

Here, de Lange and colleagues report that PHF11 (plant homeodomain finger 11) encodes a previously unknown DNA damage response factor involved in 5′ end resection, ATR signaling, and homologous recombination. Biochemical experiments demonstrated that PHF11 stimulates EXO1 by overcoming its inhibition by RPA, suggesting that PHF11 acts (in part) by promoting 5′ end resection at RPA-bound sites of DNA damage.
Keywords: PHF11, EXO1, RPA, ATR, DSB, resection, homologous recombination
Abstract
Resection of double-strand breaks (DSBs) plays a critical role in their detection and appropriate repair. The 3′ ssDNA protrusion formed through resection activates the ATR-dependent DNA damage response (DDR) and is required for DSB repair by homologous recombination (HR). Here we report that PHF11 (plant homeodomain finger 11) encodes a previously unknown DDR factor involved in 5′ end resection, ATR signaling, and HR. PHF11 was identified based on its association with deprotected telomeres and localized to sites of DNA damage in S phase. Depletion of PHF11 diminished the ATR signaling response to telomere dysfunction and genome-wide DNA damage, reduced end resection at sites of DNA damage, resulted in compromised HR and misrejoining of S-phase DSBs, and increased the sensitivity to DNA-damaging agents. PHF11 interacted with the ssDNA-binding protein RPA and was found in a complex with several nucleases, including the 5′ dsDNA exonuclease EXO1. Biochemical experiments demonstrated that PHF11 stimulates EXO1 by overcoming its inhibition by RPA, suggesting that PHF11 acts (in part) by promoting 5′ end resection at RPA-bound sites of DNA damage. These findings reveal a role for PHF11 in DSB resection, DNA damage signaling, and DSB repair.
Nuclear double-strand breaks (DSBs) activate the ATM and ATR kinase-dependent DNA damage response (DDR) pathways (for review, see Ciccia and Elledge 2010). Whereas the ATM kinase responds to the presence of dsDNA ends, activation of the ATR kinase requires the presence of ssDNA that is bound by RPA. In addition, activation of the ATR kinase requires the loading of the 9-1-1 complex on the double-stranded–single-stranded junction formed after 5′ end resection. Resection of the 5′ end of DSBs is therefore a critical step in the activation of ATR signaling.
DSB resection is also required for the initiation of several DNA repair pathways, including homologous recombination (HR) and single-strand annealing (SSA) (for review, see Symington and Gautier 2011). HR can restore the original DNA sequence using the sister chromatid as a template for repair. After 5′ end resection, HR is initiated by the BRCA2-mediated loading of the Rad51 recombinase onto the ssDNA. In the absence of HR, S-phase DSBs can become a substrate for more error-prone DSB repair, including SSA and classical or alternative nonhomologous end-joining (NHEJ).
DSB resection is initiated through the agency of BRCA1, the MRE11/RAD50/NBS1 (MRN) complex, and CtIP (for review, see Cejka 2015). The outcome is a ssDNA end with a short (∼20-nucleotide [nt]) 3′ overhang that is a substrate for further nucleolytic attack by EXO1, a dsDNA exonuclease that degrades only the 5′ strand to leave a 3′ overhang that can be thousands of nucleotides in length. In addition, an overlapping pathway involving the DNA2 nuclease acting in conjunction with the BLM and WRN RecQ helicases promotes the formation of the extended 3′ overhang (Nimonkar et al. 2011; Sturzenegger et al. 2014).
DSB resection is strictly regulated by a cell cycle-dependent competition between BRCA1 and RIF1, a regulator of resection bound to the DDR factor 53BP1 (for review, see Panier and Durocher 2013; Panier and Boulton 2014; Zimmermann and de Lange 2014). In S phase, BRCA1/CtIP-dependent resection prevails, whereas in G1, DSB resection is blocked by RIF1, thus linking the choice between NHEJ and HR to cell cycle stage.
Telomeres are a versatile model system to understand events at DNA ends. The telomeric DNA resembles a DSB with a 50- to 300-nt 3′ overhang and can therefore activate many DDR pathways, including ATM and ATR signaling, HR, and NHEJ. The telomere-specific shelterin complex represses the DDR through multiple mechanisms involving different shelterin subunits (for review, see Palm and de Lange 2008). On the one hand, the shelterin subunit TRF2 blocks c-NHEJ and ATM kinase activation, whereas the POT1 proteins in shelterin (POT1a and POT1b in mice) prevent activation of the ATR kinase at chromosome ends. Another component of shelterin, TRF1, is required to prevent replication fork stalling in the telomeric DNA (Martinez et al. 2009; Sfeir et al. 2009).
In addition to repressing ATR signaling, the POT1 proteins protect telomeres from hyperresection of the 5′ end of the telomere (Hockemeyer et al. 2006). As is the case for DSBs, EXO1 contributes to the hyperresection of the 5′ ends of dysfunctional telomeres that lack POT1a and POT1b (Wu et al. 2012; Kibe et al. 2016). However, the initial resection at telomeres does not require the CtIP/MRN processing that takes place at DSBs but is executed by the TRF2-bound nuclease Apollo (Lam et al. 2010; Wu et al. 2010, 2012; Kibe et al. 2016).
Due to the functional compartmentalization within shelterin, deletion of individual shelterin subunits activates distinct DDR signaling and repair pathways at chromosome ends. This system has allowed the identification of several novel aspects of the DDR (Zimmermann et al. 2013; Lottersberger et al. 2015; Kibe et al. 2016). Here we used dysfunctional telomeres to identify PHF11 (plant homeodomain finger 11) as a DDR factor. PHF11 is frequently methylated in Ewing's sarcoma (Alholle et al. 2013), codeleted with a cluster of genes in CLL (Parker et al. 2011), and deleted in 10%–20% of prostate cancers (cBioPortal for Cancer Genomics, http://www.cbioportal.org). PHF11 is most abundant in lymphocytes and is thought to act as a transcription factor that promotes class switching in the IgE locus in B cells (Clarke et al. 2008; Ikari et al. 2014). Polymorphisms in PHF11 are associated with atopic dermatitis (Zhang et al. 2003). However, PHF11 had not been implicated in the DDR. Our data reveal a role for PHF11 in DSB resection, ATR signaling, and DSB repair.
Results
Identification of PHF11 as a DDR factor
To identify new DDR factors, we used PICh (proteomics of isolated chromatin segments) (Dejardin and Kingston 2009) to isolate proteins enriched at telomeres lacking TRF2, which are a proxy for DSBs (Palm and de Lange 2008). PICh nominated ∼80 proteins that were enriched more than twofold at deprotected mouse telomeres (Supplemental Table S1). Comparison with the CRAPome (Mellacheruvu et al. 2013) showed that a large fraction of the candidates is likely contaminants (Supplemental Table S1), as had been noted previously (Bartocci et al. 2014). One protein, PHF11, was not an obvious contaminant. The PHF11 gene is found only in vertebrates and encodes a broadly expressed 331-amino-acid protein containing an extended PHD (plant homeodomain) finger (ePHD) and an NLS (nuclear localization signal) (Supplemental Fig. S1).
To determine whether PHF11 is a DDR factor, we examined the immunofluorescence (IF) pattern of a myc-tagged version of PHF11 before and after induction of DSBs with ionizing radiation (IR) (Fig. 1A). IR-induced PHF11 foci became prominent only at ∼2 h after irradiation, showing a delay compared with the formation of γH2AX foci (Fig. 1A,B). The focus formation by PHF11 followed a time course similar to that of RPA (Fig. 1B), suggesting that PHF11, like RPA, may be involved in a later step of the response to DSBs, perhaps when ssDNA is formed through initial resection and the ATR kinase is activated. Like RPA, PHF11 formed readily detectable foci in S phase but not in G1 (Fig. 1C) and accumulated at sites of replication stress induced by hydroxyurea (HU) or aphidicolin (Aphi), at lesions induced by camptothecin (CPT), and at telomeres lacking the protection of the shelterin proteins POT1a and POT1b (Fig. 1D,E; Supplemental Fig. S2A–E). Furthermore, like RPA, PHF11 became chromatin-bound after treatment with HU (Supplemental Fig. S2F). PHF11 also localized to telomeres lacking TRF1, which experience replication problems that lead to activation of ATR signaling (Supplemental Fig. S2G–J; Sfeir et al. 2009).
Figure 1.

PHF11 localizes to sites of DNA damage. (A) Myc and γ-H2AX IF on mouse embryonic fibroblasts (MEFs) expressing myc-tagged PHF11 (N-terminal tag) or RPA32 (C-terminal tag) at the indicated times after 3 Gy of IR. (B) Kinetics of PHF11 and RPA32 focus formation compared with γH2AX as in A. (C) IF for myc-PHF11 and 53BP1 foci in asynchronous (Asyn) and G1 cells at 2 h after 3 Gy of IR. G1 cells were collected after mitotic shake-off and plated for 4 h, with IR treatment for the last 2 h. FACS profiles of BrdU-labeled cells (30-min pulse) showed that the G1 population had 0% S-phase cells, whereas the asynchronous population had 30% S-phase cells. (D) PHF11 foci induced by 10 µg/mL Aphi for 6 h, 5 mM HU for 6 h, mitomycin C (MMC) for 6 h after 0.4 µg/mL for 2 h, or CPT for 6 h after 1 µM for 2 h. (E) PHF11 foci at telomeres in the indicated MEFs lacking POT1a/b at 72 h after Cre. (Left) IF for myc-PHF11 (red) and 53BP1 (green). (Right) IF-FISH for myc-PHF11 (red) and telomeric TTAGGG DNA (green). (Blue) DAPI DNA stain; (S) stop allele; (F) floxed allele. (F) Colocalization of RPA32-myc and HA-PHF11 (FH2-PHF11) at Aphi-induced sites of damage and at telomeres lacking POT1a/b. Cells as in E without Cre treatment (control, Aphi) or at 72 h after Cre (ΔPOT1a/b). (G) Effect of ATR on PHF11 localization to telomeres lacking POT1a/b using IF-FISH as in E at 72 h after Cre. (H) Quantification of TIFs (telomere dysfunction-induced foci) (Takai et al. 2003) formed by 53BP1 and PHF11 in ATR-proficient and ATR-deficient MEFs lacking POT1a/b at their telomeres. Averages and SDs are from three independent experiments. (****) P < 0.0001, unpaired Student's t-test.
These data showed that PHF11 localizes to a variety of DNA damage sites where RPA accumulates and ATR is activated. Nonetheless, PHF11 also localized to telomeres lacking TRF2, which do not activate ATR signaling (Supplemental Fig. S2K,L). However, in this setting, the localization was observed in only a small fraction (20%) of the cells and at only a few telomeres (Supplemental Fig. S2M,N). The accumulation of PHF11 at some of the telomeres lacking TRF2 is consistent with the identification of PHF11 in the PICh experiment. In the absence of TRF2, telomeres undergo limited ATM/CtIP-dependent resection but do not activate ATR and are not likely to contain RPA, since the telomeres retain the POT1 proteins, which exclude RPA from the single-stranded telomeric DNA (Denchi and de Lange 2007; Dimitrova and de Lange 2009; Gong and de Lange 2010; Lottersberger et al. 2013). Thus, at some of the telomeres lacking TRF2, the PHF11 foci are detectable despite the presumed absence of RPA and ATR signaling.
Recruitment of PHF11 to sites of DNA damage does not require ATR signaling
To determine whether the ATR signaling affected the formation of PHF11 foci, we used conditional knockout mouse embryonic fibroblasts (MEFs) from which ATR can be codeleted with POT1a and POT1b. Codeletion of ATR and POT1a/b showed the anticipated defect in the accumulation of 53BP1 at dysfunctional telomeres (Fig. 1H; Supplemental Fig. S3). In contrast, PHF11 bound the dysfunctional telomeres even in the absence of ATR (Fig. 1G,H; Supplemental Fig. S3A–E). However, the frequency of nuclei with a large number of telomeric PHF11 foci was slightly diminished (Supplemental Fig. S3E), suggesting a minor contribution of ATR signaling to PHF11 focus formation. Similarly, the recruitment of PHF11 to HU-induced sites of DNA damage was largely unaltered by the absence of ATR (Supplemental Fig. S3F,G). Thus, while ATR signaling may slightly enhance PHF11 focus formation, ATR is not required for PHF11 accumulation at sites of DNA damage. Tests for the possible involvement of other DDR factors (RPA, MRE11, EXO1, BRCA1, CtIP, PARP1, and Ku70/80) in the recruitment of PHF11 yielded negative results (Supplemental Fig. S4A–H).
PHF11 depletion diminishes ATR signaling
To determine whether PHF11 affects ATR signaling, we used CRISPR/Cas9 to knock out PHF11 in human epithelial RPE1/hTERT cells lacking p53/Rb and knockdown of PHF11 with shRNAs in MEFs (Supplemental Fig. S5A–F). Deletion of PHF11 from RPE1 cells led to a decrease in Chk1 phosphorylation after treatment with CPT, and this diminished ATR response was alleviated by complementation of PHF11 knockout cells with exogenous PHF11 (Fig. 2A). Immunoblotting showed that the expression of a number of DNA damage factors was unaffected by the deletion of PHF11 and that CPT-treated PHF11 knockout cells showed the increase in BLM helicase expected from their prolonged S phase (Fig. 2A). Based on the phosphorylation of Chk1, a similar effect of PHF11 deficiency on ATR signaling was observed in cells treated with IR or HU (Fig. 2B,C). Also, in this case, complementation with PHF11 restored the phosphorylation of Chk1 to normal levels (Fig. 2B,C; Supplemental Fig. S5D). In contrast to its effect on ATR signaling, PHF11-deficient cells showed an unaltered ATM response after IR (Fig. 2B,C). Furthermore, shRNA-mediated knockdown of PHF11 in mouse cells led to a significant decrease in formation of the ATR-dependent γH2AX, 53BP1, and MDC1 foci at telomeres lacking POT1a/b, and this effect was negated by expression of an shRNA-resistant form of PHF11 (Fig. 2D,E; Supplemental Fig. S5G). Cell cycle analysis showed that the effects of PHF11 deletion or depletion could not be ascribed to diminished S-phase index (Supplemental Fig. S5C,F).
Figure 2.

PHF11 promotes ATR signaling. (A) Immunoblotting for the effect of PHF11 deletion on Chk1 phosphorylation and expression of DDR factors. The indicated RPE1 cell lines lacking PHF11 (sg1 and sg2) with or without complementation with FH2-PHF11 were tested alongside the parental cells with and without CPT treatment. (B) Immunoblot for the effect of PHF11 knockout on Chk1-P and Chk2-P in response to HU. Cells were as in A. (C) Immunoblot for the effect of PHF11 knockout on phosphorylation of Chk1 and Chk2 after IR. Cells were as in A. (D) PHF11 shRNA knockdown diminishes the DDR at telomeres lacking POT1a/b. IF for γH2AX, MDC1, and 53BP1 (red) and TTAGGG FISH for telomeric DNA (green) in POT1a/b double-knockout (DKO) MEFs at 72 h after Cre. (Blue) DAPI DNA stain. (Fourth panel) Cells complemented with shRNA-resistant FH2-PHF11. (E) Quantification of the TIF response as in D. Average values and SDs are from three independent experiments. (****) P < 0.0001; (*) P < 0.05, unpaired Student's t-test.
PHF11 promotes resection at DNA ends
To understand the mechanism by which PHF11 depletion reduced ATR signaling, we examined the resection at DNA ends based on the accumulation of RPA at DNA lesions. RPE1 cells lacking PHF11 showed a significant reduction in accumulation of RPA after treatment with IR, and, as expected, this effect was observed in cells undergoing DNA replication (Fig. 3A–C). Complementation of the PHF11 knockout cells with exogenous PHF11 reversed the effect (Fig. 3A,C).
Figure 3.

PHF11 affects formation of ssDNA at sites of DNA damage. (A) IF for endogenous RPA32 and 53BP1 in the indicated RPE1 cells with and without PHF11 at 4 h after 3 Gy of IR. Cells are as in Figure 2A. (B) IF for endogenous RPA in wild-type and PHF11 knockout cells at 4 h after 3 Gy of IR. Cells were incubated with 10 mM EdU for 4 h after IR to identify S-phase cells with IR-induced RPA foci. Percentages of cells with the shown EdU pattern that contain >10 RPA foci are shown below the images. (C) Quantification of IR-induced RPA32 foci in cells with and without PHF11 (as in A). Average values and SDs are from three independent experiments. (****) P < 0.0001; (***) P < 0.001; (**) P < 0.01, unpaired Student's t-test. (D) PHF11 shRNA knockdown reduces accumulation of RPA at sites of DNA damage. MEFs expressing RPA32-myc examined by IF at 72 h after PHF11 shRNA infection. (Left) RPA foci (Myc IF; red) at telomeres (FISH; green) lacking POT1a/b at 72 h after Cre. (Middle and right) RPA foci (Myc IF; red) colocalizing with 53BP1 (53BP1 IF; green) after HU or IR treatment as indicated in cells not treated with Cre. (E) Examples of the effect of PHF11 depletion on RPA32-myc accumulation at IR-induced DSBs at the indicated time points after 10 Gy of IR (as in C). (F) Effect of PHF11 shRNA on hyperresection at telomeres lacking POT1a/b. Example of an in-gel assay for single-stranded telomeric DNA after PHF11 knockdown at the indicated times after Cre treatment of POT1a/1b double-knockout (DKO) MEFs. (G) Quantification of the telomeric overhang signals assayed as in F with two distinct shRNAs. Single-stranded TTAGGG signals were normalized to the total TTAGGG signal in the same lane. The normalized signal for cells lacking PHF11 shRNA and not treated with Cre was set to 1, and the other values are given relative to this value. Averages are from three independent experiments and SDs. (**) P < 0.01, unpaired Student's t-test. (H) Detection of BrdU in native DNA after treatment with HU in cells infected with PHF11sh9 (two examples shown) and the vector control. Cells were labeled with BrdU for 24 h and then treated with 2 mM HU for 4 h before processing for α-BrdU IF in native DNA. (I) Quantification of the effect of PHF11 depletion on detection of BrdU in native DNA after HU treatment. Assay is as in H. Values represent averages and SDs from three independent experiments. (*) P < 0.05, unpaired Student's t-test.
Similarly, when PHF11 was depleted from POT1a/b double-knockout cells, which undergo extensive resection of the telomeric 5′ end, the formation of RPA foci at telomeres was substantially diminished (Fig. 3D). Furthermore, the RPA foci in MEFs treated with HU or IR were decreased (Fig. 3D,E).
DNA end resection can be assayed in a quantitative manner at telomeres lacking POT1a and POT1b. Such POT1a/b-depleted telomeres activate the ATR kinase signaling pathway and undergo resection that is mediated by EXO1 as well as other nucleases (Kibe et al. 2016). This telomere hyperresection is readily detectable based on the increase in the amount of single-stranded TTAGGG repeats at telomeres. The relative abundance of the single-stranded TTAGGG repeats can be quantified using in-gel hybridization of a probe for the C-rich telomeric sequence to native DNA and subsequent detection of the total telomeric DNA in the same lane. PHF11 depletion with two distinct shRNAs resulted in a significant reduction in the ssDNA at dysfunctional telomeres lacking POT1a and POT1b, whereas PHF11 did not appear to affect functional telomeres (Fig. 3F,G).
In addition, PHF11 depletion diminished the appearance of ssDNA after HU treatment, as measured based on detection of BrdU in native DNA (Fig. 3H,I). Collectively, these data suggest that PHF11 promotes DSB resection in vivo.
PHF11 contributes to HR and resistance to DNA damage
We next determined whether deletion or depletion of PHF11 affects HR of DSBs as would be expected from its ability to promote resection. Indeed, RPE1 cells lacking PHF11 showed a reduced ability to execute HR based on the DR-GFP assay (Fig. 4A; Pierce et al. 1999). Expression of exogenous PHF11 complemented the HR defect in the RPE1 knockout cells (Fig. 4A).
Figure 4.

PHF11 affects DSB repair. (A) Effect of PHF11 deletion on HR efficiency measured using the DR-GFP reporter assay. The indicated cells containing the DR-GFP substrate were infected with lentiviral-I-SceI, and GFP+ cells were scored after 48 h. The percentage of GPF+ cells from PHF11 knockout cell lines sg1 and sg2 with and without complementation with FH2-PHF11 were normalized to the value for wild-type cells (set at 100%). Averages are from three independent experiments and SDs. (****) P < 0.0001; (***) P < 0.001, unpaired Student's t-test. (B) Metaphase spreads from MEFs treated with 5 µM olaparib for 18 h after treatment with PHF11 or BRCA1 shRNAs. Examples of misrejoined chromosomes are highlighted with red arrows. (C) Quantification of chromosomal abnormalities in cells as in B. (**) P < 0.01. n = 3. (D) Distributions of the frequency of chromosomal aberration (chromosome and chromatid breaks, fragments, and misrejoined chromosomes) in cells as in B and the vector control. (E) Survival of the indicated cell lines with and without PHF11 after treatment with CPT, IR, and MMC. Cells were treated in triplicate with the indicated concentrations/doses of CPT, MMC, or IR.
The efficiency of HR determines the ability of cells to efficiently repair S-phase DSBs induced by the PARP1 inhibitor olaparib (Bryant et al. 2005; Farmer et al. 2005). When HR is diminished, olaparib treatment results in unrepaired lesions, and NHEJ of these DSBs can create misrejoined chromosomes. Consistent with a role for PHF11 in resection and HR, depletion of PHF11 from MEFs resulted in a significant level of misrejoined and unrepaired lesions, although not to the same level as when BRCA1 is depleted (Fig. 4B–D).
Consistent with a role for PHF11 in DSB repair by HR, PHF11-deficient RPE1 cells showed a greater sensitivity to CPT, mitomycin C (MMC), and IR, and this effect was negated by complementation of the knockout cells with PHF11 (Fig. 4E).
PHF11 interacts with several resection factors
Given the role of PHF11 in resection, ATR signaling, and DSB repair, we searched for PHF11-interacting proteins with relevance to resection. Immunoprecipitation experiments with Flag-HA2-tagged full-length human PHF11 (FH2-hPHF11) transfected into 293T cells showed that PHF11 interacted with endogenous RPA32, the MRN complex, EXO1, DNA2, and the BRCA1 partner BARD1 but not CtIP (Fig. 5A,B). Benzonase was included in the coimmunoprecipitations to suppress spurious interactions mediated by nucleic acids. The interaction with each of these proteins was abrogated when the C terminus of PHF11 was removed (Fig. 5B). This domain has the ability to mediate homodimerization of PHF11 and is required for its association with chromatin (Fig. 5C,D), two features that might be relevant to the interaction with other proteins. The interaction of PHF11 with RPA32 was severely diminished by deletion of the C-terminal half of the ePHD finger domain, whereas all other interacting factors were more reliant on the N-terminal half of this domain (Fig. 5B).
Figure 5.
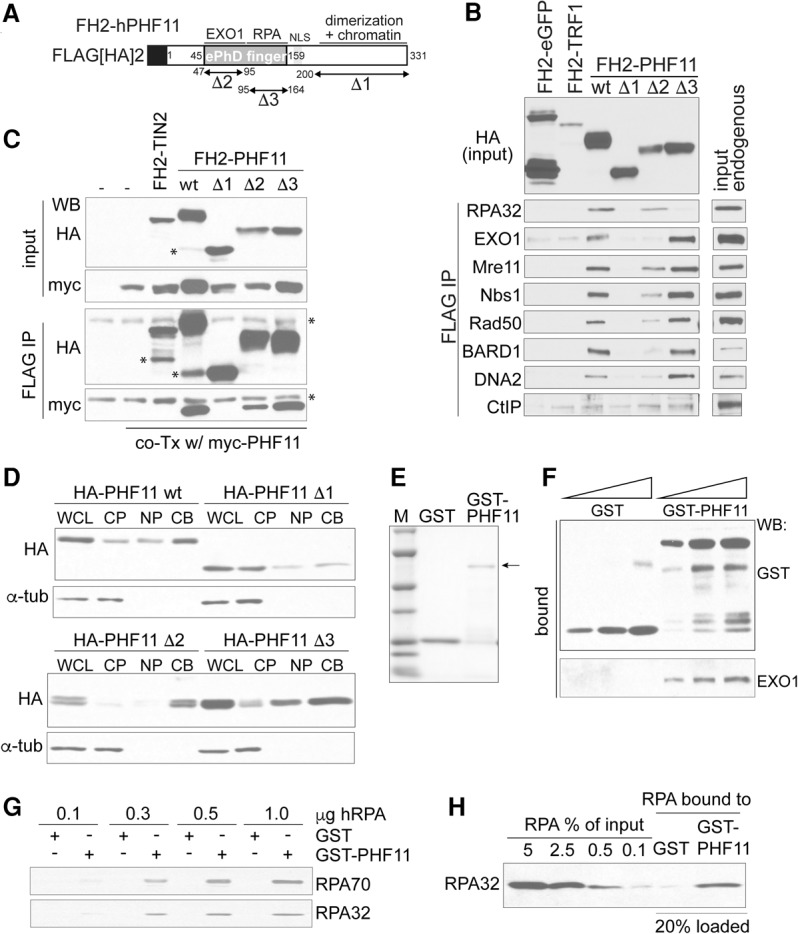
PHF11 interacts with RPA and resection factors. (A) Schematic of PHF11 and deletion mutants. (B) Coimmunoprecipitation of the indicated endogenous proteins with FH2-PHF11 (wild-type and the indicated mutants) transfected into 293T cells. FH2-eGFP and FH2-TRF1 were used as negative controls. (C) Dimerization of PHF11 through its C terminus. FH2-PHF11 and the indicated deletion mutants were cotransfected with myc-tagged PHF11. The ability of FH2-PHF11 alleles to coimmunoprecipitate the myc-tagged PHF11 was evaluated by immunoblotting after HA immunoprecipitation. FH2-tagged TIN2 served as a negative control. Asterisks indicate degradation products and nonspecific bands. (D) Deletion of the C terminus of PHF11, but not the ePHD finger domain, leads to a defect in chromatin binding. MEFs expressing the indicated PHF11 mutants were fractionated ([WCL] whole-cell lysate; [CP] cytoplasmic proteins; [NP] nuclear proteins; [CB] chromatin bound) and analyzed by immunoblotting for PHF11 and α-tubulin. (E) Coommassie-stained gel of purified recombinant GST-PHF11 and the GST control. (F) Recombinant PHF11 binds recombinant EXO1. GST-PHF11 and the GST control were used to pull down purified recombinant human EXO1, and bound proteins were evaluated by immunoblotting. (G,H) Pull-down of recombinant RPA with GST-PHF11 and the GST control. RPA70 and RPA32 were detected by immunoblotting.
The interaction of PHF11 with RPA and EXO1 was verified using purified recombinant proteins, demonstrating that these associations are due to direct protein–protein interactions (Fig. 5E–H).
PHF11 promotes in vitro EXO1 activity in the presence of RPA
The interaction of PHF11 with RPA and EXO1 is particularly relevant to the effect of PHF11 on telomere hyperresection in cells lacking POT1a/b, where RPA is detected at telomeres (Gong and de Lange 2010), and EXO1 is implicated in the processing (Kibe et al. 2016). We therefore explored this interaction further by determining whether PHF11 affected DNA end resection by EXO1 in vitro.
Since previous reports noted the inhibition of EXO1 by RPA (Genschel and Modrich 2003; Myler et al. 2016), we first examined the role of PHF11 in the context of this RPA–EXO1 interplay. When dsDNA with a 100-nt 3′ overhang (the preferred entry site for EXO1) (Cannavo et al. 2013) was examined, RPA had a strong inhibitory effect on EXO1 (Fig. 6A,B). However, addition of PHF11 to the RPA-inhibited reactions restored resection by EXO1 (Fig. 6B–D). PHF11 concentrations of 4 and 7.9 nM were sufficient to stimulate EXO1 by approximately fourfold to fivefold; the end product of EXO1 resection in the presence of PHF11 was full-length ssDNA. PHF11 itself lacked nuclease activity (Fig. 6E) and had a negligible effect on resection of the tailed duplex DNA in the absence of RPA (Fig. 6F), although the distribution of product sizes was altered (Fig. 6B). The capacity of PHF11 to stimulate resection of tailed DNA complexed with RPA was more evident at lower concentrations of EXO1: A 10-fold stimulation was elicited by 20 nM PHF11 (Fig. 6D). The effect of PHF11 is unlikely to be due to removal of RPA from the EXO1 substrate, since the inhibition of EXO1 by RPA is not negated by PHF11 when the substrate is ssDNA (Fig. 6G).
Figure 6.

PHF11 negates the inhibition of EXO1 by RPA. (A) Schematic of DNA used for resection experiments in B–F. (B) RPA inhibits EXO1-dependent resection, and PHF11 overcomes this inhibition. Resection assays with 0.25 nM EXO1 and the indicated RPA and PHF11 concentrations. (C,D) Quantification of stimulation of EXO1 (two concentrations) by PHF11 in the presence of RPA. The mean and SE are from three independent experiments. (E) Resection assays without EXO1 either with or without RPA, showing that PHF11 is not contaminated with a nuclease. (F) Quantification of experiments done without RPA showing no effect of PHF11 on EXO1-dependent resection of the 3′ tailed DNA substrate. The mean and SE are from three independent experiments. Error bars are smaller than the symbols. (G) No effect of PHF11 on inhibition of EXO1 by RPA on ssDNA.
PHF11 can act in EXO1-independent pathways
To determine whether the effect of PHF11 on ATR signaling was dependent on its ability to promote EXO1-mediated resection, we used EXO1-deficient MEFs for epistasis analysis (Fig. 7A). The EXO1−/− MEFs showed diminished Chk1 phosphorylation after HU treatment, as expected from the role of EXO1 in end resection. However, the reduction was less than that observed with an shRNA to PHF11, suggesting that PHF11 affects additional pathways leading to ATR signaling. Importantly, EXO1 deficiency did not affect resection when PHF11 was depleted, indicating that EXO1 is largely dependent on PHF11.
Figure 7.
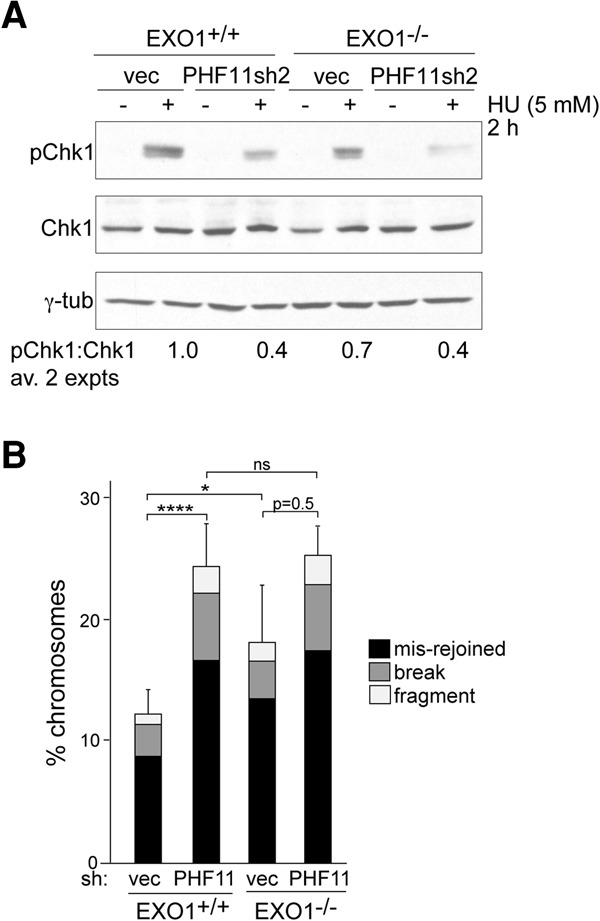
EXO1-independent effects of PHF11. (A) Immunoblot for the effect of PHF11 shRNA (sh2) knockdown in EXO1-proficient and EXO1-deficient MEFs on phosphorylation of Chk1 after HU. (B) Quantification of chromosomal abnormalities from EXO1-proficient and EXO1-deficient MEFs infected with vector or PHF11 sh2 after treatment with 2 µM olaparib for 18 h. Averages are from at least three independent experiments and SDs. (****) P < 0.0001; (*) P < 0.05, unpaired Student's t-test.
A similar conclusion emerged from the quantification of PARP inhibitor (PARPi)-induced chromosomal aberrations in EXO1-proficient and EXO1-deficient MEFs (Fig. 7B). In this setting, EXO1 deficiency significantly increased the frequency of chromosomal abnormalities. The effect of EXO1 deficiency was clearly less than that of PHF11 depletion, again pointing to additional pathways that are influenced by PHF11. Furthermore, in cells treated with PHF11 shRNA, EXO1 status did not affect the level of chromosomal abnormalities, consistent with the view that when PHF11 is compromised, EXO1 deficiency has no additional effect.
Discussion
These data establish that PHF11 promotes the resection of DNA ends in vitro and in vivo and contributes to ATR signaling and HR. The biochemical data implicate PHF11 as a factor needed to counteract the inhibition of EXO1 by RPA, but it is likely that PHF11 acts through multiple pathways. The role of PHF11 in overcoming the inhibition of EXO1 appears to be an elaboration of the regulation of DSB resection apparently not present in budding yeast, which lacks a PHF11 ortholog. Hyperresection could be more harmful in the vertebrate genomes, where repetitive sequences can lead to deletions and translocations. PHF11 may allow fine-tuning of hyperresection and thus greater protection against genome instability. It will be of interest to understand how the interplay between RPA, EXO1, and PHF11 is regulated and whether PHF11 is also involved in other resection pathways.
We were unable to determine how PHF11 is recruited to sites of DNA damage. One possibility is that PHF11 can be recruited through multiple pathways. For instance, our data do not exclude that recruitment of PHF11 could be mediated by chromatin modifications resulting from either ATM or ATR kinase signaling and also by PARP1-dependent PARsylation at sites of DNA damage. It is also possible that PHF11 binds directly to the sites of DNA damage (e.g., a DNA end or ssDNA), as is the case of the Ku70/80 heterodimer. However, such direct DNA binding is not sufficient to explain the formation of DNA damage foci by PHF11, which are readily detectable and thus likely contain a large number of PHF11 molecules. By comparison, Ku70/80 does not form readily detectable DDR foci at IR-induced DSBs.
A second unresolved issue is which resection pathways, other than those involving EXO1, are affected by PHF11. The effect of PHF11 depletion on ATR signaling and the correct repair PARPi-induced DSB is greater than that of loss of EXO1, arguing that PHF11 affects resection through a second pathway. Based on the interaction of PHF11 with DNA2, the MRN complex, and the BRCA1 partner BARD1, there are several possible settings in which PHF11 may act to promote resection. Further biochemical and genetic tests will be required to address these issues.
RPA is known to be a potent inhibitor and regulator of HR. A well-defined regulatory function of RPA in HR is manifest at the level of RAD51 filament formation: RPA binding to ssDNA prevents assembly of RAD51 nucleoprotein filaments, but this inhibition is alleviated by BRCA2 (Kowalczykowski 2015). In this regard, BRCA2 is serving as a mediator for RAD51 filament assembly (Jensen et al. 2010). Here we reveal another aspect of HR regulation by RPA, but, in this case, regulation is at the level DNA resection. In parallel to RAD51 filament formation, RPA blocks DNA degradation by EXO1, and PHF11 alleviates this inhibition. In this regard, PHF11 serves as a mediator of DNA resection. It remains to be determined how general this regulatory concept is for the DNA resection step of HR, but this idea also accommodates the role of PHF11 in processes beyond EXO1-dependent functions.
Materials and methods
PICh
PICh was performed as described (Dejardin and Kingston 2009). TRF2F/F Rosa Cre MEFs were either mock-treated (protected state) or incubated with 0.5 µM 4-hydroxytamoxifen (4-OHT) for 16 h to induce Cre activation (damaged state) and collected 72 h after addition of 4-OHT. For each sample, ∼2 × 109 MEFs were collected for PICh as described, except that nuclei were isolated (Dignam et al. 1983) before formaldehyde cross-linking, and 20 µM scrambled ssDNA (5′-GATGTGTGGATGTGGATGTGGATGTGG-3′) was added to decrease nonspecific binding during the hybridization step. Briefly, cells were harvested, washed with cold PBS, suspended in five packed cell volumes of buffer A (10 mM HEPES-KOH at pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5 mM PMSF, Roche Complete protease inhibitor), and incubated for 10 min on ice. The cell suspension was homogenized 15 times using a Wheaton homogenizer and centrifuged at 1000g for 10 min. The pellet was washed with buffer A and centrifuged again, and the resulting nuclei were cross-linked in PBS/3% formaldehyde for 30 min at room temperature.
Cell lines and expression constructs
SV40 large T antigen (SV40LT) immortalized TRF1F/F, POT1aS/FPOT1bS/F, Ku70−/−, and TRF2F/FRosaCre MEFs were reported previously (Celli et al. 2006; Hockemeyer et al. 2006; Sfeir et al. 2009; Doksani et al. 2013). SV40LT immortalized POT1aS/FPOT1bS/FATRF/F, ATRF/−, Mre11F/−, BRCA1F/F, and EXO1−/− MEFs were derived from embryonic day 12.5 (E12.5) embryos from appropriate crosses with previously reported mouse strains (Xu et al. 1999; Brown and Baltimore 2000; Wei et al. 2003; Hockemeyer et al. 2006; Deng et al. 2009). The PARP1-deficient MEFs were derived from Parp1tm1Zqw mice (The Jackson Laboratory, 002779). pBABE-myc-PHF11, pLPC-Flag-HA2-hPHF11 (FH2-hPHF11), and pWZL-HA-PHF11 were generated by PCR on full-length mouse PHF11 cDNA (Open Biosystems, MMM1013-99827056) or human PHF11 cDNA in the indicated vectors. PHF11 deletion mutants were generated by PCR using FH2-hPHF11. pWZL-RPA32-myc and pLPC-EXO1-myc encoding full-length human RPA32 and mouse EXO1 were cloned into pWZL-myc or pLPC-myc retroviral vectors, respectively. MEFs expressing PHF11 and RPA32 were generated by four retroviral infections at 12-h intervals and selected in 2–3 µg/mL puromycin after 3 d or 90 µg/mL hygromycin after 6 d.
shRNAs
Mouse PHF11 sh2 (CTCAGCCTTAAATGGAATCTT) was expressed from the pLKO.1 lentivirus (OpenBiosystems), and PHF11 sh9 (AATTCTTCAAGTTGATGGAAAC) was generated in pSuperior. For lentiviral infection, 5 × 106 293T cells were plated in 10-cm dishes 24 h before transfection and transfected with 15 µg of the appropriate plasmid DNA along with packaging plasmids (5 µg of pVSVg, 3 µg of pMDLg, 2.5 µg of pRSV) using calcium-phosphate coprecipitation. The medium was replaced after 6–8 h, and the first supernatant used for infection was harvested at 36 h. Cells were infected for a total of four to five infections at 12-h intervals. Experimental time points were defined with t = 0 set at 12 h after the first infection. For retroviral shRNA delivery, 3 × 106 Phoenix ecotropic packaging cells were plated in 10-cm dishes, and 25 µg of the appropriate plasmid DNA was introduced by calcium-phosphate coprecipitation. The retroviral infection schedules were as described for lentiviral delivery.
For quantification of mRNA transcript levels upon PHF11 knockdown, total RNA was extracted with the RNeasy kit (Qiagen) after 72 h with shRNA infection. cDNA was synthesized from 4 µg of RNA using the ThermoScript reverse transcriptase (Invitrogen). Quantitative PCR (qPCR) reactions were performed using Life Technologies SYBR Green Master Mix on an Applied Biosystems 7900HT sequence detection system. Differences between samples were calculated using QuantStudio software (Applied Biosystems) using the ΔCT method and were normalized to GAPDH. Primers used for qPCR were as follows: PHF11_FW, TGGGAAGAAAGGCAAAGGCA; PHF11_RV, ACAGCTTTTACTGGTGGTCCT; GAPDH_FW, GTGTTCCTACCCCCAATGTGT; and GAPDH _RV, ATTGTCATACCAGGAAATGAGCTT.
CRISPR/Cas9 gene editing
For CRISPR/Cas9 deletion of PHF11, the guide sequence was determined by ZiFit (http://zifit.partners.org): sgPHF11(1), 5′-TCATAAAAGAGGAGCCACCG-(PAM)-3′; and sgPHF11(2), 5′-GCAGTTCCACAGTCTGA-(PAM)-3′. Oligonucleotides were purchased from Sigma-Aldrich and cloned into the AflII-digested guide RNA (gRNA) expression vector (Addgene) by Gibson Assembly (New England Biolabs). The gRNA and hCas9 (Addgene) expression vectors were introduced by electroporation using the Kit R nucleofector kit (Lonza). Single-cell clones isolated with limiting dilution were screened by genomic DNA sequencing. TOPO-cloned PCR product (Invitrogen) was performed to verify gene editing (PHF11_fw, 5′-GAGTCCGAAAGAGAAATCACACC-3′; and PHF11_rv, 5′-TTATCCCAATCCCCCAAATGTG-3′).
Immunoblotting
Cells were lysed in 2× Laemmli buffer (100 mM Tris-HCl at pH 6.8, 200 mM dithiothreitol, 3% sodium dodecyl sulfate, 20% glycerol, 0.05% bromophenol blue), denatured for 5 min at 100°C, sheared with an insulin needle, fractionated on SDS-PAGE, and processed for immunoblotting. The antibodies used were BRCA1, McoI2 (a gift from Dr. D.M. Livingston), P-Chk2 (T68) (Cell Signaling Technology, 2661), mTRF1 (#1449), γ-tubulin (Sigma, clone GTU 488), myc (Cell Signaling Technology, 9B11), HA (Covance, HA.11), P-Chk1 (S345) (rabbit monoclonal; Cell Signaling Technology), Chk1 (Santa Cruz Biotechnology, sc-8408), Chk2 (mouse monoclonal; BD Biosciences), RPA32 (rabbit polyclonal; Bethyl Laboratories), RPA70 (rabbit polyclonal; Bethyl Laboratories), ATR (Santa Cruz Biotechnology, sc-1887), ATRIP (R&D Systems, AF1579), TopBP1 (rabbit polyclonal; Abcam), RAD1 (mouse monoclonal; Abcam), Rad9 (rabbit polyclonal; Abcam), Claspin (Bethyl Laboratories, A300-266A), Mre11 (Novus, NB100-142), RAD50 (Bethyl Laboratories, A300-184A), NBS1 (Abcam, 32074), Histone H3 (Abcam, ab1791), GST (Cell Signaling Technology, 2622), EXO1 (Santa Cruz Boitechnology, sc-199941; Abcam, ab95068), BARD1 (Millipore, ab10004), DNA2 (Abcam, ab96488), CtIP (Santa Cruz Biotechnology, sc-22838), BLM (Abcam, ab2179), and WRN (Abcam, ab200). Immunoblots for POT1a and POT1b were performed using a renaturation protocol (Loayza and de Lange 2003) with affinity-purified antibody against POT1a (#1221) and POT1b (#1223) (Hockemeyer et al. 2006). Secondary antibodies were horseradish peroxidase-conjugated donkey anti-mouse or anti-rabbit IgGs. Blots were developed with enhanced chemiluminescence (Amersham).
IF and IF-FISH
IF for γH2AX (Ab clone; Millipore, JBW301), 53BP1 (Abcam, ab175933), MDC1 (Ab clone; Millipore, P2B11), and RPA (Abcam, ab2175) was performed as described (Celli and de Lange 2005; Dimitrova et al. 2008). For IF staining of myc-PHF11, FH2-PHF11 (HA IF), and RPA32-myc (Gong and de Lange 2010), the in situ cell fractionation protocol was used (Mirzoeva and Petrini 2001). For IF-FISH staining, after the secondary antibody incubation and wash step, cells were fixed again with 2% paraformaldehyde for 5 min; dehydrated in 70%, 95%, and 100% ethanol for 5 min each; and allowed to dry completely. Hybridizing solution (70% formamide, 1 mg/mL blocking reagent [Roche], 10 mM Tris-HCl at pH 7.2, containing PNA probe FITC-OO-[CCCTAA]3 [Applied Biosystems]) was added to each coverslip, and the cells were denatured by heating for 10 min at 80°C on a heat block. After 2 h of incubation at room temperature in the dark, cells were washed twice with washing solution (70% formamide, 10 mM Tris-HCl at pH 7.2) for 15 min each and with PBS three times for 5 min each. DAPI (0.1 μg/mL) was added to the second PBS wash. Coverslips were sealed onto glass slides with embedding medium (ProLong Gold anti-fade reagent, Invitrogen). For EdU costaining, cells were incubated with RPA primary antibody using the in situ cell fractionation protocol. After the wash step, cells were processed for EdU detection (Click iT EdU imaging Alexa 647, Molecular Probes) based on manufacturer's instruction followed by incubation of FITC-conjugated secondary antibody against RPA and processing as above.
Cell cycle analysis
For FACS analysis, 1 × 106 cells were collected after a 1-h BrdU pulse, washed in PBS, fixed in ice-cold 70% ethanol by drop-wise mixing, and incubated for at least 24 h. Cells were recovered by centrifugation and resuspended in 0.5 mL of 0.5% BSA in PBS containing 5 µg of propidium-iodide and 100 µg of RNase A per milliliter. The samples were analyzed on a FACScalibur flow cytometer (Becton Dickinson) with gating. Data analysis was performed using FlowJo software. Mitotic shake-off to harvest G1 cells was performed as described previously (Gong and de Lange 2010).
Assay for telomere hyperresection
Mouse telomeric DNA was analyzed on CHEF gels as described previously (Celli and de Lange 2005). In-gel hybridization of the native DNA with 32P-γATP end-labeled [CCCTAA]4 oligonucleotides was performed, and the signal was captured. The gel was denatured in 0.5 M NaOH and 1.5 M NaCl for 30 min, neutralized twice (15 min each) with 0.5 M Tris-HCl (pH 7.5) and 3 M NaCl, prehybridized in Church mix for 1 h at 55°C, and hybridized overnight with the same probe, and the total TTAGGG signal was captured. The single-stranded signal was normalized to the total telomeric DNA in each lane. The normalized values were compared between samples.
Native BrdU staining
For detection of ssDNA generated after HU treatment, cells were preincubated with 10 μM BrdU for 24 h, washed three times with PBS, and subjected to 2 mM HU for 4 h. IF for BrdU was performed under nondenaturing conditions as described above with a primary antibody against BrdU (Millipore, MAB 3510) at 1:10,000 dilution.
DR-GFP assay
HR was assayed using the DR-GFP/I-SceI assay (Pierce et al. 1999). Nucleofection was used to introduce the DR-GFP targeting construct pHPRT-DRGFP (Addgene, 26476), and puromycin-resistant cells were selected. To measure GFP expression, cells were trypsinized and seeded at 1 × 106 to 1.5 × 106 cells per 10-cm dish. Twenty-four hours later, cells were infected with lentiviral I-SceI (pCDH1-CAG-I-SceI, a gift from Dr. Maria Jasin). After ∼48 h, cells were harvested and analyzed for GFP fluorescence on a C6 flow cytometer (Accuri) (>30,000 events per sample). Uninfected cells were used as a reference to calculate the basal level of GFP expression. GFP values were normalized to wild-type cells analyzed in parallel to derive the relative HR efficiency.
PARPi treatment and analysis of metaphase chromosomes
Cells were grown to ∼70% confluence on 10-cm dishes and incubated with 5 μM olaparib (Selleck Chemicals, AZD2281) for 16–18 h with addition of 0.2 µg/mL colcemid during the last hour. Cells were harvested by trypsinization, washed with medium containing serum and with PBS, swollen in 0.075 M KCl for 20 min at 37°C, fixed in ice-cold 3:1 methanol:acetic acid, and stored overnight at 4°C. Metaphase spreads were dropped onto slides, dried overnight, rehydrated in PBS, and stained with 0.25 μg/mL DAPI. Slides were dehydrated with 70%, 95%, and 100% ethanol for 5 min each and mounted in ProLong Gold anti-fade medium (Life Technologies).
Survival assays
Cells were plated in a 6-cm cell culture plate in triplicate at a density of 2 × 104 cells per dish. Twenty-four hours later, cells were subjected to different dosages of γ irradiation or treated with various concentrations of damage-inducing drugs (MMC or CPT) throughout the experiment. After 8 d, cell numbers were determined using the Z1 Coulter Counter (Beckman Coulter). Cell numbers after treatment were normalized to cell numbers in the untreated sample to determine the relative survival.
Coimmunoprecipitation
For coimmunoprecipitations from transiently transfected 293T cells, cells were harvested 40 h after transfection, washed with PBS, resuspended in 0.5% NP40 lysis buffer (10 mM HEPES-KOH at pH 7.9, 150 mM NaCl, 1.5 mM MgCl2, 0.5% [v/v] NP40, Roche Complete protease inhibitor, 125 U/mL benzonase), and nutated for 1 h at 4°C. After centrifugation at 16,000g for 20 min at 4°C, Flag M2-conjugated beads or anti-myc-conjugated beads were added, and the lysate was nutated for 12 h at 4°C. The pelleted beads were washed three times in same buffer, resuspended in SDS-PAGE sample buffer, boiled for 5 min, and analyzed by immunoblotting.
Protein purification and GST pull-down assay
Human PHF11 cDNA was cloned into pGEX-6p-2 and transformed in BL21 cells. At OD600 0.6, cultures were heat-shocked for 45 sec at 42°C, 500 μM IPTG was added, and a second 42°C heat shock was given for 5 min. After incubation for 12–16 h at 16°C, cells were harvested, resuspended in lysis buffer (50 mM HEPES-KOH at pH 7.9, 150 mM NaCl, 1 mM EDTA, 1% NP40, 10% glycerol, 2 mM DTT, 1 µg/mL lysozyme, 1 mM PMSF, 1× Complete protease inhibitor mix [Roche]) and sonicated on ice. The lysate was cleared by centrifugation at 40,000g for 30 min at 4°C and incubated with 500 µL of equilibrated glutathione beads for 4 h at 4°C. Beads were washed three to five times in the same buffer. Five micrograms to 10 µg of GST-hPHF11 or GST alone were incubated with various amounts of RPA or EXO1 protein purified as described previously (Henricksen et al. 1994; Blackwell et al. 2001) in binding buffer (50 mM HEPES-KOH at pH 7.9, 150 mM NaCl, 1.5 mM MgCl2, 0.5% [v/v] NP40, 1 mM DTT, 1 mM PMSF, Roche protease inhibitor mix) for 4–6 h at 4°C. Beads were collected at 500g and washed three times with binding buffer, and bound protein was eluted in 2× Laemmli buffer and analyzed by immunoblotting. MBP-tagged PHF11 was produced in baculovirus using standard procedures using an Invitrogen baculovirus vector.
Cell fractionation
Cell fractionation was performed as described previously (Mendez and Stillman 2000). Briefly, cells were harvested with trypsinization, washed with PBS, and resuspended in buffer A (10 mM HEPES at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 1 mM PMSF, Roche Complete protease inhibitor). Triton X-100 (0.1%) was added, and the mixture was incubated for 10 min on ice and centrifuged at 1300g for 4 min at 4°C to yield the cytoplasmic fractions. The pellets were washed with buffer A, resuspended in buffer B [3 mM EDTA, 0.2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetra-acetic acid (EGTA), 1 mM DTT, 1 mM PMSF, complete protease inhibitors], and incubated for 30 min on ice. The supernatants (soluble nuclear fraction) were separated from the pellets (chromatin fraction) by centrifugation at 1700g at 4°C.
DNA resection assay
A DNA substrate with a 100-nt 3′ ssDNA tail was prepared as described (Cannavo et al. 2013) and labeled at the 3′ end with [α-32P] cordycepin-5′-triphosphate using terminal deoxynucleotidyl transferase (New England Biolabs). Reactions were carried out in 20 mM HEPES-KOH (pH 7.3), 0.1 mM DTT, 5 mM MgCl2, 100 µg/ml BSA, 0.05% Triton X-100, 4 µM (nt) tailed dsDNA, 0 or 0.2 µM RPA, and the indicated amounts of PHF11 and EXO1 at 37°C. When RPA was included, it was added first and incubated for 2–5 min before subsequent addition of proteins. Reactions were started by EXO1 addition. The products of resection assays were analyzed by agarose gel electrophoresis (Nimonkar et al. 2011). Aliquots were added to stop/loading buffer (150 mM EDTA at pH 8.0, 2% SDS, 30% glycerol, 0.1% bromophenol blue, 2–4 mg/mL Proteinase K [Roche]) and were incubated for 10 min at 37°C. Products were separated using a 1% (w/v) agarose gel run in TAE (40 mM Tris acetate at pH 8.2, 1 mM EDTA) at 400 V/h and were quantified using an Amersham Biosciences Storm 860 PhosphorImager with ImageQuaNT software (version 5.2).
Supplementary Material
Acknowledgments
We thank Jerome Dejardin for advice on PICh, Anne-Claude Gingras and Dan Durocher for help with the CRAPome, Eros Lazzerini Denchi for communication of unpublished results, Maria Jasin for the lentiviral I-SceI, and Steve Elledge and Lorraine Symington for discussion. Frank Eisenhaber is thanked for verifying the absence of PHF11 in budding yeast. Katja Kratz, Fena Ochs, and Tatsuya Kibe are thanked for providing recombinant proteins. Zhe Yang is thanked for providing the RPE1/hTERT cells with CRISPR/Cas9 knockout of p53 and Rb. This work was supported by grants from the National Institutes of Health to T.d.L. (CA181090) and S.C.K. (GM62653).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.291807.116.
Freely available online through the Genes & Development Open Access option.
References
- Alholle A, Brini AT, Gharanei S, Vaiyapuri S, Arrigoni E, Dallol A, Gentle D, Kishida T, Hiruma T, Avigad S, et al. 2013. Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics 8: 1198–1204. [DOI] [PubMed] [Google Scholar]
- Bartocci C, Diedrich JK, Ouzounov I, Li J, Piunti A, Pasini D, Yates JR, Lazzerini Denchi E. 2014. Isolation of chromatin from dysfunctional telomeres reveals an important role for Ring1b in NHEJ-mediated chromosome fusions. Cell Rep 7: 1320–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell LJ, Wang S, Modrich P. 2001. DNA chain length dependence of formation and dynamics of hMutSα⋅hMutLα⋅heteroduplex complexes. J Biol Chem 276: 33233–33240. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. [DOI] [PubMed] [Google Scholar]
- Cannavo E, Cejka P, Kowalczykowski SC. 2013. Relationship of DNA degradation by Saccharomyces cerevisiae Exonuclease 1 and its stimulation by RPA and Mre11–Rad50–Xrs2 to DNA end resection. Proc Natl Acad Sci 110: E1661–E1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P. 2015. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J Biol Chem 290: 22931–22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli GB, de Lange T. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7: 712–718. [DOI] [PubMed] [Google Scholar]
- Celli GB, Lazzerini Denchi E, de Lange T. 2006. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol 8: 885–890. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke E, Rahman N, Page N, Rolph MS, Stewart GJ, Jones GJ. 2008. Functional characterization of the atopy-associated gene PHF11. J Allergy Clin Immunol 121: 1148–1154.e3. [DOI] [PubMed] [Google Scholar]
- Dejardin J, Kingston RE. 2009. Purification of proteins associated with specific genomic Loci. Cell 136: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071. [DOI] [PubMed] [Google Scholar]
- Deng Y, Guo X, Ferguson DO, Chang S. 2009. Multiple roles for MRE11 at uncapped telomeres. Nature 460: 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, de Lange T. 2009. Cell cycle dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of NHEJ in G1 and resection-mediated inhibition of NHEJ in G2. Mol Cell Biol 29: 5552–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 2008. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456: 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Wu JY, de Lange T, Zhuang X. 2013. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155: 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. [DOI] [PubMed] [Google Scholar]
- Genschel J, Modrich P. 2003. Mechanism of 5′-directed excision in human mismatch repair. Mol Cell 12: 1077–1086. [DOI] [PubMed] [Google Scholar]
- Gong Y, de Lange T. 2010. A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol Cell 40: 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, Wold MS. 1994. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem 269: 11121–11132. [PubMed] [Google Scholar]
- Hockemeyer D, Daniels JP, Takai H, de Lange T. 2006. Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell 126: 63–77. [DOI] [PubMed] [Google Scholar]
- Ikari J, Inamine A, Yamamoto T, Watanabe-Takano H, Yoshida N, Fujimura L, Taniguchi T, Sakamoto A, Hatano M, Tatsumi K, et al. 2014. Plant homeodomain finger protein 11 promotes class switch recombination to IgE in murine activated B cells. Allergy 69: 223–230. [DOI] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, Kowalczykowski SC. 2010. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467: 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibe T, Zimmermann M, de Lange T. 2016. TPP1 blocks an ATR-mediated resection mechanism at telomeres. Mol Cell 61: 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC. 2015. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YC, Akhter S, Gu P, Ye J, Poulet A, Giraud-Panis MJ, Bailey SM, Gilson E, Legerski RJ, Chang S. 2010. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J 29: 2230–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza D, de Lange T. 2003. POT1 as a terminal transducer of TRF1 telomere length control. Nature 424: 1013–1018. [DOI] [PubMed] [Google Scholar]
- Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T. 2013. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc Natl Acad Sci 110: 2146–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. 2015. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 163: 880–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. 2009. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 23: 2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. 2013. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10: 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Stillman B. 2000. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH. 2001. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol 21: 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myler LR, Gallardo IF, Zhou Y, Gong F, Yang SH, Wold MS, Miller KM, Paull TT, Finkelstein IJ. 2016. Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. Proc Natl Acad Sci 113: E1170–E1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. 2011. BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25: 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T. 2008. How shelterin protects mammalian telomeres. Annu Rev Genet 42: 301–334. [DOI] [PubMed] [Google Scholar]
- Panier S, Boulton SJ. 2014. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7–18. [DOI] [PubMed] [Google Scholar]
- Panier S, Durocher D. 2013. Push back to respond better: regulatory inhibition of the DNA double-strand break response. Nat Rev Mol Cell Biol 14: 661–672. [DOI] [PubMed] [Google Scholar]
- Parker H, Rose-Zerilli MJ, Parker A, Chaplin T, Wade R, Gardiner A, Griffiths M, Collins A, Young BD, Oscier DG, et al. 2011. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia 25: 489–497. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. 1999. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13: 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138: 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, Janscak P. 2014. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem 289: 27314–27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271. [DOI] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. 2003. DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556. [DOI] [PubMed] [Google Scholar]
- Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou HJ, Kneitz B, et al. 2003. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev 17: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, van Overbeek M, Rooney S, de Lange T. 2010. Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol Cell 39: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Takai H, de Lange T. 2012. Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. 1999. Centrosome amplification and a defective G2–M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell 3: 389–395. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Leaves NI, Anderson GG, Ponting CP, Broxholme J, Holt R, Edser P, Bhattacharyya S, Dunham A, Adcock IM, et al. 2003. Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma. Nat Genet 34: 181–186. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, de Lange T. 2014. 53BP1: pro choice in DNA repair. Trends Cell Biol 24: 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.