

ABSTRACT
The proteasome machinery is a common target of gain-of-function p53 missense mutants. Upregulation of the proteasome fosters chemoresistance to proteasome inhibitors. In triple negative breast cancer cells this resistance mechanism, namely the Nrf2-regulated “bounce-back” response to proteasome inhibitors, can be overcome by targeting p53 mutant proteins with APR-246/PRIMA-1Met.
KEYWORDS: APR-246, cancer, Chip-seq, Keap1, multi-omics, mutant p53, Nrf2, proteasome, TNBC, transcriptomics, proteomics
Point mutations affecting TP53 occur with different frequency in various tumor types, from 96% in high-grade ovarian serious carcinoma to 10% in hematopoietic malignancies, and contribute to carcinogenesis by causing the loss of wild-type p53 tumor suppressor activities, exerting dominant negative effects over the wild-type allele, and providing mutant p53 with novel oncogenic gain of function (GOF) properties that promote cell proliferation, survival, and invasion.1 Indeed, among many functions, mutant p53 proteins are involved in inactivation of p63/p73 tumor suppressors and upregulation of cyclins, nucleotide metabolism, steroid synthesis, integrin recycling, and the Warburg effect.2
We performed comparative analyses of the DNA-interactomic (ChIP-sequencing), transcriptomic, and proteomic data obtained from a triple negative breast cancer (TNBC) cell line, and of the transcriptomes orchestrated by 5 different GOF mutants in the context of their respective TNBC cell line. Our data revealed transcription of 20S/26S proteasome/immunoproteasome genes as a process commonly upregulated by missense GOF mutant p53 variants. Accordingly, in several cancer models and in TNBC patients with missense mutant p53 we observed elevated proteasome activity and transcription of proteasome genes.3 Interestingly, in TNBC cells upregulation of the proteasome activity leads to degradation of the KSRP microRNA maturation factor and downregulation of its tumor suppressive activities.3 Together with the recently described mutant p53-mediated inhibition of Drosha complex,4 our evidence supports and extends knowledge on the role of mutant p53 in global destabilization of miRNA homeostasis.
As GOF missense p53 mutant does not recognize DNA through a specific sequence its transactivation activity is presumed to occur mostly through interactions with DNA-bound transcription factors such as SREBP1/2, ETS2, or NF-YA.1 We found that activation of the proteasome by GOF mutant p53 variants in TNBC is mediated by Nrf2 (NFE2L2).3
The Nrf2 transcription factor is the master regulator of the oxidative stress response and its driving role in tumorigenesis has rapidly gained recognition in recent years.5 Unlike another family member Nrf1 (NFE2L1), which is also involved in regulation of antioxidant systems of the cell, Nrf2 has previously been reported to cooperate with various oncogenes (KRAS, C-MYC, BRAF), resulting in cytoprotective activities and a reduction in reactive oxygen species (ROS) levels in cancer cells.6
In order to be activated, Nrf2 must detach from its inhibitor Keap1 that binds it directly and sequesters it in the cytoplasm, where Nrf2 is ubiquitinated and directed to proteasomaldegradation. When the Nrf2/Keap1 binding is disturbed (e.g., by oxidative stress), Nrf2 shuttles to the nucleus, binds the promoters of its target genes, and activates transcription.5 Mounting evidence gathered in the last years has shown that Nrf2 transcriptional targets are not only genes involved in the oxidative stress response such as HMOX1/HO-1 (heme oxygenase-1), NQO1 (NAD(P)H quinone dehydrogenase 1), GCLM (glutamate-cysteine ligase modifier subunit), or TXN1 (thioredoxin).5 Rather, large-scale experiments have begun to unravel a broader impact of this potent transcription factor on the homeostasis of both normal and transformed cells, through processes as varied as drug detoxification, proliferation, inflammation, cell differentiation, and tissue regeneration.7 However, since the activity of Nrf2 is strongly dependent on the cell context (tumor versus normal) and the intracellular level of ROS and subsequent oxidative damage, a comprehensive picture of the role of Nrf2 in cancer progression is still far from being obtained.5
Our findings of an interplay between mutant p53 and this transcription factor add new pieces to the puzzle regarding the ability of various oncogenes to hijack the Nrf2-antioxidant pathway.5,8 Although different GOF p53 mutants were shown to act as co-activators of Nrf2 in proteasome gene transcription, they also exhibit co-repressor activity toward Nrf2 in the HMOX1 antioxidant system,3 as also reported by others.9 This raises an intriguing hypothesis involving a role of mutant p53 in regulating specific subsets of the Nrf2 oncogenic program. GOF p53 mutants could act as a molecular switch, turning on or off particular components of the Nrf2 transcriptional program.
A better understanding of the interplay between mutant p53 and Nrf2 will shed light on the long-debated dual role of Nrf2 as both an oncosuppressor and an oncoprotein5 and may offer an interesting opportunity for therapeutically leveraging the dependency of cancer cells on the mutant p53–Nrf2 axis.
Our study supports the hypothesis that concomitant inhibition of mutant p53 with APR-246/PRIMA1-Met and inhibition of the proteasome with carfilzomib might be an effective strategy to overcome the “bounce-back” chemoresistance response to proteasome inhibitors induced by mutant p53–Nrf2 (Fig. 1).3 Of note, APR-246 has recently been reported to contribute to cancer cell killing by inducing oxidative stress,10 further sustaining the hypothesis that targeting the Nrf2-regulated oxidative stress response pathway might be a promising anticancer strategy.8
Figure 1.
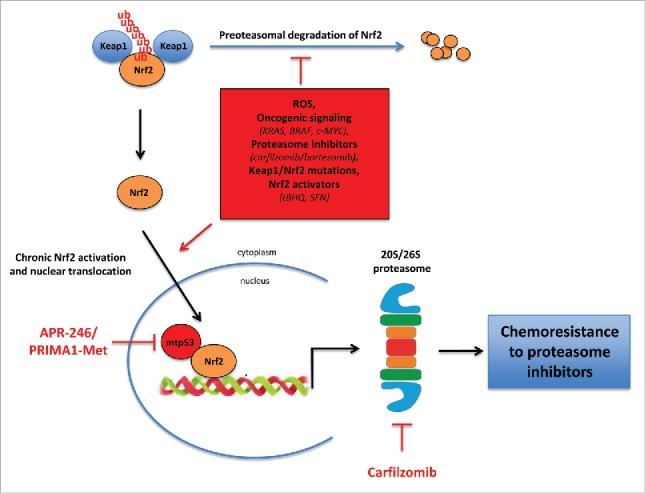
A scheme of the mutant p53–Nrf2–proteasome pathway with chemotherapeutic intervention targeted at mutant p53 and the proteasome.
Although more studies are needed to determine the full potential of targeting the mutant p53–Nrf2 axis in patients, our results could pave the way to defining better strategies to combat chemoresistant tumors bearing mutant p53.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Italian Association for Cancer Research (AIRC) Special Program Molecular Clinical Oncology “5 per mille” (grant no. 10016) and the Italian Ministry of Health (RF-2011-02346976) to G.D.S.
References
- 1.Walerych D, Lisek K, Sal DG. Mutant p53: One, no one, and one hundred thousand. Front Oncol 2015; 5:289; PMID:26734571; http://dx.doi.org/23321033 10.3389/fonc.2015.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D'Incalci M, Piccolo S, Veronesi A, Zambelli A, Del Sal G. Targeting triple negative breast cancer: Is p53 the answer? Cancer Treat Rev 2013; 39:541-50; PMID:23321033; http://dx.doi.org/ 10.1016/j.ctrv.2012.12.001 [DOI] [PubMed] [Google Scholar]
- 3.Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, Rajkowska K, Gaweda-Walerych K, Ingallina E, Tonelli C, et al.. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. 2016; 18:897-909; PMID:27347849; http://dx.doi.org/ 10.1038/ncb3380 [DOI] [PubMed] [Google Scholar]
- 4.Garibaldi F, Falcone E, Trisciuoglio D, Colombo T, Lisek K, Walerych D, Del Sal G, Paci P, Bossi G, Piaggio G, et al.. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene 2016; 35:3760-70; PMID:26996669; http://dx.doi.org/ 10.1038/onc.2016.51 [DOI] [PubMed] [Google Scholar]
- 5.Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med 2016; xx:1-16; PMID:27263465; http://dx.doi.org/24647116 10.1016/j.molmed.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 6.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al.. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2012; 475:106-9; PMID:21734707; http://dx.doi.org/24647116 10.1038/nature10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014; 39:199-218; PMID:24647116; http://dx.doi.org/ 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 8.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12:931-47; PMID:24287781; http://dx.doi.org/ 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- 9.Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P, et al.. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J Cell Sci 2012; 125:5578-86; PMID:22899716; http://dx.doi.org/ 10.1242/jcs.106815 [DOI] [PubMed] [Google Scholar]
- 10.Bykov VJN, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol 2016; 6:21; PMID:26870698; http://dx.doi.org/ 10.3389/fonc.2016.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]