

Abstract
Development of potent and isoform selective nitric oxide synthase (NOS) inhibitors is challenging owing to the structural similarity in the heme active sites. One amino acid difference between NOS isoforms, Asp597 in rat nNOS versus Asn368 in bovine eNOS, has been identified as the structural basis for why some dipeptide amide inhibitors bind more tightly to nNOS than eNOS. We now have found that the same amino acid variation is responsible for substantially different binding modes and affinity for a new class of aminopyridine based inhibitors.
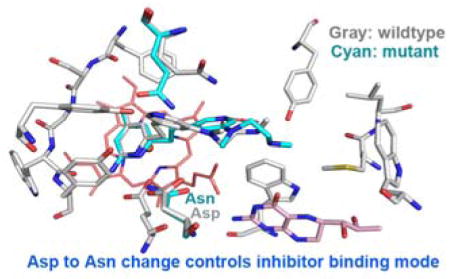
Graphical Abstract
Nitric oxide (NO) is involved in a wide range of physiological functions, including the cardiovascular, neuronal, and immune systems.1 Mammals have three nitric oxide synthase (NOS) isoforms that convert L-arginine to NO and L-citrulline.2 NO produced by neuronal NOS (nNOS) participates in neural transmission. NO produced by endothelial NOS (eNOS) regulates blood pressure, and NO produced by inducible NOS (iNOS) in macrophage cells is used as part of the immune defense system. NO biosynthesis must be tightly regulated because uncontrolled NO production is associated with various pathological conditions, such as neurodegenerative diseases,3 arthritis,4 septic shock,5 and atherosclerosis.6 As a result, NOSs are important drug targets.
One of the important issues in NOS inhibitor development is selectivity. Indiscriminate inhibition of all three NOS isoforms could be detrimental. Of particular importance is the avoidance of eNOS inhibition, given the important role this isoform plays in the cardiovascular system. Once the crystal structures of the heme containing domain for all three NOS isoforms were solved it became possible to use structure-based drug design approaches to target specific NOS isoforms.7 This was a challenging problem, given the very close similarity in active site structures. Nevertheless, we found that a single amino acid difference between all mammalian nNOS’ and eNOS’, Asp597 in rat nNOS vs. Asn368 in bovine eNOS, is responsible for the much better affinity of several dipeptide inhibitors for nNOS over eNOS.8 While dipeptide inhibitors (e.g. compound 1 in Figure 1) adopt a curled conformation in nNOS, allowing the α-amino group to optimally interact with Glu592 and Asp597, the same inhibitors adopt an extended conformation in eNOS because Asp597 is replaced by Asn368 in eNOS. Consequently, the more electronegative nNOS active site binds these positively charged inhibitors more tightly.
Figure 1.

Inhibitors discussed in this study.
We recently reported an nNOS selective aminopyridine-based inhibitor 2 (Figure 1) that binds to nNOS and eNOS in very different conformations.9 In nNOS, inhibitor 2 anchors to the active site via H-bonds from its aminopyridine nitrogen atoms to the Glu592 side chain (Figure 2A). However, its central pyridine ring adopts an unusual upward binding position, where it H-bonds with Tyr562. To do so, the side chain of Gln478 must adopt an alternate rotamer position. The ethylenediamine tail approaches the water site between the H4B and heme propionate A without making any H-bond. In contrast, in eNOS the central pyridine ring does not point upward but, rather, it extends out near heme propionates. Two additional inhibitors, 3 and 6 derived from 2, also have the central pyridine oriented quite differently in nNOS and eNOS (Figure 3).10 The central pyridine in 3 and 6 H-bonds with Tyr562 of nNOS, as with 2, and the central pyridine of 3 and 6 approaches heme propionates in eNOS as does 2.
Figure 2.

Two different binding modes observed for 2 in the structures of nNOS (A, PDB code 4UH1) and eNOS (B, 4UH8). Major H-bonds are depicted with dashed lines.
Figure 3.

Different binding mode of compound 3 bound to rat nNOS (A, PDB code 5FVQ) compared with bovine eNOS (C, 5FVY); compound 6 bound to nNOS (B, 5FVT) compared with eNOS (D, 5FVZ). Major hydrogen bonds are depicted with dashed lines.
It is likely that the reason these inhibitors bind more tightly to nNOS than to eNOS is due to the different orientation of the central pyridine. As noted earlier, Asp597 in nNOS (Asn368 in eNOS) has been shown to play a major role in isoform selectivity. We thus reasoned that Asp597 provides additional electrostatic stabilization to the central pyridine when oriented up, where it can be stabilized more effectively by Asp597 in nNOS relative to Asn468 in eNOS. Another possible contributor to selectivity is Met336 in nNOS (Val106 in eNOS), which interacts with the tail end of these inhibitors. As in earlier studies we explored the effect of the nNOS D597N or D597N/M336V mutants on the binding of inhibitors 2, 3, and 6 using in vitro inhibitory assays, crystal structures, and theoretical calculations.
EXPERIMENTAL PROCEDURES
Protein Purification and Crystal Preparation
D597N and D597N/M336V rat nNOS mutants were expressed and purified following the same protocols used for the wild type protein.11 The heme domain protein for crystallization was generated by limited trypsin digestion with the partially purified full-length nNOS mutant protein and further cleaned by a Superdex 200 gel filtration column. The crystal growth conditions are identical to the sitting drop vapor diffusion methods used for wild-type nNOS.11 Fresh crystals were passed through a series of cryo-protectant soaking solutions and then soaked with 10 mM of inhibitors for 4–6 h before being flash cooled with liquid nitrogen and stored until X-ray data collection.
X-ray Diffraction Data Collection and Structure Determination
The cryogenic (100 K) X-ray diffraction data were collected remotely at the Stanford Synchrotron Radiation Lightsource (SSRL) or Advanced Light Source (ALS) through data collection control software Blu-Ice12 and a crystal mounting robot. A Q315r CCD detector was used, 100° of data were collected with 0.5° per frame. Raw CCD data frames were indexed and integrated using MOSFLM13 and scaled with Aimless.14
The binding of inhibitors was detected by initial difference Fourier maps calculated with REFMAC.15 The inhibitor molecules were then modeled in COOT and refined using REFMAC or PHENIX.16 Water molecules were added in PHENIX and checked by COOT.17 The TLS protocol18 was implemented in the refinements with each subunit as one TLS group. The omit Fo – Fc density maps were calculated by running one round of PHENIX_REFINE including a simulated annealling protocol (2000 K initial temperature) with inhibitor coordinates removed from the input PDB file to generate the map coefficients DELFWT and PHDELWT. The refined structures were validated in COOT before deposition in the protein data bank. The crystallographic data collection and structure refinement statistics are summarized in Table 1 with the PDB accession codes included.
Table 1.
Crystallographic data collection and refinement statistics.
| Data set 1 | nNOS D597N-2 | nNOS D597N-3 | nNOS D597N/M336V-6 |
|---|---|---|---|
| Data collection | |||
|
| |||
| PDB code | 5G0N | 5G0O | 5G0P |
|
| |||
| Space group | P212121 | P212121 | P212121 |
|
| |||
| Cell dimensions a, b, c (Å) | 51.8 110.2 164.2 | 51.7 110.7 164.2 | 51.6 111.2 164.3 |
|
| |||
| Resolution (Å) | 1.94 (2.00–1.94) | 1.85 (1.90–1.85) | 2.10 (2.19–2.10) |
|
| |||
| Rmerge2 | 0.107 (1.230) | 0.119 (2.463) | 0.155 (1.998) |
| Rpim3 | 0.053 (0.664) | 0.058 (1.208) | 0.105 (1.353) |
| CC 1/24 | 0.997 (0.610) | 0.998 (0.659) | 0.995 (0.451) |
|
| |||
| I/σI | 9.1 (1.1) | 7.1 (0.7) | 6.8 (0.8) |
|
| |||
| No. unique reflections | 70,703 | 81,212 | 55,639 |
|
| |||
| Completeness (%) | 99.6 (93.4) | 99.9 (100.0) | 99.3 (99.1) |
|
| |||
| Redundancy | 5.0 (4.2) | 5.1 (5.1) | 5.4 (5.5) |
|
| |||
| Refinement | |||
|
| |||
| Resolution (Å) | 1.94 | 1.85 | 2.10 |
|
| |||
| No. reflections used | 70,518 | 80,907 | 55,529 |
|
| |||
| Rwork/Rfree 5 | 0.175/0.216 | 0.193/0.233 | 0.200/0.245 |
|
| |||
| No. atoms | |||
|
| |||
| Protein | 6683 | 6687 | 6684 |
|
| |||
| Ligand/ion | 173 | 175 | 173 |
|
| |||
| Water | 498 | 460 | 332 |
|
| |||
| R.m.s. deviations | |||
|
| |||
| Bond lengths (Å) | 0.007 | 0.008 | 0.009 |
|
| |||
| Bond angles (deg) | 1.14 | 1.17 | 1.17 |
See Figure 1 for the inhibitor chemical formula.
I is the observed intensity and <I> is the average intensity over multiple symmetry related observations.
Precision indicating R factor.
CC ½ values are calculated by splitting the data randomly in half. The RMS Correlation Ratio (RCR) is calculated from a scatter plot of pairs of DeltaI from the two subsets (halves) by comparing the RMS value (excluding extremes) projected on the line with slope = 1 (“correlation”) with the RMS value perpendicular to this (“error”).
Rfree was calculated with the 5% of reflections set aside throughout the refinement.
Inhibitor Ki Determinations
The hemoglobin capture assay19 was used to measure nitric oxide production. The assay was performed at 37 °C in HEPES buffer (100 mM, with 10% glycerol, pH 7.4) in the presence of 10 μM L-arginine. Also included were 100 μM NADPH, 0.83 mM CaCl2, approximately 320 units/mL of calmodulin (≈40,000 units/mg), 10 μM tetrahydrobiopterin, and human oxyhemoglobin (3 μM). This assay was performed in 96-well plates using a Biotek Gen5TM microplate reader. NO production was read by monitoring the absorbance at 401 nm (resulting from the conversion of oxyhemoglobin to methemoglobin). Kinetic readouts were recorded for 6 min. Each compound was assayed at least in duplicate, and seven to nine concentrations (50 nM – 200 μM) were used to construct dose-response curves. IC50 values were calculated by non-linear regression using GraphPad Prism software, and Ki values were obtained using the Cheng-Prusoff equation [Ki = IC50/(1+[S]/Km)] with the following Km values: 1.3 μM (rat nNOS), 1.7 μM (rat nNOS D597N mutant), and 1.9 μM (rat nNOS D597N/M336V mutant).
Computational Approaches
The relative free energy of binding of various NOS inhibitors was estimated with MM-PBSA20 as implemented in Amber using procedures developed in our earlier studies with NOS inhibitors.21 A single energy minimized structure was used for the calculations and as others have done22 the change in solute entropy was ignored. Given that we are comparing exactly the same inhibitor bound to different active sites, ignoring solute entropy introduces little error, although only relative and not absolute free energies can be compared. Inhibitor parameters were assigned using the GAFF force field23 and AM1-BCC charge scheme,24, 25 as implemented in the Antechamber module in Amber. Heme parameters developed for cytochrome P450 were provided by Dr. Dan Harris.26
RESULTS AND DISCUSSION
As shown in Figure 4, when inhibitor 2 binds to the nNOS D597N mutant, its central pyridine ring is no longer oriented upward to make a H-bond with Tyr562. Instead, the central pyridine sits right above the two heme propionates with its ring nitrogen forming a H-bond with the heme propionate D. This pyridine ring position resembles what was found in the eNOS-2 structure. The only difference is that in eNOS the ring flips almost 180° so that the ring nitrogen does not make any H-bond with the heme. As a result, the diamine tail of 2 has different locations. In nNOS D597N, the tail amine H-bonds with the water bridging H4B and heme propionate A (Figure 4). But in eNOS, the tail juts out farther and thus does not H-bond with any protein residue (Figure 2B).
Figure 4.

Stereo-view of structure of compound 2 bound to nNOS D597N mutant. The omit Fo – Fc density is shown at the 2.5 σ contour level. Major H-bonds are depicted with dashed lines.
In the structure of nNOS D597N mutant with compound 3 bound (Figure 5A), the electron density for the central pyridine is weak. However, there are two reasons for concluding that the central pyridine adopts a conformation similar to that found in the eNOS-3 complex (Figure 3C). First, at low contour level (≈ 0.6 σ) the pyridine ring is visible. Second, Gln478 does not adopt the alternate rotamer found in the WT nNOS-3 complex, a movement required to accommodate the pyridine ring in the up position. The two piperazine N atoms here also interact with heme propionate D and Asn569 (equivalent to Asn340 in eNOS). Whereas in the structure of nNOS D597N/M336V in complex with compound 6, the central pyridine is positioned between the two propionates (Figure 5B). This is different from the eNOS-6 structure where the central pyridine and aminopyridine are almost parallel (Figure 3D). The long amine tail is also disordered and does not H-bond with heme propionate D as observed in the eNOS-6 structure (Figure 3D).
Figure 5.

Structures of compound 3 bound to nNOS D597N mutant (A) and compound 6 bound to nNOS D597N/M336V double mutant (B). The omit Fo – Fc map is displayed at the 2.5 σ contour level. Major hydrogen bonds are depicted with dashed lines.
The general pattern is clear that when Asp597 is mutated to Asn, the central pyridine ring of 2, 3, or 6 no longer orients upward to H-bond with Tyr562. Apparently, it is the electrostatic interactions between the negatively charged Asp597 and the central pyridine of the inhibitor that determine whether the pyridine can achieve its upward position, even though the distance between the two is 4.6 Å. As in previous studies, it also is possible that Met336 (Val106 in eNOS) plays a role in selectivity. If the charge of Asp597 and the size of Met336 are important to inhibitor binding, the Ki should increase with the mutants. The Ki values for inhibitors 2, 3, 4, 5, and 6 against single and double mutant rat nNOS are shown in Table 2, and the selectivities of n/e are included for comparison. All of the compounds exhibited a drop in potency with both the single and double mutants, ranging from 18-fold for 3 to 3-fold for 5 relative to wild-type rat nNOS. This, again, supports the importance of those residues for potency and selectivity. However, the mutant/WT ratios are smaller than the n/e selectivities, which suggests that the Asp/Asn difference alone is not fully responsible for n/e selectivity. One additional important factor contributing to selectivity is how the tail end of the inhibitors interacts with the heme. Comparing the structures of 2 complexed to eNOS and the D597N nNOS mutant, the central pyridine is positioned parallel to the heme rather than pointing up as in WT nNOS. Note, however, (Figure 4) that in the nNOS D597N mutant, the central pyridine is about 3.1 Å from the heme propionate D but in eNOS the central pyridine is oppositely oriented and about 3.7 Å from heme propionate A with very poor H-bond geometry. As a result, the central pyridine establishes better interactions in the D597N nNOS mutant than in eNOS. Precisely why this difference occurs is not easily understood, although in previous studies we found that nNOS is more flexible than eNOS and can possibly better adapt to the tail end of many of these inhibitors. 27
Table 2.
Inhibition of Mutant rat nNOSs by Compounds 3, 4, 5, and 6.
| Comp. | nNOS Ki (nM)a | Selectivity | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| D597N | D597N/M336V | WT | D597N/WTb | (D597N/M336V)/WTb | n/ec | |
| 2 | 139.4 | 116.5 | 17.0 | 8 | 7 | 759 |
|
| ||||||
| 3 | 748 | 736 | 42.6 | 18 | 17 | >700 |
| 4 | 641 | 458 | 72.3 | 9 | 6 | 283 |
| 5 | 46.7 | 65.3 | 16.4 | 3 | 4 | 809 |
| 6 | 97.5 | 108 | 25.2 | 4 | 4 | 659 |
Ki values are calculated directly from IC50 values. IC50 values are the average of at least two replicates. All experimental standard error values are less than 15%.
Values are calculating by the ratios of respective Ki values of nNOS single mutant or double mutant nNOS to those with WT rat nNOS.
n/e selectivity values are calculated by the inverse of the ratios of respective Ki values measured with nNOS and eNOS.
To verify that the electrostatic interaction is the driving force behind the inhibitor’s binding preference, we also carried out a series of mm_pbsa calculations on 2 and 6. We focused on these two inhibitors since the electron density is well resolved for the entire inhibitor. Since NOS is a dimer, the relative free energy for each active site was calculated, and the average of the two is reported in Table 3. The exception is inhibitor 2, where only the monomer B for nNOS was included since the tail end of the inhibitor in molecule A is partially disordered. The calculated value reported in Table 3 is PBtotal, which is the total energy excluding solute entropy. PBtotal was calculated assuming that the central pyridine is protonated (total +3 charge on the inhibitor) or not (total +2 charge on the inhibitor). The 50% protonated state was taken as the average of pyridine-protonated and -unprontated values. The “in silico” values for the nNOS D597N mutant used the nNOS-inhibitor wild-type structure where Asp597 was changed to Asn597. This provides information on the importance of the Asp597 negative charge. To compare PBtotal to experimental ΔG values derived from Ki measurements, PBtotal was normalized to the wild-type nNOS experimental ΔG. For example, ΔG = −10.6 kcal/mol for 2 while PBtotal for the +3 model is −55.82 kcal/mol so the normalization factor is 10.6/55.82. The in silico D597N mutant exhibits a large drop in PBtotal, which, as expected, shows that Asp597 is quite important for inhibitor binding and is most important using the fully protonated pyridine model. The predicted ΔG for the eNOS-2 complex agrees best with the experimental value, −6.6 kcal/mol, using the 50% protonated model, which gives −6.4 kcal/mol. Similar results are obtained with inhibitor 6. As expected, removing the negative charge on Asp597 has a dramatic effect; but, somewhat unexpectedly, removing the proton from the central pyridine does not have much of an effect on PBtotal in nNOS. This suggests that Asp597 provides longer range electrostatic stabilization besides stabilizing the central pyridine. The tail end of the inhibitor and aminopyridine each carry a +1 charge, and although Asp597 is far from these groups, 7.4 Å and 8.4 Å, respectively, the lack of solvent dielectric screening in the active site very likely enhances longer range electrostatic interactions.
Table 3.
MM_PBSA relative free energy values computed for inhibitors 2 and 6. All values are in kcal/mol.
| Protein | PBtotal +3 | PBtotal +2 | PBtotal av | ΔGexp | ΔGnorm+3 | ΔGnorm+2 | ΔGnorm av |
|---|---|---|---|---|---|---|---|
| nNOS WT-2 | −55.82 | −53.74 | −54.79 | −10.6 | −10.6 | −10.6 | −10.6 |
| nNOS D597N-2 in silico | −33.29 | −48.06 | |||||
| nNOS D597N-2 crystal | −42.86 | −42.83 | −42.85 | −9.3 | −8.1 | −8.4 | −8.4 |
| eNOS WT-2 | −28.80 | −37.41 | −33.10 | −6.6 | −5.5 | −7.4 | −6.4 |
| nNOS WT-6 | −64.04 | −60.09 | −62.06 | −10.3 | −10.3 | −10.3 | −10.3 |
| nNOS D597N-6 in silico | −41.38 | −52.67 | −47.05 | ||||
| eNOS WT-6 | −31.87 | −39.83 | −35.85 | −5.5 | −5.14 | −6.84 | −5.96 |
In summary, a single amino acid difference, Asp597 of nNOS versus Asn368 of eNOS, controls the affinity and binding mode of a new class of nNOS selective inhibitors. In nNOS the central pyridine points up toward Tyr562, and calculations indicate that the pyridine is at least partially protonated for optimal electrostatic interactions with Tyr562, even though the pyridine pKa ≈ 6. The calculations also show that Asp597 must provide additional electrostatic stabilization to the rest of the inhibitor, because neutralizing the pyridine ring results in only a small drop in the relative free energy. This underscores the importance of long-range electrostatic effects that can be achieved in enzyme active sites where solvent dielectric screening is limited and is an important consideration in drug design.
Acknowledgments
This work was supported by NIH grants GM57353 (TLP) and GM049725 (RBS)
We wish to thank the SSRL and ALS beamline staff for their support during remote X-ray diffraction data collection.
Abbreviations
- NO
nitric oxide
- nNOS
neuronal nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- H4B
tetrahydrobiopterin
- MM-PBSA
molecular mechanics Poisson Bolzmann surface area
- GAFF
general Amber force field.
Footnotes
Coordinates and structure factors have been deposited in the Protein Data Base under accession numbers 5G0N, 5G0O, and 5G0P.
References
- 1.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Stuehr DJ, Griffith OW. Mammalian nitric oxide synthases. Adv Enzymol Relat Areas Mol Biol. 1992;65:287–346. doi: 10.1002/9780470123119.ch8. [DOI] [PubMed] [Google Scholar]
- 3.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 4.Bingham CO., 3rd The pathogenesis of rheumatoid arthritis: pivotal cytokines involved in bone degradation and inflammation. J Rheunatol Suppl. 2002;65:3–9. [PubMed] [Google Scholar]
- 5.Vallance P, Moncada S. Role of endogenous nitric oxide in septic shock. New Horiz. 1993;1:77–86. [PubMed] [Google Scholar]
- 6.Anderson TJ. Nitric oxide, atherosclerosis and the clinical relevance of endothelial dysfunction. Heart Fail Rev. 2003;8:71–86. doi: 10.1023/a:1022199021949. [DOI] [PubMed] [Google Scholar]
- 7.Poulos TL, Li H. Structural basis for isoform-selective inhibition in nitric oxide synthase. Acc Chem Res. 2013;46:390–398. doi: 10.1021/ar300175n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 9.Kang S, Li H, Tang W, Martasek P, Roman LJ, Poulos TL, Silverman RB. 2-Aminopyridines with a Truncated Side Chain To Improve Human Neuronal Nitric Oxide Synthase Inhibitory Potency and Selectivity. J Med Chem. 2015;58:5548–5560. doi: 10.1021/acs.jmedchem.5b00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H-YQY, Li H, Roman LJ, Martasek P, Poulos TL, Silverman RB. Potent and Selective Human Neuronal Nitric Oxide Synthase Inhibition by Optimization of the 2-Aminopyridine-based Scaffold with a Pyridine Linker. J Med Chem. 2016 doi: 10.1021/acs.jmedchem.6b00273. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-alkyl-N′-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 12.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 13.Leslie AGW, Powell HR. Processing Diffraction Data with Mosflm. Evolving Meth Macromol Crystallogr. 2007;245:41–51. [Google Scholar]
- 14.Evans PR. Scaling and assessment of data quality. Acta Cryst. 2006;D62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 15.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Cryst. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 17.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 19.Murphy ME, Noack E. Nitric oxide assay using hemoglobin method. Methods Enzymol. 1994;233:240–250. doi: 10.1016/s0076-6879(94)33027-1. [DOI] [PubMed] [Google Scholar]
- 20.Massova I, Kollman PA. Computational alanine scanning to probe protein-protein interactions: A novel approach to evaluate binding free energies. J Amer Chem Soc. 1999:8133–8143. [Google Scholar]
- 21.Igarashi J, Li H, Jamal J, Ji H, Fang J, Lawton GR, Silverman RB, Poulos TL. Crystal structures of constitutive nitric oxide synthases in complex with de novo designed inhibitors. J Med Chem. 2009;52:2060–2066. doi: 10.1021/jm900007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown SP, Muchmore SW. High-througput calculation of protein-ligand binding affinities: Modification and adaption of the MM-PBSA protocol to enterprise grdi computing. J Chem Inf Model. 2006;46:999–1005. doi: 10.1021/ci050488t. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case D. Development and testing of a general Amber force field. J Amer Chem Soc. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 24.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atom charges. AM1-BCC model: I. Method. J Comp Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 25.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atom charges. AM1-BCC model: II. Parameterization and validation. J Comp Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 26.Harris DL, Park JY, Gruenke L, Waskell L. Theoretical study of the ligand-CYP2B4 complexes: effect of structure on binding free energies and heme spin state. Proteins. 2004;15:895–914. doi: 10.1002/prot.20062. [DOI] [PubMed] [Google Scholar]
- 27.Delker SL, Xue F, Li H, Jamal J, Silverman RB, Poulos TL. Role of zinc in isoform-selective inhibitor binding to neuronal nitric oxide synthase. Biochemistry. 2010;49:10803–10810. doi: 10.1021/bi1013479. [DOI] [PMC free article] [PubMed] [Google Scholar]