

ABSTRACT
The nuclear lamina is a critical structural domain for the maintenance of genomic stability and whole-cell mechanics. Mutations in the LMNA gene, which encodes nuclear A-type lamins lead to the disruption of these key cellular functions, resulting in a number of devastating diseases known as laminopathies. Cardiomyopathy is a common laminopathy and is highly penetrant with poor prognosis. To date, cell mechanical instability and dysregulation of gene expression have been proposed as the main mechanisms driving cardiac dysfunction, and indeed discoveries in these areas have provided some promising leads in terms of therapeutics. However, important questions remain unanswered regarding the role of lamin A dysfunction in the heart, including a potential role for the toxicity of lamin A precursors in LMNA cardiomyopathy, which has yet to be rigorously investigated.
KEYWORDS: cardiomyocyte, cardiomyopathy, LINC complex, LMNA, mechanotransduction, nuclear lamina, prelamin A
Introduction
LMNA encodes the intermediate filament proteins lamins A and C which are generated by alternative splicing.1 While lamin C is translated as a mature protein, lamin A is translated as a precursor, prelamin A, and requires extensive C-terminal processing to reach maturation. The B-type lamins are generated from 2 genes B1 from the LMNB2 gene, and B2 and B3 from LMNB3.2 A-type lamins polymerise into high order lattice structures with the B-type lamins to form the nuclear lamina (NL). The NL lies directly adjacent to the inner nuclear membrane (INM) on the nucleoplasmic side and forms a physical complex with SUN domain proteins and nesprins, which together comprise the nuclear envelope (NE) spanning LInkers of the Nucleoskeleton to Cytoskeleton (LINC) complex.3 On the cytoplasmic face of the outer nuclear membrane (ONM) nesprins link to cytoskeletal components, predominantly F-actin, and provide a structural link between the nucleus and cytoplasm.4 This structural link can be viewed as reaching as far as the extracellular matrix (ECM) if the sequential links between F-actin and focal adhesion proteins are considered. This tethering of the lamina with NE spanning proteins via the LINC complex is crucial for the integrity of whole-cell mechanics, as well as mechano-transduction to the nucleus from the cytoplasmic and extracellular domains.5
Inside the nucleus, lamins associate with LEM proteins (LAP2, Emerin, MAN) and heterochromatin.6 They serve a scaffolding function in order to facilitate the correct expression of genes as well as enable efficient DNA damage repair. When the integrity of the NL is compromised, these processes become dysregulated with potentially devastating results. Patients harbouring mutations in LMNA can develop a number of different tissue-specific syndromes collectively termed laminopathies, many of which can be broadly defined as ‘premature aging disorders’ and include Hutchinson Gilford progeria syndrome (HGPS). One of the most common diseases caused by LMNA mutations is cardiomyopathy, which can occur as an isolated phenotype or frequently in combination with a skeletal muscle dystrophy such as Emery Dreifuss muscular dystrophy (EDMD) or limb girdle muscular dystrophy.7-10 It has also been described in parallel with a number of other laminopathies (Table 1).11-16
Table 1.
Laminopathies identified to have overlapping cardiomyopathy phenotypes.
| Laminopathy | Heart involvement | Ref |
|---|---|---|
| Dilated cardiomyopathy with conduction defects | Left ventricle dilatation, systolic dysfunction, atrioventricular conduction block, arrhythmia, congestive heart failure | 7 |
| Emery Dreifuss muscular dystrophy | Atrioventricular conduction block, arrhythmia, systolic dysfunction, congestive heart failure | 8,9 |
| Limb girdle muscular dystrophy | Atrioventricular block, progressive left ventricle dysfunction, arrhythmia | 10 |
| Variant progeroid syndrome with right ventricular cardiomyopathy | Right atrium and ventricle dilatation, tricusped valve dilatation | 11 |
| Atypical progeroid syndrome with cardiomyopathy | Right ventricle dilatation, arrhythmia, tricusped valve regurgitation | 11,12 |
| Familial partial lipodystrophy of dunningan type 2 | Left ventricle dilatation, systolic dysfunction, atrioventricular block, complete left bundle branch block | 13 |
| Lipodystrophy with hypertrophic cardiomyopathy | Left ventricle hypertrophy, aortic valve calcification, stenosis and regurgitation | 15 |
| Charcot Marie Tooth type 2 axonal neuropathy | Left ventricle dilatation, systolic dysfunction, | 15 |
| Severe metbolic syndrome* | Left ventricle dilatation, systolic dysfunction, ventricular extra systole | 16 |
Caused by a ZMPSTE24 mutation
Two key processes have been proposed to account for cardiac dysfunction. The mechanical hypothesis proposes that disruption to and uncoupling of structural proteins at the NL leads to increased pathologic susceptibility to mechanical stress. Consequently, tissues that endure high levels of mechanical stress, i.e. striated muscle and heart, are most susceptible to disease. The gene expression hypothesis implies that structural changes to the NL not only lead to impaired transduction of signals from the extracellular and cytoplasmic domains, but also disrupted chromatin organization which impacts directly on gene transcription. However, the molecular events immediately downstream of lamin dysfunction are not well understood, especially in the context of the whole organ. Moreover these 2 mechanisms are unlikely to be mutually exclusive (Fig. 1), so it is not clear which, if either, is the key trigger. Importantly, the mechanisms that promote premature aging associated with accumulation of lamin A variants, such as increased levels of DNA damage and senescence, as observed in other tissues, have not been studied in the context of LMNA induced cardiomyopathy. This review explores the role of A-type nuclear lamins in the context of the clinical features of cardiomyopathies caused by mutations in LMNA and discusses the body of knowledge regarding pathologic mechanisms, and future areas for development.
Figure 1.
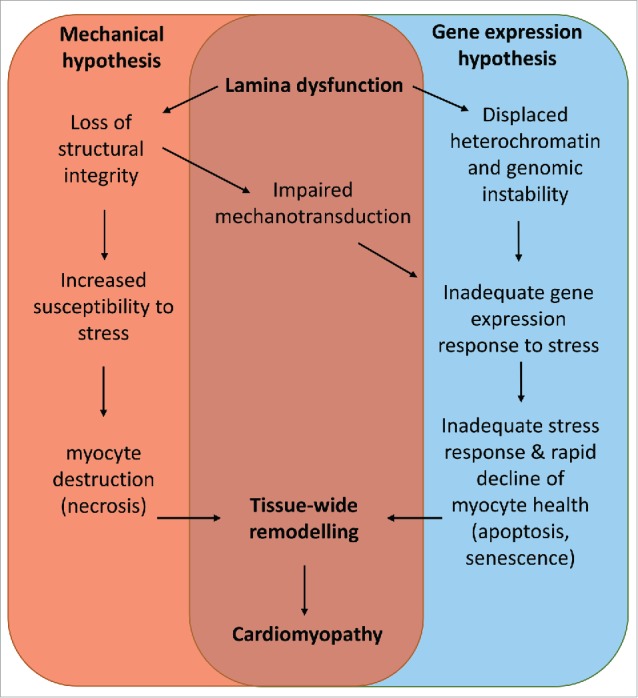
A model unifying the known mechanisms of LMNA cardiomyopathy.
Cardiomyopathy
Cardiomyopathies are characterized by cardiomyocyte (CM) dysfunction and tissue-wide remodelling of the myocardium leading to functional decline.17 They are mainly caused by familial mutations in structural proteins, but are also caused by somatic de novo mutations or external causes such as myocarditis,.18,19 toxin exposure,20 chemotherapy21 and autoimmunity.22 They also arise due to age related changes to the vasculature leading to pressure overload of the heart. Cardiomyopathies eventually progress to heart failure, the point at which the heart is no longer able to pump sufficient blood through the body to meet the metabolic demands of the respiring tissues (Fig. 2).
Figure 2.
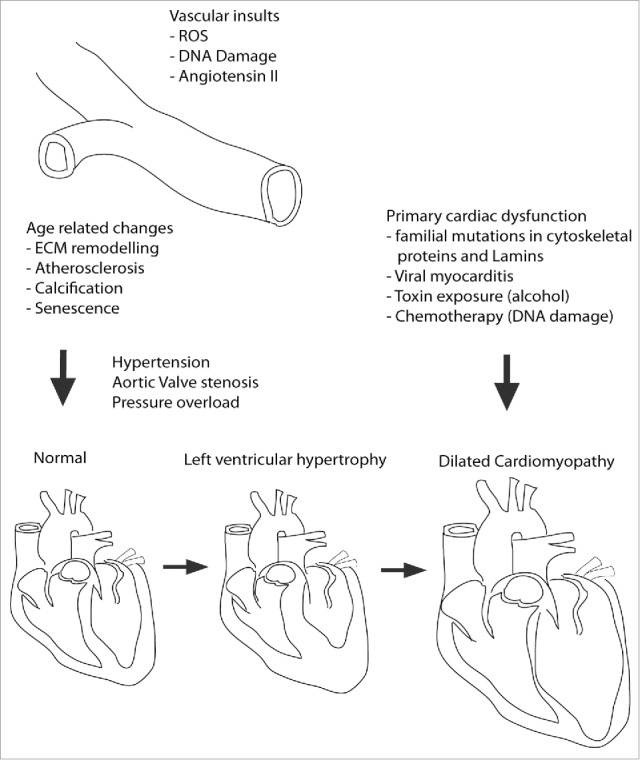
Etiology of Cardiomyopathy onset and progression. DCM is caused in primary and secondary fashion. In secondary it is due to excessive remodelling of the myocardium because of sustained pressure overload caused by vascular remodelling leading to increased overall blood pressure or aortic valve stenosis (hardening). This usually occurs via an intermediate step in which the heart tissue becomes thicker, known as hypertrophy. Primary DCM is caused predominantly by mutations in proteins of the sarcomere, cytoskeleton, or those involved in Ca2+ handling. Additionally, viruses and toxins such as alcohol or chemotherapy agents can initiate DCM independent of vascular remodelling. In this context hypertrophy is bypassed.
Cardiomyopathies are classified according to their functional and morphological features.23 The foremost classifications are dilated- (DCM), hypertrophic- (HCM) and arrhythmogenic right ventricular- (ARVC).24
Dilated cardiomyopathy
DCM is characterized by dilation of one, or both, ventricular chambers. Functionally, it is accompanied by reduced force of contraction leading to reduced cardiac output. Structurally, DCM is characterized by ventricular wall thinning, resulting from cardiomyocyte death and myocardial fibrosis. DCM can be caused by mutations in genes encoding proteins of the sarcomere, sarcolemma desmosome and NE. Upwards of 40 disease genes have been identified, most of which are autosomal dominant mutations.25
DCM mechanisms can be categorised into mechanical dysfunction, structural dysfunction and dysregulation of Ca2+ handling. In mechanical dysfunction mutations in genes encoding sarcomeric proteins such as β myosin heavy chain (β-MHC), reduce contractility whereas mutations in genes encoding actin thin filaments cause a reduction in Ca2+ sensitivity by attenuating the affinity of myofilaments to calcium, resulting in reduced generation of force in the myocyte. Structural deficiencies are caused by mutations in genes encoding cytoskeletal components, and include proteins of the sarcomere and the costamere, which links the sarcomere to the sarcolemma and ECM.26-28 Mutations also occur in intermediate filament proteins such as desmin, which link the sarcomere to the cell periphery and also to the nucleus via the LINC complex (Fig. 3).29 Disrupting any one of these compartments inhibits transmission of force throughout the myocyte. Dysregulation of Ca2+ handling is caused by mutations in PLN, the gene encoding phospholamban, which regulates the sarcoplasmic reticulum Ca2+ ATPase (SERCA) activity. The PLN mutation p.Arg14del, that causes DCM with arrhythmia, inhibits SERCA, resulting in a reduction of Ca2+ reuptake in diastole.30
Figure 3.
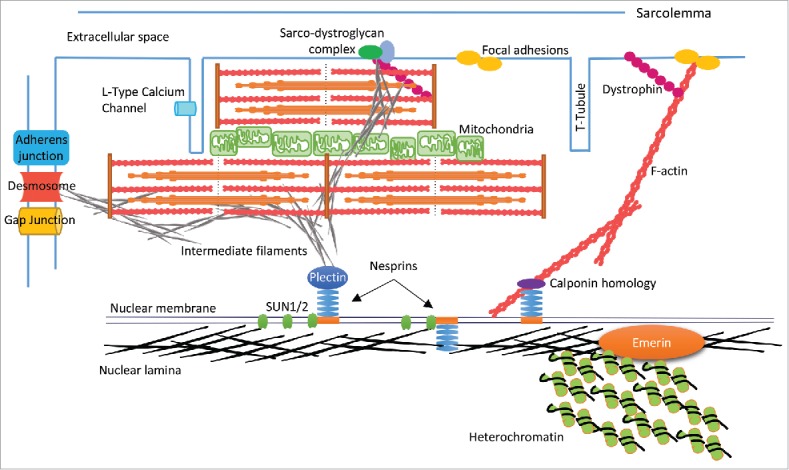
Schematic representation of CM structure. The nuclear lamina associates with LEM proteins i.e., Emerin and heterochromatin on the inner nuclear membrane (INM). Lamins also bind with SUN proteins and nesprins to form the LINC complex, which spans the nuclear envelope (NE), and links to cytoskeletal components (actin, intermediate filaments) via binding domains such as plectin and calponin homology. F-actin extends to the cell periphery and links to membrane anchors such as dystrophin and focal adhesion proteins (e.g. vinculin) thereby creating a mechanical links between the nucleus and ECM. In the context of CMs there may also be interactions with sarcomere structures, which are currently undefined. IFs such as desmin may knit the NE-sarcomere-sarcolemma via the sarco-dystroglycan complex, and provide a mechanical link from the nucleus to the intercalated disc via the cytoskeletal components of the sarcomere and IFs.
The LMNA gene is commonly mutated in DCM and accounts for approximately 6–8% of DCM cases in humans.7,31 The resultant DCM phenotype is complicated by conduction defects resulting in arrhythmias.32 Genotype-phenotype correlation is poor and the recurrence of common DCM causing mutations in LMNA is low; one study reported 165 unique DCM causing mutations to LMNA,33 occurring in all 12 exons of the gene. This diversity makes mechanistic analysis challenging since it is difficult to hypothesize a common mechanism based on specific lamin protein domains and the interactions that might be disturbed by causal mutations. However, clues to lamin mediated mechanisms may be evident in unique aspects of LMNA cardiomyopathy, such as conduction defects.
The cardiac conduction system and conduction defects
Electrical activity in the heart is controlled by specialized ‘pacemaker’ cardiomyocytes residing in the Sino-Atrial node in the atria, which receive signals from the autonomic nervous system. They initiate the propagation of electrical current through the myocardium of the atria causing the cells to contract. At the same time current flows to the Atrio-Ventricular node which initiates the propagation of current down the septum via fibers termed the bundle of His, and Purkinje fibers, which pass current from the apex, upwards and side-wards through the ventricular myocardium.34 Cardiomyocytes are excitable cells and have a negative resting membrane potential, allowing current to be propagated cell to cell via gap junctions made up of high conductance channels termed connexins.35 Connexins operate at the polar ends of cardiomyocytes in a junctional complex with the adherens junction (AJ) and desmosome, termed the intercalated disc (ID). Conduction defects could result because of a malfunction at any point during this process.
Notably, almost half of LMNA cardiomyopathy patients succumb to sudden cardiac death as a result of a fatal arrhythmia,36 and conduction defects associated with LMNA mutations can substantially precede the onset of DCM symptoms, meaning subtle but fatal arrhythmias may occur before any noticeable change in function. This makes diagnosis difficult in probands who only display cardiac disease.37 Patients displaying muscular dystrophy phenotypes with known LMNA mutations can be fitted with cardiac pacemakers/devices as a pre-emptive measure.
Increasingly, patients with LMNA mutations also present with nuanced cardiac defects that show features of HCM and ARVC. Therefore, another means of identifying LMNA mechanisms may be to look into the pathways commonly identified in HCM and ARVC which are unique from DCM.
Hypertrophic cardiomyopathy
HCM is an autosomal dominant disease characterized by hypertrophy of ventricular myocardium which is not explained by pressure overload.24 There is associated fibrosis and myocyte disarray as well as high prevalence of arrhythmia and is a common cause of sudden cardiac death in young athletes.38
Like DCM, mechanisms involve mechanical and Ca2+ handling changes, although cytoskeletal involvement is less prominent. HCM is usually considered as a disease of the sarcomere. Of 10 genes identified as causal, 9 encode sarcomeric proteins.39,40 The 2 most important are MYBPC3 encoding cardiac myosin-binding protein C (cMyBP-C), and MYH7 encoding β-MHC, and account for the majority of cases.41 Mutations mostly lead to substitution of single amino acids, though half of mutations to MYBPC3 are known to cause truncations to the protein product and lead to haploinsufficiency.42,43 Altered myosin kinetics and increased calcium sensitivity of thin-filaments lead to increased contractility,44-46 and activate hypertrophic signaling pathways. Mutations in troponin T lead to elevated sarcoplasmic reticulum Ca2+ content in diastole,47 predisposing the myocardium to arrhythmia,48 leading to aberrant downstream signaling.49
Ca2+ handling is disrupted by 2 main mechanisms. Firstly, mutations in cMyBP-C and the troponin complex increases the sensitivity of troponin C to Ca2+.50 Troponin is the principal calcium buffer in the SR, therefore, increased affinity should increase calcium levels in diastole.51,52 Contractile inefficiency can also compromise the energetics of the cardiomyocyte. Sarcomeric mutations that alter cross-bridge formation kinetics may cause a deficit in ATP availability and could impact upon other ATP requiring processes within the cell, such as Ca2+ uptake via SERCA during diastole.53 A myocardial energetics hypothesis is supported by mutations in the γ2 subunit of AMP-activated protein kinase, involved in energy sensing, which lead to HCM.54
The LMNA C591F and LMNA R644C mutations lead to phenotypes consistent with HCM.14,55 How LMNA mutations can influence HCM related mechanisms is not entirely clear. The loss of lamins A/C in isolated cardiomyocytes does not impact Ca2+ transients, but the shortening of cardiomyocytes is reduced.56 This implies that while the function of SERCA appears to be normal, the activation of myofilaments is hindered and may point to a mechanism involving reduced availability of ATP to the myofilaments.
Arrhythmogenic right ventricular cardiomyopathy
ARVC is characterized by pathological remodelling of the right ventricle, dilatation of cardiac chambers and systolic dysfunction57 in which myocyte death, inflammation and fibrofatty replacement of the myocardium are prominent features.58 Patients are also prone to arrhythmia and ARVC is known to be another common cause of sudden cardiac death.59,60
Most ARVC mutations occur in desmosomal genes.61 The desmosome anchors the IFs of one cell to the cytoplasmic membrane of another at the ID in order to create a lattice structure that provides mechanical strength to tissue. Mutations in desmoplakin, plakoglobin, plakophilin-2, and desmoglein-2 all cause ARVC.62-65 The molecular mechanisms that regulate the progression to ARVC are unclear, but 2 main hypotheses exist. Firstly, desmosomal mutations compromise the integrity of cell-cell interactions and as such make the tissue structure susceptible to mechanical stress, leading to cell detachment and necrosis, causing an inflammatory response. In this setting, fibroadipose deposition is a reparative response to injury. Second is the transdifferentiation model, which proposes that desmosomal perturbations can dysregulate the Wnt/β-catenin signaling pathway leading to activation of adipogenic and fibrogenic genes and a switch of cell fate from cardiomyocyte to adipocyte.66,67
Recently, patients presenting with ARVC were found to have LMNA mutations.11,68,69 It remains unclear how NL disruption could be a cause of ARVC, but it is plausible that disruption of the lamina could lead to the destabilisation of cell contacts since the cell membrane and nucleus are linked by a ‘molecular daisy chain’ of structural proteins. With respect to the transdifferentiation of fibrofatty tissue, emerin, a lamin A binding partner, has a known association with β-catenin and is thought to regulate its nuclear localization.70 Hypothetically, LMNA mutations which cause ARVC could interfere with the interaction of emerin with β-catenin and cause dysregulation of β-catenin signaling leading to aberrant cell differentiation.
Though phenotypically divergent, the mechanistic processes leading to DCM, HCM and ARVC share important core principles, i.e., structural instability and remodelling of tissue and ECM. LMNA mutations are likely to contribute to these processes via disruption of the links between the nucleus and cytoplasm. The mechanisms by which this occurs have been subjected to investigation in a number of animal models.
Modeling LMNA cardiomyopathy
In vivo models
Murine models with modified Lmna genes have enabled insights into the possible pathological mechanisms driving LMNA induced cardiomyopathies and are summarised in Table 2.
Table 2.
Clinical LMNA mutations and associated mouse model phenotypes
| LMNA Mutation | Human Disease | Mouse Model | Disease phenotype | Survival |
|---|---|---|---|---|
| N195K | DCM-CD | LmnaN195K/N195K | DCM and Heart failure | 12–14 weeks |
| H222P | EDMD | LmnaH222P/H222P | DCM and heart failure | Males: 4–9 months Females: 9–13 months |
| G608G | HGPS | LmnaG609G/G609G | Progeria - LQT and arrhythmia | 3–4 months |
| M371K | EDMD | MHC-LmnaM371K | Acute and subacute heart failure | embryonic lethal and 2–7 weeks |
| ΔK32 | L-CMD | LmnaΔK32/+ | DCM and heart failure | 5–6 weeks |
| L530P | EDMD | LmnaL530P/L530P | Progeria- with cardiac remodelling | 3–7 weeks |
| E82K | DCM-CD | MHC-LmnaE82K | DCM | long lived |
LmnaH222P/H222P mice
Global LmnaH222P/H222P mice were originally designed to study EDMD, which the human LMNA H222P mutation causes. DCM is acquired secondary to EDMD in the human clinic and the LmnaH222P/H222P homozygous knockin mice have proven a good model of LMNA cardiomyopathy71 Born without phenotype, they progress to a DCM phenotype at 16 weeks, leading to heart failure and death in males at 5–9 months and females at 7–13 months. They have reduced cardiac function and cardiac fibrosis.71
In one study, hearts from young pre-phenotypic mice were subjected to gene expression microarray analysis, which identified upregulation of MAPK signaling pathways.72 Extracellular signal regulated kinase (ERK1/2), c Jun N-terminal kinase (JNK), as well as p38 branches of MAPK signaling were all upregulated. Subsequent inhibition of JNK and ERK1/2 in mice significantly improved LV functional parameters and led to a reduction in fibrosis.73-75 The case for hyperactivation of ERK1/2 was also supported by post-mortem analysis of human heart tissue expressing 2 distinct mutations, LMNA ΔK261 and LMNA IVS9 + 1 g>a.76 Additionally, hyperactivation of the mammalian Target Of Rapamycin (mTOR) signaling pathway inhibited autophagy, a crucial housekeeping process that facilitates the degradation of unwanted proteins and organic components of the cell during severe stress.77 Treatment of LmnaH222P/H222P mice with temsirolimus, an inhibitor of mTOR, led to activation of autophagy, and amelioration of cardiac decline.78 Treatment with angiotensin converting enzyme (ACE) inhibitors also improved myocardial function in these mice. ACE converts inactive angiotensin I to angiotensin II which stimulates sympathetic activation leading to increases in heart rate and vascular tone resulting in pressure overload.79 These data imply that by lowering mechanical stress in vivo, LMNA cardiomyopathy can be delayed or attenuated.
LmnaN195K/N195K mice
The LMNA N195K mutation is known to cause DCM with conduction defects in humans.80 Accordingly, global LmnaN195K/N195K mice developed DCM with associated conduction defects and died at 2–3 months old as a result of arrhythmia.81 Abnormal desmin localization was observed alongside a reduction in mRNA of HF1b/Sp4, a transcription factor that is crucial in the development of the cardiac conduction system, suggesting a possible mechanism for the early presentation of conduction defects in patients. Loss and mislocalisation of connexins 40 and 43, leading to reduced propagation of electrical current through the myocardial tissue, was also observed. These findings point toward a mechanism for conduction defects that may be linked to mechanical susceptibility, as they implicate lamina dysfunction as a cause for the disruption of cell-cell contacts. The observation that multiple components of the cardiac conduction system are dysregulated in LmnaN195K/N195K mice may help to explain why conduction defects are so prevalent in LMNA cardiomyopathy.82
LmnaΔK32/+ mice
In man, LMNA disease mutations are mostly heterozygous, leading to dominant negative phenotypes. Many model systems of LMNA cardiomyopathy in mice show phenotypic changes only when the mutation is homozygous, meaning the contribution of wildtype lamin in disease progression is overlooked. However, global LmnaΔK32/+ mice were investigated and found to be pathogenic.83 In humans deletion of lysine at position 32 in the lamin A amino acid sequence causes severe congenital muscular dystrophy (L-CMD) phenotypes.84 Mice with this mutation developed DCM that occurred in a 2-step process resulting in death between 35 and 70 weeks. Initially the toxic accumulation of ΔK32-lamin was avoided by proteasomal degradation. However, this resulted in reduced lamin A/C expression, which then initiated the process of cardiac remodelling and DCM. After DCM was established dysfunction of the ubiquitin proteasome system occurred, leading to toxic increases in ΔK32-lamin. These mice also showed earlier onset DCM as a result of exercise- induced stress.85
Other models of LMNA mutations
In man, the LMNA M371K mutation causes EDMD.86 In MHC-LmnaM371K mice, cardiac specific overexpression of LMNA M371K led to mice with very low survival at birth. Mice that survived died between 2–7 weeks from cardiac defects.87 This study suggests that accumulation of mutant M371K lamin A is toxic in the presence of endogenous wildtype lamins in a cardiomyocyte specific setting. Importantly, this study also investigated overexpression of wildtype lamin A/C in the heart. These mice displayed no phenotypic defects and were long lived, supporting the idea that mutant LMNA is a toxic driver of disease phenotypes, perhaps operating by disrupting lamin structure and function, by dysregulation of lamin processing and binding, or even by disrupting the balance of lamin isoform expression.
The DCM causing mutation, LMNA E82K, has also been investigated in vivo in a cardiac specific manner. MHC-LmnaE82K mice showed evidence of cardiac dysfunction at 6 months of age indicated by a reduction in cardiac function and myocardial remodelling.88 Fas and mitochondrial pathways of apoptosis were identified as the mechanisms responsible for cardiac decline in this model.
In the clinic, the LMNA L530P mutation leads to EDMD. In vivo global LmnaL530P/L530P mice had a progeria phenotype and also showed a cardiac phenotype, described as displaying features consistent with pulmonary hypertension. They had enlarged hearts and fibrosis, suggesting a program of pathological cardiac remodelling was induced.89 Moreover, when modeled in C. elegans the corresponding mutation Lmn-1 L535P caused a muscular dystrophy characterized by increased resistance of muscle nuclei to mechanical strain alongside structural disorganisation of muscle actin filaments.90
Lmna knockout models
Though not clinically relevant, global Lmna−/− mice die between 6–8 weeks of age because of DCM and heart failure56 and share mechanistic traits with LmnaH222P/H222P mice including impaired autophagy.91 Intervention with the mTOR inhibitor rapamycin significantly improves cardiac function and survival in Lmna−/− mice, and strengthens the argument for aberrant mTOR activation in LMNA cardiomyopathy.
Further investigation showed that Lmna−/− cardiomyocytes were structurally compromised. This occurs via the disruption of nesprin1α resulting in uncoupling of the nucleus from the cytoskeleton leading to mechanical instability and defective transmission of force.92 In support of this finding, desmin was also mislocalised.56 Activation of hypertrophic genes was attenuated, suggesting that Lmna−/− mice were unable to adapt to DCM progression with compensatory hypertrophy, accounting for rapid disease progression. Connexins were also mislocalised contributing to attenuated contractility and also arrhythmia.93,94
Lmna haploinsufficiency was also investigated. Lmna+/− mice displayed early onset cardiac conduction system disease and late onset DCM,95 which could be alleviated by exercise and β-blockers.96 Application of pathological hemodynamic stress led to a blunted hypertrophic response due to impaired activation of the mechanosensitive gene, Egr1,97 and provided evidence that mechanotransduction signaling pathways and pathophysiological adaptations to stress can be inhibited by lamina disruption.
In vivo models have been invaluable in the observation of critical molecular events, which underpin the pathology of LMNA cardiomyopathy, especially with regard to conduction defects. However, a complete understanding of mechanisms will likely require rigorous investigation of early, pre-phenotypic timepoints to identify the earliest possible molecular changes immediately downstream of lamin dysfunction.
In vitro models
Analysis of LMNA cardiomyopathy mutants in single cell models have also provided important mechanistic insights into disease mechanisms at the molecular level.
Evidence for the mechanical hypothesis
Studies performed in fibroblasts showed that the DCM causing LMNA mutations M371K, ΔK32 and N195K failed to restore nuclear stiffness after deformation, and the mutant lamin proteins were more soluble than wild type lamin A. Moreover, ΔK32 and N195K mutant lamin proteins failed to assemble into filaments in vitro, instead forming aggregates, and mimicked the loss of lamin function.98
Structural analysis of DCM mutations E161K and R190W showed alterations in secondary and tertiary structures leading to perturbed intrinsic self-assembly of high order lamin structures.99 At the level of electron microscopy, the lamin lattice networks showed substantial organisational changes and reduced elasticity. Another LMNA DCM mutation S143P, common in Finnish patients, was also found to undergo reduction in self-assembly behavior in vitro and was associated with an elevated unfolded protein response according to whole genome analysis.100
An hypothesis has been put forward to explain the impact of these findings. In fully differentiated mechanical structures such as striated muscle, lamins A and C are highly expressed. It is thought that their flexible rheological (gel-like) behavior, acts as a ‘valve’ for the B-type lamins which, in contrast, resist deformation.101 Therefore, if a proportion of the A-type lamins are mutant, and the ability of the nucleus to soften in response to strain is reduced, then the lamina network is likely to collapse in response to a relatively low mechanical stress threshold. Accordingly, a number of myopathic LMNA mutations have been tested in an in situ model of D. melanogaster body wall muscle, and an in vitro model of mouse embryonic fibroblasts, which found increases in nuclear strain and decreases in nuclear displacements in response to mechanical stretch.98
Evidence for the gene expression hypothesis
A number of studies have investigated the role of lamin dysfunction on mechanotransduction pathways—signaling mediated by physical interactions—by examining gene expression responses. Investigation of Lmna−/− mouse fibroblasts found impaired activation of mechanosensitive genes Iex1 and Egr1 in single cell models of mechanical stretch.102,103 It has also been shown that the NL may be able to detect perturbations in force and convert these into adaptive or pathological gene expression responses. For example, disruption of the NL by Lmna deficiency attenuated NF-kB mediated transcriptional response to mechanical or cytokine stimulation despite increased transcription factor binding, implying that lamins are crucial for transcriptional activation.104-106
In vitro studies have also provided detailed insight on the impact of LMNA disruption on the mechanical properties of cells and have identified that these can elicit abnormal gene expression and signaling responses. However, despite the wealth of investigations into LMNA cardiomyopathy mutations in isolated models, analyses have rarely been undertaken in isolated cardiomyocytes; thus the cardiomyocyte specific effects of these mutations are largely undefined. One study has addressed this by investigating induced pluripotent stem cell derived cardiomyocytes from a patient harbouring the LMNA R225X mutation and found that when cardiomyocyte contraction was initiated by electrical stimulation, the cells underwent apoptosis.107
In summary, data from the clinic and model systems suggests that structural defects drive the onset of disease by contributing to defective electrical signaling, and also by inhibiting efficient molecular signaling responses to mechanical stress. In addition, a common theme in models investigating LMNA cardiomyopathy is the activation of cellular stress responses—apoptosis, autophagy and the unfolded protein response—indicating that certain lamin mutants may be toxic to cells, a feature which has until now been understudied.
Lamin toxicity in cardiomyopathy
One model yet to be tested in the context of LMNA cardiomyopathy is the prelamin A toxicity model. Prelamin A toxicity is central to a number of laminopathy sub-types, primarily the premature aging disorders. In HGPS, for example, the final enzymatic cleavage of prelamin A, performed by ZMPSTE24, is abolished and the protein remains permanently farnesylated, meaning it cannot be inserted efficiently or completely into the NL.108 In the most common form of HGPS, this occurs because of a mutation that leads to the deletion of 50 amino acids which contain the ZMPSTE24 cleavage site,109-111 resulting in a truncated prelamin A mutant protein called progerin. Loss of function mutations to ZMPSTE24 lead to prelamin A accumulation and can also drive HGPS phenotypes.112 Prelamin A accumulation is also an important mediator in normal aging in a number of tissues, including the vasculature.113 It is not known, however, whether this is relevant in myocardial aging.
Progerin and prelamin A are thought to drive disease phenotypes by disrupting nuclear morphology and heterochromatin distribution as well as DNA damage repair pathways resulting in premature senescence.114-116 Heterochromatin instability appears to be partly responsible for the defective recruitment of repair factors to sites of DNA damage.117,118 In addition, prelamin A accumulation causes nuclear pore complex dysfunction which also impairs recruitment of DNA repair proteins.119 Interestingly, one study investigated the LMNA L306R mutation, which caused a premature aging syndrome with severe ARVC presentation; cells with this mutation had dysmorphic nuclei, elevated levels of DNA damage and underwent premature cellular senescence.11 Moreover, stem cell derived cardiomyocytes with the LMNA R225X mutation displayed nuclear morphology defects and premature senescence under stress.107 These studies provide the clearest evidence yet that premature aging mechanisms may be partly responsible for LMNA cardiomyopathy phenotypes.
There is evidence to suggest that prelamin A/progerin accumulation is important in the establishment of cardiac disease phenotypes. For example, HGPS patients were observed to suffer cardiomegaly and cardiac dilatation toward the end of life120 and HGPS patients who survived to older ages displayed cardiac remodeling and atrial enlargement.121 A mouse model of HGPS has also shown evidence of cardiac dysfunction.122 Recently, a mutation in ZMPSTE24 was identified that caused a substantial reduction in ZMPSTE24 activity and led to DCM associated with metabolic syndrome.16 Meanwhile, Zmpste24−/− mice showed evidence of myocardial disruption at 3 months of age.123 The LMNA p.T655fsX49 mutation caused accumulation of non-farnesylated prelamin A, leading to cardiac conduction defects in humans.13 Accordingly, accumulation of non-farnesylated prelamin A led to late onset DCM in mice expressing a homozygous ‘non-farnesylated prelamin A only’ allele.124 DCM mutations have also been shown to accumulate prelamin A in model systems, as expression of the LMNA R89L mutant caused accumulation of prelamin A in vitro.125 Moreover, some of the murine LMNA cardiomyopathy models have shown nuclear morphology defects and heterochromatin disorganisation, both hallmarks of prelamin A/progerin toxicity. Further investigation is now required to determine how prevalent the accumulation of lamin A precursors is in cardiomyopathy patient hearts.
Therapeutic potential
Modulation of mTOR signaling currently provides the most promising way forward for the treatment of LMNA cardiomyopathy specifically.126,127 For laminopathies as a whole, exon skipping128 and influencing the splicing of LMNA toward lamin C to avoid the dominant negative effects of lamin A mutants,129 are being investigated as potential therapies and may have relevance to LMNA cardiomyopathies. In prelamin A accumulating diseases such as HGPS, farnesyl transferase inhibitors (FTIs) have been through clinical trials with modest success.130 This was followed up by trials of a combination therapy of FTIs with statins and bisphosphonates, which inhibit upstream processing in the Acetyl-CoA pathway of cholesterol synthesis and protein prenylation (which includes farnesylation), but again showed limited benefit.131 Inhibition of prelamin A processing enzymes such as ICMT, which controls carboxymethylation of prelamin A, may instead be beneficial in the context of prelamin A toxicity, via regulation of mTOR.132 Moreover, Remodelin, a small molecule inhibitor of N-acetyltransferase 10 (NAT10), alleviates many cellular abnormalities in HGPS, potentially operating via a novel mechanism involving microtubule reorganisation, and shows much promise at the pre-clinical stage.133
Future directions
The gene expression and mechanical hypotheses have been well investigated in LMNA cardiomyopathy, and important cell signaling pathways have been established. However, questions regarding the role of Ca2+ signaling and myocardial energetics remain unanswered. Moreover, the pleiotropic effects of single LMNA mutations, such as the R644C mutation involved in EDMD, DCM, HCM and ARVC progression,134 suggest that external factors may be important in the establishment of disease. The nature of these stimuli may impact the direction of disease progression, implying that the study of epigenetics and intercellular communication, for example, may also be important in LMNA cardiomyopathy.
The toxicity of prelamin A or other lamin variants is also potentially important and requires further investigation. Associated pathways involving DNA damage and premature senescence should also be considered both in the context of cardiomyopathies and normal myocardial aging. Therapies for laminopathies driven by accumulation of unprocessed prelamin A or truncated lamin mutants are currently under investigation with some having been to clinical trials. If LMNA cardiomyopathy is driven by toxic accumulation of lamin variants, these therapies may also be important in the future treatment of some LMNA cardiomyopathies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I would like to thank Elizabeth Halton and Andrew Cobb for their critical appraisal of the manuscript.
Funding
This work was supported by the British Heart Foundation. Grant code PG/15/93/31834.
References
- [1].Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem 1993; 268:16321-6; PMID:8344919 [PubMed] [Google Scholar]
- [2].Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Science 2001; 114:4557-65; PMID:11792820 [DOI] [PubMed] [Google Scholar]
- [3].Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 2006; 172:41-53; PMID:16380439; http://dx.doi.org/ 10.1083/jcb.200509124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mejat A, Misteli T. LINC complexes in health and disease. Nucleus 2010; 1:40-52; PMID:21327104; http://dx.doi.org/ 10.4161/nucl.1.1.10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol 2009; 10:63-73; PMID:19197333; http://dx.doi.org/ 10.1038/nrm2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wilson KL, Foisner R. Lamin-binding Proteins. Cold Spring Harb Perspect Biol 2010; 2:a000554; PMID:20452940; http://dx.doi.org/ 10.1101/cshperspect.a000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, et al.. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003; 41:771-80; PMID:12628721; http://dx.doi.org/ 10.1016/S0735-1097(02)02954-6 [DOI] [PubMed] [Google Scholar]
- [8].Bonne G, Leturcq F, Ben Yaou R. Emery-Dreifuss Muscular Dystrophy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, et al., eds. GeneReviews(R) Seattle (WA), 1993 [PubMed] [Google Scholar]
- [9].Zhang L, Shen H, Zhao Z, Bing Q, Hu J. Cardiac effects of the c.1583 C–>G LMNA mutation in two families with Emery-Dreifuss muscular dystrophy. Mol Med Rep 2015; 12:5065-71; PMID:26165385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chang SH, Tsai CT, Lai LP, Lei MH. Identification of a lamin A/C gene mutation in a Taiwanese family with limb girdle muscular dystrophy and cardiomyopathy. Inter J Cardiol 2010; 145:598-9; PMID:20615561; http://dx.doi.org/ 10.1016/j.ijcard.2010.06.014 [DOI] [PubMed] [Google Scholar]
- [11].Alastalo TP, West G, Li SP, Keinanen A, Helenius M, Tyni T, Lapatto R, Turanlahti M, Heikkila P, Kaariainen H, et al.. LMNA Mutation c.917T>G (p.L306R) Leads to Deleterious Hyper-Assembly of Lamin A/C and Associates with Severe Right Ventricular Cardiomyopathy and Premature Aging. Hum Mutat 2015; 36:694-703; PMID:25820511; http://dx.doi.org/ 10.1002/humu.22793 [DOI] [PubMed] [Google Scholar]
- [12].Guo X, Ling C, Liu Y, Zhang X, Zhang S. A Case of Novel Lamin A/C Mutation Manifesting as Atypical Progeroid Syndrome and Cardiomyopathy. Can J Cardiol 2016; 32:1166 e29–31 [DOI] [PubMed] [Google Scholar]
- [13].Andre P, Schneebeli S, Vigouroux C, Lascols O, Schaaf M, Chevalier P. Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A. Am Heart J 2015; 169:587-93; PMID:25819867; http://dx.doi.org/ 10.1016/j.ahj.2014.12.021 [DOI] [PubMed] [Google Scholar]
- [14].Araújo-Vilar D, Lado-Abeal J, Palos-Paz F, Lattanzi G, Ma Bandín, Bellido D, Domínguez-Gerpe L, Calvo C, Pérez O, Ramazanova A, et al.. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin Endocrinol 2008; 69:61-8; PMID:18031308; http://dx.doi.org/ 10.1111/j.1365-2265.2007.03146.x [DOI] [PubMed] [Google Scholar]
- [15].Duparc A, Cintas P, Somody E, Bieth E, Richard P, Maury P, Delay M. A cardio-neurological form of laminopathy: dilated cardiomyopathy with permanent partial atrial standstill and axonal neuropathy. Pacing Clin Electrophysiol 2009; 32:410-5; PMID:19272076; http://dx.doi.org/ 10.1111/j.1540-8159.2008.02254.x [DOI] [PubMed] [Google Scholar]
- [16].Galant D, Gaborit B, Desgrouas C, Abdesselam I, Bernard M, Levy N, Merono F, Coirault C, Roll P, Lagarde A, et al.. A Heterozygous ZMPSTE24 Mutation Associated with Severe Metabolic Syndrome, Ectopic Fat Accumulation, and Dilated Cardiomyopathy. Cells 2016; 5; PMID:27120622; http://dx.doi.org/ 10.3390/cells5020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart A, et al.. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113:1807-16; PMID:16567565; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.106.174287 [DOI] [PubMed] [Google Scholar]
- [18].Yajima T. Viral myocarditis: potential defense mechanisms within the cardiomyocyte against virus infection. Future Microbiol 2011; 6:551-66; PMID:21585262; http://dx.doi.org/ 10.2217/fmb.11.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation 2009; 119:2615-24; PMID:19451363; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.108.766022 [DOI] [PubMed] [Google Scholar]
- [20].Walker RK, Cousins VM, Umoh NA, Jeffress MA, Taghipour D, Al-Rubaiee M, Haddad GE. The good, the bad, and the ugly with alcohol use and abuse on the heart. Alcohol Clin Exp Res 2013; 37:1253-60; PMID:23527963; http://dx.doi.org/ 10.1111/acer.12109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Raj S, Franco VI, Lipshultz SE. Anthracycline-induced cardiotoxicity: a review of pathophysiology, diagnosis, and treatment. Curr Treat Options Cardiovasc Med 2014; 16:315; PMID:24748018; http://dx.doi.org/ 10.1007/s11936-014-0315-4 [DOI] [PubMed] [Google Scholar]
- [22].Yoshikawa T, Baba A, Nagatomo Y. Autoimmune mechanisms underlying dilated cardiomyopathy. Circ J 2009; 73:602-7; PMID:19246813; http://dx.doi.org/ 10.1253/circj.CJ-08-1151 [DOI] [PubMed] [Google Scholar]
- [23].Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Eng J Med 2011; 364:1643-56; PMID:21524215; http://dx.doi.org/ 10.1056/NEJMra0902923 [DOI] [PubMed] [Google Scholar]
- [24].Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, et al.. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93:841-2 [DOI] [PubMed] [Google Scholar]
- [25].Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, Vatta M, Pinamonti B, Muntoni F, Caforio AL, et al.. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol 1999; 34:181-90; http://dx.doi.org/ 10.1016/S0735-1097(99)00172-2 [DOI] [PubMed] [Google Scholar]
- [26].Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, et al.. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 2003; 42:2014-27; PMID:14662268; http://dx.doi.org/ 10.1016/j.jacc.2003.10.021 [DOI] [PubMed] [Google Scholar]
- [27].Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, Bowles NE, Towbin JA. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000; 106:655-62; PMID:10974018; http://dx.doi.org/ 10.1172/JCI9224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Franz WM, Cremer M, Herrmann R, Grunig E, Fogel W, Scheffold T, Goebel HH, Kircheisen R, Kubler W, Voit T, et al.. X-linked dilated cardiomyopathy. Novel mutation of the dystrophin gene. Ann N Y Acad Sci 1995; 752:470-91; PMID:7755293; http://dx.doi.org/ 10.1111/j.1749-6632.1995.tb17457.x [DOI] [PubMed] [Google Scholar]
- [29].Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, et al.. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007; 115:1244-51; PMID:17325244; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.106.644013 [DOI] [PubMed] [Google Scholar]
- [30].Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW 2nd, et al.. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A 2006; 103:1388-93; PMID:16432188; http://dx.doi.org/ 10.1073/pnas.0510519103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, et al.. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 2008; 156:161-9; PMID:18585512; http://dx.doi.org/ 10.1016/j.ahj.2008.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, et al.. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol 2008; 52:1250-60; PMID:18926329; http://dx.doi.org/ 10.1016/j.jacc.2008.06.044 [DOI] [PubMed] [Google Scholar]
- [33].Tesson F, Saj M, Uvaize MM, Nicolas H, Ploski R, Bilinska Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol J 2014; 21:331-42; PMID:24846508; http://dx.doi.org/ 10.5603/CJ.a2014.0037 [DOI] [PubMed] [Google Scholar]
- [34].van Weerd JH, Christoffels VM. The formation and function of the cardiac conduction system. Development 2016; 143:197-210; PMID:26786210; http://dx.doi.org/ 10.1242/dev.124883 [DOI] [PubMed] [Google Scholar]
- [35].Peters NS. Gap junctions and clinical cardiology: from molecular biology to molecular medicine. Eur Heart J 1997; 18:1697-702; PMID:9402442; http://dx.doi.org/ 10.1093/oxfordjournals.eurheartj.a015162 [DOI] [PubMed] [Google Scholar]
- [36].van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, et al.. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med 2005; 83:79-83; PMID:15551023; http://dx.doi.org/ 10.1007/s00109-004-0589-1 [DOI] [PubMed] [Google Scholar]
- [37].Brodt C, Siegfried JD, Hofmeyer M, Martel J, Rampersaud E, Li D, Morales A, Hershberger RE. Temporal relationship of conduction system disease and ventricular dysfunction in LMNA cardiomyopathy. J Card Fail 2013; 19:233-9; PMID:23582089; http://dx.doi.org/ 10.1016/j.cardfail.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Maron BJ, Thompson PD, Puffer JC, McGrew CA, Strong WB, Douglas PS, Clark LT, Mitten MJ, Crawford MH, Atkins DL, et al.. Cardiovascular preparticipation screening of competitive athletes. A statement for health professionals from the Sudden Death Committee (clinical cardiology) and Congenital Cardiac Defects Committee (cardiovascular disease in the young), American Heart Association. Circulation 1996; 94:850-6; PMID:8772711; http://dx.doi.org/ 10.1161/01.CIR.94.4.850 [DOI] [PubMed] [Google Scholar]
- [39].Bonne G, Carrier L, Richard P, Hainque B, Schwartz K. Familial hypertrophic cardiomyopathy: from mutations to functional defects. Circ Res 1998; 83:580-93; PMID:9742053; http://dx.doi.org/ 10.1161/01.RES.83.6.580 [DOI] [PubMed] [Google Scholar]
- [40].Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet 2000; 355:58-60; PMID:10615904; http://dx.doi.org/ 10.1016/S0140-6736(99)06187-5 [DOI] [PubMed] [Google Scholar]
- [41].Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, et al.. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107:2227-32; PMID:12707239; http://dx.doi.org/ 10.1161/01.CIR.0000066323.15244.54 [DOI] [PubMed] [Google Scholar]
- [42].Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 2009; 105:219-22; PMID:19574547; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.202440 [DOI] [PubMed] [Google Scholar]
- [43].van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, et al.. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009; 119:1473-83; PMID:19273718; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.108.838672 [DOI] [PubMed] [Google Scholar]
- [44].Redwood CS, Moolman-Smook JC, Watkins H. Properties of mutant contractile proteins that cause hypertrophic cardiomyopathy. Cardiovasc Res 1999; 44:20-36; PMID:10615387; http://dx.doi.org/ 10.1016/S0008-6363(99)00213-8 [DOI] [PubMed] [Google Scholar]
- [45].Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 2001; 104:557-67; PMID:11239412; http://dx.doi.org/ 10.1016/S0092-8674(01)00242-2 [DOI] [PubMed] [Google Scholar]
- [46].Kirschner SE, Becker E, Antognozzi M, Kubis HP, Francino A, Navarro-Lopez F, Bit-Avragim N, Perrot A, Mirrakhimov MM, Osterziel KJ, et al.. Hypertrophic cardiomyopathy-related beta-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am J Physiol Heart Circ Physiol 2005; 288:H1242-51; PMID:15550524; http://dx.doi.org/ 10.1152/ajpheart.00686.2004 [DOI] [PubMed] [Google Scholar]
- [47].Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res 2003; 92:428-36; PMID:12600890; http://dx.doi.org/ 10.1161/01.RES.0000059562.91384.1A [DOI] [PubMed] [Google Scholar]
- [48].Huke S, Knollmann BC. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 2010; 48:824-33; PMID:20097204; http://dx.doi.org/ 10.1016/j.yjmcc.2010.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. Ann N Y Acad Sci 2005; 1047:86-98; PMID:16093487; http://dx.doi.org/ 10.1196/annals.1341.008 [DOI] [PubMed] [Google Scholar]
- [50].Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res 2007; 101:1266-73; PMID:17932326; http://dx.doi.org/ 10.1161/CIRCRESAHA.107.156380 [DOI] [PubMed] [Google Scholar]
- [51].Kataoka A, Hemmer C, Chase PB. Computational simulation of hypertrophic cardiomyopathy mutations in troponin I: influence of increased myofilament calcium sensitivity on isometric force, ATPase and [Ca2+]i. J Biomech 2007; 40:2044-52; PMID:17140583; http://dx.doi.org/ 10.1016/j.jbiomech.2006.09.026 [DOI] [PubMed] [Google Scholar]
- [52].Smith GA, Dixon HB, Kirschenlohr HL, Grace AA, Metcalfe JC, Vandenberg JI. Ca2+ buffering in the heart: Ca2+ binding to and activation of cardiac myofibrils. Biochem J 2000; 346 Pt 2:393-402; PMID:10677358; http://dx.doi.org/ 10.1042/bj3460393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ashrafian H, Redwood C, Blair E, Watkins H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet 2003; 19:263-8; PMID:12711218; http://dx.doi.org/ 10.1016/S0168-9525(03)00081-7 [DOI] [PubMed] [Google Scholar]
- [54].Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 2001; 10:1215-20; PMID:11371514; http://dx.doi.org/ 10.1093/hmg/10.11.1215 [DOI] [PubMed] [Google Scholar]
- [55].Mercuri E, Brown SC, Nihoyannopoulos P, Poulton J, Kinali M, Richard P, Piercy RJ, Messina S, Sewry C, Burke MM, et al.. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve 2005; 31:602-9; PMID:15770669; http://dx.doi.org/ 10.1002/mus.20293 [DOI] [PubMed] [Google Scholar]
- [56].Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, et al.. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest 2004; 113:357-69; PMID:14755333; http://dx.doi.org/ 10.1172/JCI200419448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sen-Chowdhry S, McKenna WJ. The utility of magnetic resonance imaging in the evaluation of arrhythmogenic right ventricular cardiomyopathy. Curr Opin Cardiol 2008; 23:38-45; PMID:18281826; http://dx.doi.org/ 10.1097/HCO.0b013e3282f2c96e [DOI] [PubMed] [Google Scholar]
- [58].Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Ann Rev Med 2010; 61:233-53; PMID:20059337; http://dx.doi.org/ 10.1146/annurev.med.052208.130419 [DOI] [PubMed] [Google Scholar]
- [59].McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 1994; 71:215-8; PMID:8142187; http://dx.doi.org/ 10.1136/hrt.71.3.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, Chevalier P, Bouvagnet P. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003; 108:3000-5; PMID:14662701; http://dx.doi.org/ 10.1161/01.CIR.0000108396.65446.21 [DOI] [PubMed] [Google Scholar]
- [61].Kirchhof P, Fabritz L, Zwiener M, Witt H, Schafers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, et al.. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006; 114:1799-806; PMID:17030684; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.106.624502 [DOI] [PubMed] [Google Scholar]
- [62].Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2007; 81:964-73; PMID:17924338; http://dx.doi.org/ 10.1086/521633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 2004; 13:185-94; PMID:15210133; http://dx.doi.org/ 10.1016/j.carpath.2004.03.609 [DOI] [PubMed] [Google Scholar]
- [64].Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol 2005; 16:927-35; PMID:16101641; http://dx.doi.org/ 10.1111/j.1540-8167.2005.40842.x [DOI] [PubMed] [Google Scholar]
- [65].Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2007; 50:1813-21; PMID:17980246; http://dx.doi.org/ 10.1016/j.jacc.2007.08.008 [DOI] [PubMed] [Google Scholar]
- [66].Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest 2006; 116:2012-21; PMID:16823493; http://dx.doi.org/ 10.1172/JCI27751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].MacRae CA, Birchmeier W, Thierfelder L. Arrhythmogenic right ventricular cardiomyopathy: moving toward mechanism. J Clin Invest 2006; 116:1825-8; PMID:16823481; http://dx.doi.org/ 10.1172/JCI29174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Larsen MK, Nissen PH, Berge KE, Leren TP, Kristensen IB, Jensen HK, Banner J. Molecular autopsy in young sudden cardiac death victims with suspected cardiomyopathy. Forensic Sci Int 2012; 219:33-8; PMID:22177269; http://dx.doi.org/ 10.1016/j.forsciint.2011.11.020 [DOI] [PubMed] [Google Scholar]
- [69].Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, Muir A, Pantazis A, McKenna WJ, Elliott PM. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Euro Heart J 2011; 33(9):1128-36; PMID:22199124 [DOI] [PubMed] [Google Scholar]
- [70].Markiewicz E, Tilgner K, Barker N, van de Wetering M, Clevers H, Dorobek M, Hausmanowa-Petrusewicz I, Ramaekers FC, Broers JL, Blankesteijn WM, et al.. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J 2006; 25:3275-85; PMID:16858403; http://dx.doi.org/ 10.1038/sj.emboj.7601230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, et al.. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet 2005; 14:155-69; PMID:15548545; http://dx.doi.org/ 10.1093/hmg/ddi017 [DOI] [PubMed] [Google Scholar]
- [72].Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest 2007; 117:1282-93; PMID:17446932; http://dx.doi.org/ 10.1172/JCI29042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet 2009; 18:241-7; PMID:18927124; http://dx.doi.org/ 10.1093/hmg/ddn343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wu W, Shan J, Bonne G, Worman HJ, Muchir A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim Biophys Acta 2010; 1802:632-8; PMID:20388542; http://dx.doi.org/ 10.1016/j.bbadis.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-Activated Protein Kinase Inhibitors Improve Heart Function and Prevent Fibrosis in Cardiomyopathy Caused by Mutation in Lamin A/C Gene. Circulation 2010; 123:53-61; PMID:21173351; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.110.970673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Muchir A, Sa Reilly, Wu W, Iwata S, Homma S, Bonne G, Worman HJ. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res 2012; 93:311-9; PMID:22068161; http://dx.doi.org/ 10.1093/cvr/cvr301 [DOI] [PubMed] [Google Scholar]
- [77].Choi JC, Wu W, Muchir A, Iwata S, Homma S, Worman HJ. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J Biol Chem 2012:1-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med 2012; 4:144ra02; ; http://dx.doi.org/ 10.1126/scitranslmed.3003875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Muchir A, Wu W, Sera F, Homma S, Worman HJ. Mitogen-activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochem Biophys Res Commun 2014; 452:958-61; PMID:25218145; http://dx.doi.org/ 10.1016/j.bbrc.2014.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, HJ Vidaillet Jr., Spudich S, De Girolami U, et al.. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999; 341:1715-24; PMID:10580070; http://dx.doi.org/ 10.1056/NEJM199912023412302 [DOI] [PubMed] [Google Scholar]
- [81].Mounkes LC, Kozlov SV, Rottman JN, Stewart CL. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet 2005; 14:2167-80; PMID:15972724; http://dx.doi.org/ 10.1093/hmg/ddi221 [DOI] [PubMed] [Google Scholar]
- [82].Nguyen-Tran VT, Kubalak SW, Minamisawa S, Fiset C, Wollert KC, Brown AB, Ruiz-Lozano P, Barrere-Lemaire S, Kondo R, Norman LW, et al.. A novel genetic pathway for sudden cardiac death via defects in the transition between ventricular and conduction system cell lineages. Cell 2000; 102:671-82; PMID:11007485; http://dx.doi.org/ 10.1016/S0092-8674(00)00089-1 [DOI] [PubMed] [Google Scholar]
- [83].Cattin M-E, Bertrand AT, Schlossarek S, Le Bihan MC, Skov Jensen S, Neuber C, Crocini C, Maron S, J Lainé, Mougenot N, et al.. Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum Mol Genet 2013; 22(15):3152-64 [DOI] [PubMed] [Google Scholar]
- [84].Quijano-Roy S, Mbieleu B, Bonnemann CG, Jeannet PY, Colomer J, Clarke NF, Cuisset JM, Roper H, De Meirleir L, D'Amico A, et al.. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008; 64:177-86; PMID:18551513; http://dx.doi.org/ 10.1002/ana.21417 [DOI] [PubMed] [Google Scholar]
- [85].Cattin ME, Ferry A, Vignaud A, Mougenot N, Jacquet A, Wahbi K, Bertrand AT, Bonne G. Mutation in lamin A/C sensitizes the myocardium to exercise-induced mechanical stress but has no effect on skeletal muscles in mouse. Neuromuscul Disord 2016; 26(8):490-9; PMID:27287550 [DOI] [PubMed] [Google Scholar]
- [86].Bonne G, Mercuri E, Muchir A, Urtizberea A, Becane HM, Recan D, Merlini L, Wehnert M, Boor R, Reuner U, et al.. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol 2000; 48:170-80; PMID:10939567; http://dx.doi.org/ 10.1002/1531-8249(200008)48:2%3c170::AID-ANA6%3e3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- [87].Wang Y, Herron AJ, Worman HJ. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum Mol Genet 2006; 15:2479-89; PMID:16825283; http://dx.doi.org/ 10.1093/hmg/ddl170 [DOI] [PubMed] [Google Scholar]
- [88].Lu D, Lian H, Zhang X, Shao H, Huang L, Qin C, Zhang L. LMNA E82K mutation activates FAS and mitochondrial pathways of apoptosis in heart tissue specific transgenic mice. PLoS One 2010; 5:e15167; PMID:21151901; http://dx.doi.org/ 10.1371/journal.pone.0015167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003; 423:298-301; PMID:12748643; http://dx.doi.org/ 10.1038/nature01631 [DOI] [PubMed] [Google Scholar]
- [90].Zuela N, Zwerger M, Levin T, Medalia O, Gruenbaum Y. Impaired mechanical response of an EDMD mutation leads to motility phenotypes that are repaired by loss of prenylation. J Cell Sci 2016; 129:1781-91; PMID:27034135; http://dx.doi.org/ 10.1242/jcs.184309 [DOI] [PubMed] [Google Scholar]
- [91].Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, et al.. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Medi 2012; 4:144ra03; http://dx.doi.org/ 10.1126/scitranslmed.3003802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nikolova-Krstevski V, Leimena C, Xiao X-H, Kesteven S, Tan JC, Yeo LS, Yu Z-Y, Zhang Q, Carlton A, Head S, et al.. Nesprin-1 and actin contribute to nuclear and cytoskeletal defects in lamin A/C-deficient cardiomyopathy. J Mol Cell Cardiol 2011; 50:479-86; PMID:21156181; http://dx.doi.org/ 10.1016/j.yjmcc.2010.12.001 [DOI] [PubMed] [Google Scholar]
- [93].Chen SC, Kennedy BK, Lampe PD. Phosphorylation of connexin43 on S279/282 may contribute to laminopathy-associated conduction defects. Exp Cell Res 2013; 319:888-96; PMID:23261543; http://dx.doi.org/ 10.1016/j.yexcr.2012.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Frock RL, Chen SC, Da D-F, Frett E, Lau C, Brown C, Pak DN, Wang Y, Muchir A, Worman HJ, et al.. Cardiomyocyte-specific expression of lamin a improves cardiac function in lmna(−/−) mice. PloS One 2012; 7:e42918; PMID:22905185; http://dx.doi.org/ 10.1371/journal.pone.0042918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, et al.. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol 2008; 44:293-303; PMID:18182166; http://dx.doi.org/ 10.1016/j.yjmcc.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Chandar S, Yeo LS, Leimena C, Tan J-C, Xiao X-H, Nikolova-Krstevski V, Yasuoka Y, Gardiner-Garden M, Wu J, Kesteven S, et al.. Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circulat Res 2010; 106:573-82; PMID:20019332; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.204388 [DOI] [PubMed] [Google Scholar]
- [97].Cupesi M, Yoshioka J, Gannon J, Kudinova A, Stewart CL, Lammerding J. Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J Mol Cell Cardiol 2010; 48:1290-7; PMID:19913544; http://dx.doi.org/ 10.1016/j.yjmcc.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet 2013; 22:2335-49; PMID:23427149; http://dx.doi.org/ 10.1093/hmg/ddt079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bhattacharjee P, Banerjee A, Banerjee A, Dasgupta D, Sengupta K. Structural alterations of Lamin A protein in dilated cardiomyopathy. Biochemistry 2013; 52:4229-41; PMID:23701190; http://dx.doi.org/ 10.1021/bi400337t [DOI] [PubMed] [Google Scholar]
- [100].West G, Gullmets J, Virtanen L, Li SP, Keinanen A, Shimi T, Mauermann M, Helio T, Kaartinen M, Ollila L, et al.. Deleterious assembly of mutant p.S143P lamin A/C causes ER stress in familial dilated cardiomyopathy. J Cell Sci 2016; 129(14):2732-43; PMID:27235420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Banerjee A, Rathee V, Krishnaswamy R, Bhattacharjee P, Ray P, Sood AK, Sengupta K. Viscoelastic behavior of human lamin A proteins in the context of dilated cardiomyopathy. PLoS One 2013; 8:e83410; PMID:24386194; http://dx.doi.org/ 10.1371/journal.pone.0083410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem 2006; 281:25768-80; PMID:16825190; http://dx.doi.org/ 10.1074/jbc.M513511200 [DOI] [PubMed] [Google Scholar]
- [103].Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A / C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113:370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol 2005; 170:781-91; PMID:16115958; http://dx.doi.org/ 10.1083/jcb.200502148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lammerding J, Lee RT. The nuclear membrane and mechanotransduction: impaired nuclear mechanics and mechanotransduction in lamin A/C deficient cells. Novartis Found Symp 2005; 264:264-73; discussion 73–8; PMID:15773759; http://dx.doi.org/ 10.1002/0470093765.ch18 [DOI] [PubMed] [Google Scholar]
- [106].Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113:370-8; PMID:14755334; http://dx.doi.org/ 10.1172/JCI200419670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Siu CW, Lee YK, Ho JC, Lai WH, Chan YC, Ng KM, Wong LY, Au KW, Lau YM, Zhang J, et al.. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging (Albany NY) 2012; 4:803-22; PMID:23362510; http://dx.doi.org/ 10.18632/aging.100503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, et al.. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; PMID:12702809; http://dx.doi.org/ 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- [109].De Sandre-Giovannoli A, Levy N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog Mol Subcell Biol 2006; 44:199-232; PMID:17076270; http://dx.doi.org/ 10.1007/978-3-540-34449-0_9 [DOI] [PubMed] [Google Scholar]
- [110].De Sandre-giovannoli AD, Cau P, Navarro C, Amiel J, Lyonnet S, Stewart CL, Munnich A, Merrer ML, Le N. Lamin A Truncation in Hutichnson-Gilford Progeria. Science 2003; 300:21702; http://dx.doi.org/ 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- [111].Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al.. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293-8; PMID:12714972; http://dx.doi.org/ 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Shackleton S, Smallwood DT, Clayton P, Wilson LC, Agarwal aK, Garg a, Trembath RC. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet 2005; 42:e36; PMID:15937076; http://dx.doi.org/ 10.1136/jmg.2004.029751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010; 121:2200-10; PMID:20458013; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.109.902056 [DOI] [PubMed] [Google Scholar]
- [114].Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, et al.. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2004; 101:8963-8; PMID:15184648; http://dx.doi.org/ 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci 2006; 119:4644-9; PMID:17062639; http://dx.doi.org/ 10.1242/jcs.03263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J 2008; 22:603-11; PMID:17848622; http://dx.doi.org/ 10.1096/fj.07-8598com [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, JD Huang, Li KM, Chau PY, Chen DJ, et al.. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11:780-5; PMID:15980864; http://dx.doi.org/ 10.1038/nm1266 [DOI] [PubMed] [Google Scholar]
- [118].Misteli T, Scaffidi P. Genome instability in progeria: when repair gets old. Nat Med 2005; 11:718-9; PMID:16015360; http://dx.doi.org/ 10.1038/nm0705-718 [DOI] [PubMed] [Google Scholar]
- [119].Cobb AM, Larrieu D, Warren DT, Liu Y, Srivastava S, Smith AJ, Bowater RP, Jackson SP, Shanahan CM. Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging cell 2016; 15:1039–1050. PMID:27464478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, et al.. Cardiovascular Pathology in Hutchinson-Gilford Progeria: Correlation With the Vascular Pathology of Aging. Arterioscler Thromb Vasc Biol 2010; 30:2301-9; PMID:20798379; http://dx.doi.org/ 10.1161/ATVBAHA.110.209460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Gerhard-Herman M, Smoot LB, Wake N, Kieran MW, Kleinman ME, Miller DT, Schwartzman A, Giobbie-Hurder A, Neuberg D, Gordon LB. Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 2012; 59:92-7; PMID:22083160; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.111.180919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C, Rivera J, Tazi J, Guzman G, Varela I, et al.. Splicing-Directed Therapy in a New Mouse Model of Human Accelerated Aging. Sci Transl Med 2011; 3:106ra7-ra7; http://dx.doi.org/ 10.1126/scitranslmed.3002847 [DOI] [PubMed] [Google Scholar]
- [123].Pendás AM, Zhou Z, Cadiñanos J, Freije JMP, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, et al.. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94-9 [DOI] [PubMed] [Google Scholar]
- [124].Davies BSJ, Barnes RH, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG, et al.. An accumulation of non-farnesylated prelamin A causes cardiomyopathy but not progeria. Hum Mol Genet 2010; 19:2682-94; PMID:20421363; http://dx.doi.org/ 10.1093/hmg/ddq158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Brodsky GL, Bowersox JA, Fitzgerald-miller L, Miller LA, Maclean KN. The prelamin A pre-peptide induces cardiac and skeletal myoblast differentiation. Biochem Biophys Res Commun 2007; 356:872-9; PMID:17389141; http://dx.doi.org/ 10.1016/j.bbrc.2007.03.062 [DOI] [PubMed] [Google Scholar]
- [126].Cattin ME, Muchir A, Bonne G. 'State-of-the-heart' of cardiac laminopathies. Curr Opin Cardiol 2013; 28:297-304; PMID:23455585; http://dx.doi.org/ 10.1097/HCO.0b013e32835f0c79 [DOI] [PubMed] [Google Scholar]
- [127].Lu JT, Muchir A, Nagy PL, Worman HJ. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Models Mech 2011; 4:562-8; PMID:21810905; http://dx.doi.org/ 10.1242/dmm.006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Scharner J, Figeac N, Ellis JA, Zammit PS. Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon-skipping therapy for laminopathies. Gene Ther 2015; 22:503-15; PMID:25832542; http://dx.doi.org/ 10.1038/gt.2015.8 [DOI] [PubMed] [Google Scholar]
- [129].Lee JM, Nobumori C, Tu Y, Choi C, Yang SH, Jung HJ, Vickers TA, Rigo F, Bennett CF, Young SG, et al.. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest 2016; 126:1592-602; PMID:26999604; http://dx.doi.org/ 10.1172/JCI85908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, et al.. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2012; 109:16666-71; PMID:23012407; http://dx.doi.org/ 10.1073/pnas.1202529109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gordon LB, Kleinman ME, Massaro J, RB D'Agostino Sr, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, et al.. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation 2016; 134:114-25; PMID:27400896; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.116.022188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Ibrahim MX, Sayin VI, Akula MK, Liu M, Fong LG, Young SG, Bergo MO. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013; 340:1330-3; PMID:23686339; http://dx.doi.org/ 10.1126/science.1238880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 2014; 344:527-32; PMID:24786082; http://dx.doi.org/ 10.1126/science.1252651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Rankin J, Auer-Grumbach M, Bagg W, Colclough K, Nguyen TD, Fenton-May J, Hattersley A, Hudson J, Jardine P, Josifova D, et al.. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet Part A 2008; 146A:1530-42; PMID:18478590; http://dx.doi.org/ 10.1002/ajmg.a.32331 [DOI] [PubMed] [Google Scholar]