W, Kagawa A, Saito K, et al. Identification of a second DNA binding site in the human Rad52 protein. J Biol Chem. 2008;283(35):24264–24273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy-Lahad E. Fanconi anemia and breast cancer susceptibility meet again. Nat Genet. 2010;42(5):368–369. [DOI] [PubMed] [Google Scholar]
- 9.Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124(18):2812–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lok BH, Powell SN. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin Cancer Res. 2012;18(23):6400–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIlwraith MJ, West SC. DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol Cell. 2008;29(4):510–516. [DOI] [PubMed] [Google Scholar]
- 12.Gaines WA, Godin SK, Kabbinavar FF, et al. Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat Commun. 2015;6:7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Z, Scott SP, Bussen W, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108(2):686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32(30):3552–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.