

In this Resource/Methodology, Ingouff et al. developed a new method to investigate DNA methylation reprogramming in plants. Using Arabidopsis, the authors developed sensors that reported CG and non-CG methylation with single-cell resolution.
Keywords: DNA methylation, sensors, reprogramming, reproduction
Abstract
Cytosine methylation is a key epigenetic mark in many organisms, important for both transcriptional control and genome integrity. While relatively stable during somatic growth, DNA methylation is reprogrammed genome-wide during mammalian reproduction. Reprogramming is essential for zygotic totipotency and to prevent transgenerational inheritance of epimutations. However, the extent of DNA methylation reprogramming in plants remains unclear. Here, we developed sensors reporting with single-cell resolution CG and non-CG methylation in Arabidopsis. Live imaging during reproduction revealed distinct and sex-specific dynamics for both contexts. We found that CHH methylation in the egg cell depends on DOMAINS REARRANGED METHYLASE 2 (DRM2) and RNA polymerase V (Pol V), two main actors of RNA-directed DNA methylation, but does not depend on Pol IV. Our sensors provide insight into global DNA methylation dynamics at the single-cell level with high temporal resolution and offer a powerful tool to track CG and non-CG methylation both during development and in response to environmental cues in all organisms with methylated DNA, as we illustrate in mouse embryonic stem cells.
Methylation of cytosine residues on the DNA molecule plays important biological functions in many eukaryotes (Law and Jacobsen 2010), including maintenance of genome stability, imprinting, and repression of gene expression. Its faithful propagation is thus critical for proper development. In mammals, DNA methylation is subjected to dramatic reprogramming (resetting) from one generation to the next in both the germline and the early embryo (Feng et al. 2010; Hajkova 2011; Seisenberger et al. 2013; Heard and Martienssen 2014). These reprogramming phases are critically important for the proper acquisition of totipotency in the zygote and to erase epigenetic marks that might have accumulated during its progenitors’ life cycle.
In plants, little is known about DNA methylation dynamics during reproduction (Hsieh et al. 2009; Schoft et al. 2009; Calarco et al. 2012; Jullien et al. 2012; Moreno-Romero et al. 2016), owing largely to the technical difficulty of isolating plant germ cells and the very rapid changes affecting chromatin marks during plant reproduction (Ingouff et al. 2010; She et al. 2013). Contrary to animals, which establish germlines during early embryo development, plant gametes originate in the adult plant from somatic precursors (Kawashima and Berger 2014). In the male (the anthers) and female (the ovules) reproductive organs, somatic cells shift identity to form precursor cells analogous to animal primordium germ cells. In the ovule primordium, typically a single somatic cell differentiates into a megaspore mother cell (MMC), which will undergo meiosis to produce four haploid spores. Only one spore (the functional megaspore) usually survives and produces through three mitoses and differentiates into a haploid embryo sac or female gametophyte containing typically two female gametes (the egg and the central cell) and accessory cells. In the anthers, groups of microspore mother cells (MiMCs) differentiate, each producing four haploid microspores after meiosis. Each microspore undergoes two mitotic divisions to form a mature pollen grain or male gametophyte, two sperm cells enclosed in the vegetative cell. Both sperm cells participate in a double fertilization of the egg and central cells, giving rise to the embryo and the endosperm, respectively.
In plants, cytosine methylation is found in three contexts: CG, but also CHG and CHH (with H = A, T, or C). Current evidence globally suggests long-term stability of methylation patterns in Arabidopsis, particularly for CG methylation. Inheritance of methylated cytosine (mC) has been studied in both experimental and natural Arabidopsis populations as well as in maize (Martienssen and Colot 2001; Regulski et al. 2013). In wild-type Arabidopsis accessions in both natural and laboratory conditions, the results indicate a remarkable level of stability of mC in all three contexts over many generations, with epigenetic changes accumulating in a clock-like manner that matches DNA mutation rates (Becker et al. 2011; Schmitz et al. 2011; Hagmann et al. 2015).
On the other hand, alterations to DNA methylation patterns induced by life history such as environmental stresses are mitotically stable in individual plants but, save for rare exceptions (Weigel and Colot 2012), are usually not meiotically inherited (Dowen et al. 2012; Secco et al. 2015), thus suggesting active resetting of altered methylation marks. Interestingly, epialleles are relatively easy to generate in experimental populations using mutants with hypomethylated genomes (Vongs et al. 1993; Johannes et al. 2009; Reinders et al. 2009). Some of these epialleles are stably inherited once wild-type activity is restored, but a subset undergoes progressive remethylation, reaching wild-type levels after a limited number of generations. This process is dependent on RNAi and occurs during reproduction (Teixeira et al. 2009), indicating that the Arabidopsis methylome can be actively modified during reproductive development. This was confirmed for male gametophyte development (Calarco et al. 2012), where cell type-specific methylomes (Schoft et al. 2009; Calarco et al. 2012) indicate that, contrary to mammals, sperm cells retain CG and CHG methylation during their differentiation. Although a low level of CHH methylation is detected in the microspore after meiosis and remains in the sperm cells upon mitoses, a higher methylation level is restored by de novo DNA methyltransferase activity in the vegetative nucleus prior to fertilization and then in the embryo after fertilization (Calarco et al. 2012). Thus, there is some evidence that at least CHH methylation, but not CG or CHG, is labile during male gametogenesis. Much less is known about methylation dynamics during female gamete development. Genetic evidence indicates that at least CG methylation is stable during the three haploid divisions leading to the formation of the egg cell (Saze et al. 2003) but is likely lower upon its maturation (Jullien et al. 2012). However, virtually nothing is known about the dynamics of mC during premeiotic and meiotic development and for non-CG contexts during ovule development. The timing, extent, and mechanisms of such modifications are unclear and remain difficult to dissect with current methods for methylation analysis because they occur in a limited number of reproductive cells deeply embedded in several layers of somatic cells.
In Arabidopsis, mapping of cytosine methylation at single-nucleotide resolution by genome-wide bisulfite sequencing (GWBS) and the integrative analysis of the genomic distribution of DNA methylation, histone modifications, and histone variants have generated a highly refined model for the structure of the epigenome (Cokus et al. 2008; Lister et al. 2008; Roudier et al. 2011; Yelagandula et al. 2014). While the applications of GWBS are rapidly improving, including at the single-cell level (Lau and Bergmann 2015; Clark et al. 2016), they still typically require a significant amount of cells to achieve reasonable coverage and would be challenging to apply on a long developmental series during plant reproduction. Thus, complementary approaches are needed to refine our understanding of mC dynamics in addition to bulk analysis of often heterogeneous cell types. Particularly important is the development of tools to decipher the temporal dynamic of the epigenome during development and in response to exogenous cues during development and growth. Such tools would be well suited to analyze the changes occurring during reproductive development. Here, we developed genetically encoded fluorescent dynamic sensors of DNA methylation (DYNAMETs) that selectively report, with high temporal resolution, CG and CHH methylation in the model plant Arabidopsis thaliana. They provide access to cell-specific information regarding DNA methylation in plant germ cells and allowed us to analyze changes in real time during reproduction. They reveal distinct and sex-specific dynamics for these two contexts of methylation. We further documented the functionality of a mCG sensor in mouse embryonic stem cells (mESCs), suggesting that similar dynamics could be examined during mammalian development.
Results
Evaluation of potential DYNAMETs
To generate DYNAMETs in Arabidopsis, we expanded a strategy relying on a genetically encoded methyl-CpG-binding domain (MBD) fused to a fluorescent protein to report in real time CG methylation states in mammals (Okada et al. 2010; Yamagata 2010). We performed a literature search to find domains or proteins with affinity for mCs in any of three sequence contexts encountered in the plant epigenome (CG, CHG, and CHH). We identified the MBD of Arabidopsis MBD6 that binds specifically to symmetrical mCG in vitro (Zemach and Grafi 2003) as well as SUVH4/KRYPTONITE and SUVH9, two Arabidopsis suppressor of variegation 3-9 homologous (SUVH) proteins that contain SET and RING finger-associated (SRA) domains that bind mCHG and mCHH sequences, respectively, in vitro (Johnson et al. 2007, 2008).
To test their capacity to report DNA methylation states in planta, we generated DYNAMET cassettes consisting of the MBD of MBD6, the SRA domain of SUVH4 (SUVH4Δ-SRA), and SUVH9 (SUVH9Δ-SRA) fused to a nuclear localization signal (NLS) in-frame with a fluorescent protein (Supplemental Fig. S1A; see the Supplemental Material for details regarding the optimization of the reporters). Each cassette was placed under the control of a ubiquitous HISTONE THREE-RELATED 5 (HTR5) promoter that is strongly active in most cell types of the reproductive organs except in the central cell of mature ovules (Figs. 1–3; Supplemental Fig. S2A,B). Although all three DYNAMET fusion proteins were detected in transient assays in tobacco cells (Supplemental Fig. S1B,C), fluorescence was visible only in stably transformed Arabidopsis plants carrying pHTR5:MBD-Venus (Fig. 1A) but never in transgenic lines for pHTR5:SUVH4Δ-SRA-ECFP or pHTR5:SUVH9Δ-SRA-Venus. We thus used the entire SUVH4 and SUVH9 proteins fused to fluorescent proteins and driven by the HTR5 promoter (Supplemental Fig. S1A). Fluorescence of both DYNAMET fusions was similarly detectable in transient assays in tobacco cells (Supplemental Fig. S1D,E), but only the SUVH9-Venus fusion protein could be detected in stable transgenic plants (Fig. 1B). Transgenic lines expressing MBD-Venus and SUVH9-Venus showed no detectable phenotypic alterations, suggesting that the DYNAMET protein fusions are not toxic. In conclusion, we obtained two sensors putatively targeting CG (MBD-Venus) and non-CG (SUVH9-Venus) methylation but not CHG.
Figure 1.

The DYNAMETs MBD-Venus and SUVH9-Venus are real-time reporters of DNA methylation status. (A,B) Representative nuclear distribution of MBD-Venus and SUVH9-Venus fluorescence in wild-type and ddm1 genetic backgrounds. Aggregate profiles of ChIP-seq (chromatin immunoprecipitation [ChIP] followed by sequencing) reads for MBD-Venus and SUVH9-Venus over transposable elements (C) and protein-coding sequences (D). Living-cell dynamic of MBD-Venus (E–H) and SUVH9-Venus (I–L) during male gametogenesis. Tetrads (TET) of haploid microspores (E,I), early microspores (EMS) (F,J), late microspores (LMS) (G,K), and bicellular pollen grains (BCP) (H,L) with a large vegetative cell and a smaller generative cell. Bars: A,B, 5 μm; E,I, 10 μm; F–H,J–L, 25 μm. Images are maximum intensity projections of Z-stacks. (M) Quantification of DYNAMET fluorescence intensity (FI) relative to DAPI during male gametogenesis. Cells were immunostained with an Atto 488-conjugated anti-GFP/YFP antibody. Error bars correspond to standard deviation. The number of nuclei analyzed (n) is indicated in each bar. (gen) Generative cell nucleus; (veg) vegetative cell nucleus. (**) P < 0.01. Only statistically significant pairwise differences are indicated.
Figure 2.
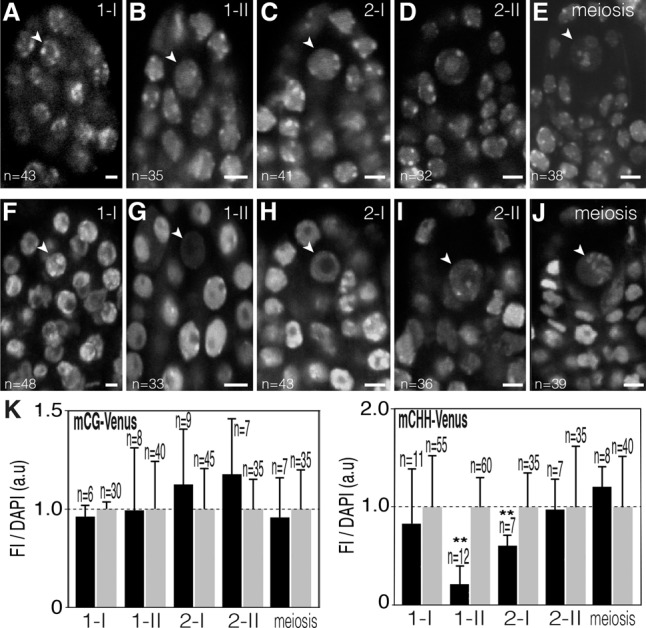
CHH methylation is reprogrammed during MMC differentiation. Representative confocal images of mCG-Venus (A–E) and mCHH-Venus (F–J) in living ovule primordia with differentiating MMCs (arrowhead) at stage 1-I (A,F), stage 1-II (B,G), stage 2-I (C,H), and stage 2-II (D,I) and with MMCs undergoing meiosis (E,J). Bars, 5 μm. (K) Ratio of DYNAMET fluorescence intensity (FI) relative to DAPI counterstain in developing MMCs (black bars) relative to the surrounding nucellar cells (gray bars). Flower buds were immunostained with an Atto 488-conjugated anti-GFP/YFP antibody. (a.u) Arbitrary units; (n) number of nuclei analyzed in each bar. Error bars correspond to standard deviation. (**) P < 0.01. Only statistically significant pairwise differences are indicated. Images are maximum intensity projection of Z-stacks.
Figure 3.
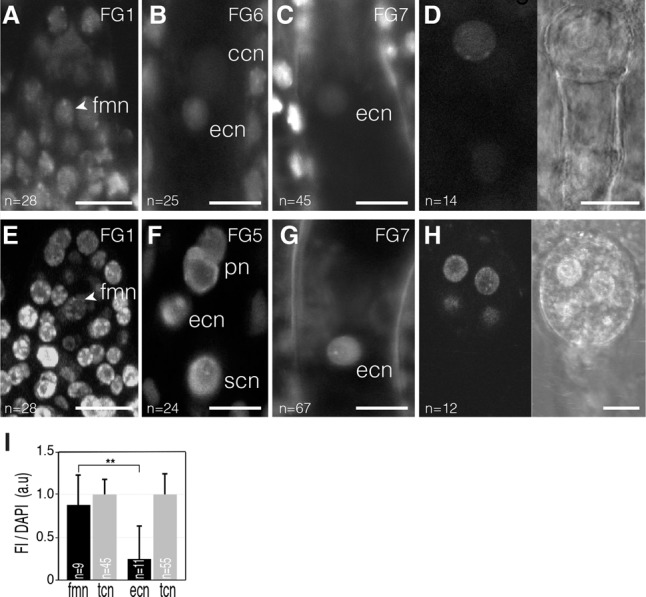
CG methylation is reduced in mature female gametophytes. Confocal images of mCG-Venus (A–D) and mCHH-Venus (E–H) in developing female gametophytes (A–C,E–G) and early embryo development (D,H). (A) The post-meiotic functional megaspore nucleus (fmn; arrowhead) shows a mCG-Venus pattern similar to surrounding integument cell nuclei (tcn). (B) mCG-Venus fluorescence is barely detectable in the egg cell nucleus (ecn) of the maturing female gametophyte (stage FG6). (C) A weak fluorescence is detectable in the egg cell nucleus in the mature female gametophyte (stage FG7). (D) A typical mCG-Venus nuclear pattern is restored at the one-cell stage embryo. (E) The nucleus of the functional megaspore (fmn; arrowhead) generated following meiosis shows a typical mCHH-Venus pattern. (F) This pattern is maintained in the nucleus of all cell types of the maturing female gametophyte (stage FG5). (G) mCHH-Venus pattern is detectable in the egg cell nucleus (ecn) in the mature female gametophyte (stage FG7). (H) mCHH-Venus pattern is detectable in the nuclei of an octant stage embryo. (I) Bars: A–C,E–G, 10 µm; D,H, 15 µm. (I) Fluorescence intensity (FI) of mCG-Venus relative to DAPI in arbitrary units (a.u) in developing female gametophytes of ovules immunostained with an Atto 488-conjugated anti-GFP/YFP antibody. Error bars correspond to standard deviation. The number of nuclei analyzed (n) is indicated in each bar. (**) P < 0.01. Only statistically significant pairwise differences are indicated. (ccn) Central cell nucleus; (pn) polar nuclei; (scn) synergid cell nucleus. Images are maximum intensity projections of Z-stacks.
DYNAMETs report context-specific DNA methylation states at CG and CHH sites
To validate the sensors, we first analyzed the subnuclear distribution of MBD-Venus and SUVH9-Venus in transgenic plants. As expected, the fluorescence of both DYNAMETs colocalized with DAPI-stained constitutive heterochromatin (Fig. 1A,B), where most cytosine methylation accumulates in Arabidopsis (Zhang et al. 2006; Lister et al. 2008). To confirm colocalization of MBD-Venus with heterochromatin, we introgressed the reporter in a ddm1 (decrease in DNA methylation 1) mutant background. This led to a diffuse pattern of fluorescence (Fig. 1A), consistent with the global loss of mCG in heterochromatin in ddm1 (Lippman et al. 2004; Stroud et al. 2014).
To determine the affinity of the DYNAMET reporters for the different methylation contexts in planta, we performed chromatin immunoprecipitation (ChIP) followed by sequencing (ChIP-seq). Genome-wide distribution of MBD-Venus and SUVH9-Venus in wild-type plants accurately recapitulated CG and non-CG (CHG/CHH) methylation patterns, respectively, as defined previously by GWBS (Fig. 1C,D; Supplemental Fig. S3; Cokus et al. 2008; Lister et al. 2008). To evaluate the specificity of the DYNAMET sensors, we then used published GWBS data sets for all three mC contexts and analyzed correlations with the binding of both reporters in transposable elements (TEs) and genes (Supplemental Fig. S4). We found that while MBD-Venus shows strong enrichment for CG gene body methylation (Supplemental Fig. S4J), we could not detect any enrichment of SUVH9-Venus on the same loci (Supplemental Fig. S4D). This indicates a limited affinity of the SUVH9-Venus reporter for mCG sites. Conversely, while SUVH9-Venus is significantly enriched for the small set of CHH (but not CG) methylated genes (Supplemental Fig. S4F), there was no enrichment of MBD-Venus for these loci (Supplemental Fig. S4L), suggesting that the latter has very limited affinity for mCHH. We then evaluated DYNAMET binding quantitatively. The data for SUVH9-Venus over TEs (Supplemental Fig. S4C) suggest a good correlation between CHH methylation levels and binding intensity (R2 = 0.68). For MBD-Venus, while stronger enrichment in highly methylated TEs is highly significant, we observed enrichment even for low levels of methylation (<10%, Supplemental Fig. S4G). The same was true for gene body methylation. Thus, SUVH9-Venus is a better quantitative tool than MBD-Venus.
To complement these analyses, we then estimated the correlation between the ChIP data for MBD-Venus and MeDIP-seq (methylated DNA immunoprecipitation [MeDIP] followed by sequencing) data from Arabidopsis (Supplemental Fig. S5), which is technically similar to our ChIP experiment. The comparison with MBD-Venus is relevant, as MeDIP recognizes all mCs, with mCG methylation being predominant in Arabidopsis. The results indicate a high correlation (R2 = 0.91) between both data sets.
In Arabidopsis, DNA methylation is initially catalyzed by a 24-nucleotide (nt) siRNA-dependent DNA methylation (RdDM) pathway involving the DOMAINS REARRANGED METHYLASEs (DRM1 and DRM2). CG and CHG methylation are then maintained by DNA METHYLTRANSFERASE1 (MET1) and CHROMOMETHYLASE3 (CMT3), respectively (Law and Jacobsen 2010). CHH methylation is maintained either through the RdDM pathway (Matzke et al. 2015) or by the CMT2 DNA methyltransferase (Zemach et al. 2013; Stroud et al. 2014). To further define the methylation specificity of SUVH9-Venus, we analyzed its genome-wide distribution in different DNA methyltransferase mutants by ChIP-seq. Enrichment in TEs was reduced in the drm2 mutant (Supplemental Fig. S6A,C) and in the cmt2 mutant on both short TEs and for the central domain of longer TEs (Supplemental Fig. S6B), both of which are known targets of CMT2 (Zemach et al. 2013). Enrichment was eliminated in the drm1 drm2 cmt2 cmt3 quadruple mutant (Supplemental Fig. S6D), which abolishes CHH methylation (Stroud et al. 2014). These experiments collectively indicate that SUVH9-Venus recognizes DRM2- and CMT2-dependent CHH methylation.
We next tested whether MBD6-Venus and SUVH9-Venus DYNAMET lines could monitor changes in methylation states in planta. We thus analyzed DYNAMET fluorescence during male gametogenesis, where GWBS data are available (Calarco et al. 2012). Similarly, we observed in living cells that MBD-Venus patterns were stable throughout male gametogenesis, with a slight reduction in the vegetative nucleus (Fig. 1E–H) likely reflecting active DNA demethylation (Calarco et al. 2012; Ibarra et al. 2012). In contrast, SUVH9-Venus reported rapid and massive demethylation after the differentiation of the functional microspore, followed by remethylation upon the first mitotic division (Fig. 1I–L). These results are in agreement with the methylome data from microspores and cell types of mature pollen (Calarco et al. 2012; Ibarra et al. 2012) and further suggest that SUVH9-Venus reports specifically CHH methylation but not CHG methylation, which is known to be stable in the microspore. We confirmed these observations using immunolocalization experiments with an anti-YFP antibody as well as DAPI counterstaining to precisely quantify fluorescence against chromatin content (Fig. 1M; Supplemental Fig. S7). The results are in good agreement with living-cell observations. They notably confirmed the striking difference observed between early and late microspores for SUVH9-Venus.
Next, we evaluated the DYNAMET reporters as living-cell sensors by performing time-lapse imaging. First, we imaged MBD-Venus dynamics during mitosis in root cells over 30 min, with one acquisition every 2 min (Supplemental Movie 1; Supplemental Fig. S8). The reporter remained tightly bound to segregating chromosomes, which is compatible with CG methylation being a stable epigenetic mark (Law and Jacobsen 2010). We then tracked SUVH9-Venus in male meiocytes during meiotic prophase 1 (Supplemental Movie 2) and early sporogenesis (Supplemental Movie 3) using a multiphoton microscope with time intervals of 30 min. Imaging over a 16-h period revealed highly dynamic chromatin movements in meiocytes and microspores. These experiments showed that the sensors can be used to image DNA methylation over relatively long periods of time.
Altogether, we conclude that DYNAMETs function as live-imaging reporters of DNA methylation states for the symmetrical CG (MBD-Venus) and asymmetrical CHH (SUVH9-Venus) contexts with high temporal resolution. For the sake of clarity, the sensors are referred to as mCG-Venus and mCHH-Venus DYNAMETs here.
We further tested the capacity of our mCG reporter to detect CG methylation in living mESCs in both wild-type cells and cells defective for the Dnmt1 maintenance DNA methyltransferase (Supplemental Fig. S9A–D). The cytological patterns observed in wild-type mESCs (Supplemental Fig. S9B) are consistent with the typical patterns obtained with mC antibodies (Li et al. 2015). In addition, we noted a significant decrease of signal intensity in dnmt1-deficient cells with similar illumination settings (Supplemental Fig. S9C,D), consistent with the partial (∼80%) loss of mCG typically detected in these cells (Li et al. 2015). These observations suggest that the mCG reporter is functional in mammalian ESCs.
Global DNA methylation changes occur during female sporogenesis and gametogenesis and early embryo development
We next studied the DNA methylation pattern using DYNAMET lines during the course of male and female germline differentiation. During female sporogenesis, starting from the differentiation of the germ cell precursors and until the formation of the functional haploid spores upon meiosis, we observed a steady nuclear distribution of mCG-Venus (Fig. 2A–E). Similarly, a stable mCG pattern was observed during all stages of female gametogenesis, from the definition of the haploid megaspore until female gametes are formed (Fig. 3A,B). However, mCG-Venus fluorescence decreased to a lower level in the mature egg cell nucleus when compared with either the surrounding tissues (Fig. 3C) or the functional megaspore (Fig. 3I). As mCG-Venus binds only symmetrically methylated CG in vitro (independently confirmed in Supplemental Fig. S10A,B; Zemach and Grafi 2003), this suggests either lower CG methylation or significant hemimethylation in the mature egg cell. This latter hypothesis is consistent with a previous report suggesting passive demethylation in this cell due to the absence of the maintenance DNA methyltransferase MET1 (Jullien et al. 2012). However, following fertilization, a typical distribution of mCG-Venus fluorescence was restored in the embryo, indicating that symmetrical mCG is rapidly re-established upon fertilization (Fig. 3D).
In contrast to the relative stability of mCG, mCHH-Venus fluorescence became undetectable in the developing female germline (the MMC) at stage 2-I during female sporogenesis (stages as described in She et al. 2013) and prior to DNA replication (cf. Figs. 2G and 3F) and gradually reappeared between stage 2-II and the onset of meiosis (Fig. 2H–J). These dynamic changes correlate with the establishment of a peculiar but transient chromatin structure devoid of the centromeric H3 variant CenH3 and linker H1 histones (She et al. 2013) but still with condensed centromeric 180-base-pair (bp) repeats (Supplemental Fig. S11).
In contrast, mCHH-Venus patterns appeared stable throughout female gametogenesis (Fig. 3E,F) even in the mature egg cell (Fig. 3G). Although the mature egg cell chromatin structure resembled that of stage 2-I MMCs (lack of linker and centromeric H3 histone and with condensed centromeric repeats) (Supplemental Fig. S12; Ingouff et al. 2007; Yelagandula et al. 2014), it retained a typical mCHH pattern that is also visible after fertilization in the early embryo (Fig. 3H). Finally, we also monitored mCG and mCHH-Venus patterns during male sporogenesis and observed stable levels for both sensors (Supplemental Fig. S13).
To confirm both patterns, we performed immunolocalization experiments with an anti-GFP/YFP antibody and DAPI counterstaining (Figs. 2K and 3I; Supplemental Figs. S14, S15). The results confirmed the live-cell observations, with mCG-Venus remaining broadly invariant during sporogenesis but significantly reduced in the egg cell, and mCHH-Venus being significantly hypomethylated at stage 1–2 during female sporogenesis. Collectively, our data therefore indicate distinct dynamics for both methylation contexts: mCHH is highly labile and reprogrammed by demethylation/remethylation during both male gametogenesis and female sporogenesis, while mCG is globally maintained in male and female gametic precursors and male gametes. Its highly significant decrease in the egg cell might be due to either demethylation or loss of symmetrical methylation in the CG context but is restored following fertilization.
Establishment of CHH methylation in the egg cell relies on a DRM2 and RNA polymerase V (Pol V)-dependent but Pol IV 24-nt siRNA-independent pathway
To identify the DNA methyltransferases controlling mCHH dynamics during female gametogenesis, we analyzed mCHH-Venus expression in genetic backgrounds defective for non-CG DNA methyltransferase activity, including CMT2, CMT3, and DRM2 (Fig. 4). Only a null mutant for DRM2 completely lost mCHH-Venus fluorescence, specifically in the mature egg cell. These observations are in agreement with the fact that DRM1 and DRM2 are the sole DNA methyltransferases detected in the mature egg cell (Jullien et al. 2012). Loss of fluorescence in drm2-2 eggs suggested that mCHH-Venus is targeted for degradation when unbound to its targets. Accordingly, treating whole inflorescences with syringolin A, which inhibits proteasome activity (Groll et al. 2008), increased accumulation of mCHH-Venus compared with mock treatment (Supplemental Fig. S16). Consistent with the absence of mCHH-Venus in the egg cell, a maternally provided hypomethylated CHH methylome from a drm2-2 plant led to pleiotropic developmental abnormalities in the young embryo (Supplemental Fig. S17A,B). Interestingly, drm2 mutation transmitted from heterozygous plants showed increased penetrance relative to the expected value, possibly indicating effects from the maternal sporophytic tissues, as described previously for MET1 and CMT3 (FitzGerald et al. 2008; Pillot et al. 2010).
Figure 4.
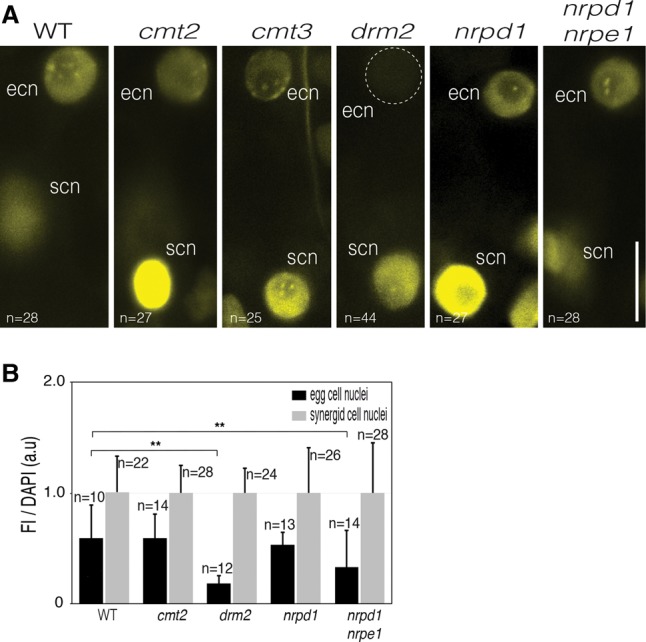
A noncanonical RdDM pathway controls CHH methylation in the egg cell. (A) Methylated heterochromatin foci were detected in the egg cell nucleus (ecn) with mCHH-Venus in the wild type. A marked loss of fluorescence was observed in the egg cell of a null mutant for DRM2, the key enzyme of the RdDM pathway. (scn) Synergid cell nucleus. Bar, 10 µm. (B) Fluorescence intensity (FI) of mCHH-Venus relative to DAPI (arbitrary units [a.u]) in egg cell nuclei relative to synergid cell nuclei of mature wild-type and mutant female gametophytes. Whole-mount immunofluorescences were performed on flower buds with an Atto 488-conjugated anti-GFP/YFP antibody. A significant decrease was observed for drm2 and nrpd1/nrpe1 mutants. Error bars correspond to standard deviation. The number of nuclei analyzed (n) is indicated in each bar. (**) P < 0.01. Only statistically significant pairwise differences are indicated. Images are maximum intensity projections of Z-stacks.
We next analyzed mCHH-Venus in plants deficient for Pol IV (Pol IV/NRDP1) and Pol V (Pol V/NRDPE1) activity that, in somatic tissues, generates the 24-nt siRNA and long noncoding RNA, respectively, which are responsible for targeting DRM2 to RdDM targets. Unexpectedly, the mCHH fluorescence pattern was similar to the wild type in the egg cells of both mutants (Fig. 4A). To better evaluate the effect of these mutations, we performed immunolocalization experiments combined with DAPI counterstaining. Interestingly, quantifications confirmed the observations made on fresh tissues but also revealed a significant fluorescence decrease in the double nrpd1/nrpe1 mutant but not in the single nrpd1 mutant (Fig. 4B; Supplemental Fig. S18). Thus, NRPE1, but not NRPD1, likely contributes to DNA methylation maintenance in the egg cell.
To better understand these observations, we monitored the expression pattern of CMT2, of the catalytic subunits of Pol IV (NRPD1) and Pol V (NRPE1), and of their common catalytic subunit NRP(D/E)2A in transgenic lines expressing the corresponding genomic fusions with fluorescent proteins (Fig. 5A–D). Although all protein fusions were detected in the ovule integuments, only the tagged Pol V subunits Venus-NRPE1 and Venus-NRP(D/E)2A were conspicuously detected in the egg cell (Fig. 5C,D). Most remarkably, tagged CMT2 and the Pol IV catalytic subunit NRPD1 were absent from the egg cell (Fig. 5A,B; Jullien et al. 2012), an observation that we confirmed using a NRPD1-specific antibody (Supplemental Fig. S18). Thus, establishment of mCHH in the egg cell is dependent on DRM2 and Pol V but likely independent of Pol IV.
Figure 5.
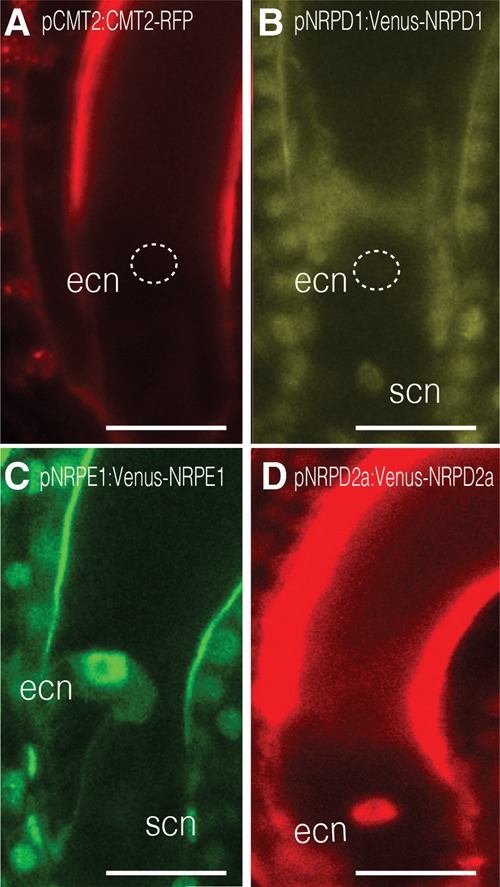
Expression pattern of actors of CHH methylation in the female gametophyte. Expression of pCMT2:CMT2-RFP (A) and the Pol IV catalytic subunit pNRPD1:Venus-NRPD1 (B) is not detectable in the egg cell. Only the expression of the two catalytic subunits of Pol V—pNRPE1:Venus-NRPE1 (C) and pNRP(D/E)2a:Venus-NRP(D/E)2a (D)—is detected in the egg cell nucleus (ecn). (scn) Synergid cell nucleus. Bar, 15 µm. (n > 25) Number of observations for each sample. Images are maximum intensity projections of Z-stacks.
Discussion
Here, we generated live-imaging DYNAMETs for the CG and CHH sequence contexts. We showed that DYNAMETs can be used to track DNA methylation dynamics in individual cells in Arabidopsis plants. These sensors provide access to a global cell-specific methylation pattern with high temporal resolution. This temporal dimension is particularly critical when studying reproductive cells, where chromatin remodeling occurs rapidly (Ingouff et al. 2010; She et al. 2013).
Our study indicates that DNA methylation reprogramming occurs differently in plants and mammals (Supplemental Fig. S12). While CG methylation is extensively reprogrammed in mammals (Feng et al. 2010; Hajkova 2011; Seisenberger et al. 2013; Heard and Martienssen 2014), numerous genetic and inheritance experiments in Arabidopsis revealed the stability of CG methylation patterns (Paszkowski and Grossniklaus 2011; Schmitz and Ecker 2012; Weigel and Colot 2012; Heard and Martienssen 2014). Our results confirmed that CG methylation is, in general, stable in the male and female germlines in Arabidopsis. The egg cell seems to be the exception, as we observed a significant decrease of signal upon its maturation, which might be due to either demethylation or loss of symmetrical methylation resulting from lack of MET1 activity (Jullien et al. 2012). The methylome of the egg cell in rice was published recently (Park et al. 2016) and revealed a CG methylation pattern typical of somatic cells. Assuming that these results are also valid in the Arabidopsis egg cell, this might suggest significant hemimethylation rather than demethylation in the egg cell. Importantly, fluctuation of mCG in the mature egg cell is not accompanied by loss of mCHH. In contrast, we observed highly dynamic changes affecting CHH methylation, which undergoes waves of demethylation/remethylation during both female sporogenesis (this study) and male gametogenesis (Calarco et al. 2012; Ibarra et al. 2012; this study). Interestingly, we could not identify cell types that lost both contexts simultaneously. Supplemental Figure S20 provides a summary of our observations.
In the absence of methylome data for egg cells and early embryos in Arabidopsis, it is difficult to assess the relative importance of CG, CHG, and CHH methylation for genome stability in these cells. However, mutants affecting CHG (Pillot et al. 2010) or CHH (this study) methylation and, even more profoundly, CG methylation (Xiao et al. 2006) have embryo patterning defects of varying severity (milder in CHH and highest in CG), suggesting that all three contexts are developmentally important. This is in sharp contrast to somatic tissues, where CG methylation defects severely affect plant development, while non-CG methylation alterations have only limited phenotypic consequences (Henderson and Jacobsen 2007). Thus, proper de novo establishment of CHH methylation patterns in the egg cell is likely important for transcriptional control in the early embryo. Similar observations with respect to H3K9me2 (Autran et al. 2011) suggest a more general role for maternally controlled non-CG methylation for embryo transcriptional control.
We further demonstrated here that the DYNAMETs represent powerful tools for genetic analyses, allowing cell-specific screening. In particular, our genetic evidence showed that global CHH methylation in the egg cell was dependent on DRM2 and Pol V, where both proteins might have cell-autonomous activity, but independent of Pol IV activity, which is responsible for 24-nt heterochromatic siRNA production and is required to guide DRM2 to RdDM targets in somatic cells (Matzke et al. 2015). Accordingly, we observed that Pol IV was absent from the egg cell. This suggests that alternative as yet unidentified sources of small RNAs might be recruited in the egg cell to establish DNA methylation. One possibility is that RNA Pol II substitutes for Pol IV as a source of small RNAs to induce DRM-dependent methylation (Matzke et al. 2015; Cuerda-Gil and Slotkin 2016), acting either cell-autonomously or non-cell-autonomously. Such Pol IV-independent small RNAs could originate from surrounding cells of the maternal sporophyte or accessory cells of the female gametophyte and be transported to the egg cell, as shown between accessory cells and target sperm cells (Martinez et al. 2016). Alternatively, as yet unknown actors might be required, and identifying components of such a pathway will be of major interest.
By revealing the temporal aspects of mC changes, the DYNAMETs significantly expand the toolkit available for DNA methylation research. They reveal highly dynamic patterns of DNA methylation during plant reproduction and suggest that the regulatory pathways controlling methylation in somatic cells are likely distinct in reproductive cells.
Materials and methods
Plant material, growth conditions
Arabidopsis mutant alleles and reporter lines used in this study are summarized in Supplemental Table 1. After 2 d at 4°C in the dark, the seeds were germinated and grown on soil in a growth chamber (16 h light/8 h night at 20°C). For mutant analyses, a single transgenic event for SUVH9-Venus was introgressed into the drm2-2, cmt2-3, nrpd1-1, and nrpd1-1/nrpe1-1 mutant stocks.
Cloning of DYNAMET cassettes and genomic fusions
Each DYNAMET contained a methylcytosine-binding domain fused to a nuclear localization signal (NLS) in-frame with a fluorescent protein that was placed under the control of a histone3.3 HTR5 (At4g40040) promoter (pHTR5). The domain targeting mCs in the CG, CHG, and CHH sequence contexts corresponded to the MBD of MBD6 (Zemach and Grafi 2003), the SRA domain of SUVH4, and the SRA domain of SUVH9, respectively (Johnson et al. 2007, 2008). The full-length SUVH4 mutated in its SET domain and SUVH9 were also tested to report for CHG and CHH methylation, respectively. All of the DYNAMET constructs were generated by gene synthesis (GenScript). The sequence of each DYNAMET cassette is in the Supplemental Material. The sequence corresponding to SUH4-mSET was cloned by GoldenGate cloning (Engler et al. 2009; Lampropoulos et al. 2013) to generate the pMI108 vector. This binary vector also comprised a second molecular construct with a histone H2B (At5g22880) fused to a RFP mCherry (H2B-mCherry) under the control of the HTR5 promoter. All of the other cassettes were cloned into a pCAMBIA2300 (pCB2300) binary vector to generate the recombinant plasmids pCB2300-pHTR5:MBD-Venus (MBD-Venus), pCB2300-pHTR5:SUVH4Δ-SRA-Venus (SUVH4Δ-SRA-Venus), pCB2300-pHTR5:SUVH9Δ-SRA-Venus (SUVH9Δ-SRA-Venus), and pCB2300-pHTR5:SUVH9-Venus (SUVH9-Venus). Detailed information regarding the optimization of the various DYNAMET constructs is in the Supplemental Material.
The HTR5 promoter was amplified with primers pHTR5-FWDattB4 and pHTR5-REVattB1 and was used in a BP reaction with pDONRP4-P1R (Invitrogen). The resulting entry clone pEN-L4-pHTR5-R1 was subsequently combined with pEN-L1-NF-L2, pEN-R2-S*-L3, and the destination vector pK7m34GW,0 in a three-fragment LR reaction (Karimi et al. 2007). The final vector pHTR5:NLS-GUS-GFP consisted of the HTR5 promoter controlling the expression of a nuclear-localized GFP-GUS fusion protein (NLS-GUS-GFP). The transgenic plants were generated in Col 0 accession by floral dipping (Clough and Bent 1998) and selected on MS solid medium (Duchefa) with the appropriate selective agent.
All of the NRPD1, NRPE1, and NRP(D/E)2a promoter and gene sequences were obtained by synthesis (GenScript). Genomic fusions for pNRPD1:Venus-NRPD1, pNRPE1:Venus-NRPE1, and p NRP(D/E)2a:Venus-NRP(D/E)2a were generated using Multisite Gateway technology. The detailed cloning procedures are available on request. Genomic fusion of pCMT2:CMT2-RFP was generated by LR recombination using pDONR-pCMT2:CMT2 (Jullien et al. 2012) and pAlli2-GW-RFP1 (Ingouff et al. 2006). For mESCs, the MBD of MBD6 was codon-optimized for Mus musculus. Two consecutive MBDs followed by an alanine linker in-frame with three SV40 NLS and Venus were cloned with NcoI/XbaI restriction enzymes into the pEF plasmid (Invitrogen) backbone to result in pEF-MBD2x-3xNLS-Venus. The NcoI/XbaI insert was synthetized by GenScript. pEF.myc.ER-E2-Crimson was a gift from Benjamin Glick (Addgene, plasmid no. 38770).
ESC culture and transfection
Mouse embryonic fibroblast (MEF) feeder cells to support stem cell growth were obtained from MTI Global Stem (GSC-6001G). Wild-type J1 (American Type Culture Collection, SCRC-1010) and J1 Dnmt1 knockout (Dnmt1tm1Enl, MGI:1857601) ESCs were grown in knockout-DMEM (GIBCO/Thermo Fisher Scientific), 1000 U/mL LIF (ESGRO/Millipore), 0.1 mM 2-βmercaptoethanol, 15% FBS (Stem Cell Technologies), 1 mM MEM nonessential amino acids, and 20 mM L-glutamine. ESCs (4.6 × 106 cells) were transfected with 4 µg of pEF-MBD2x-3xNLS-Venus plasmid DNA using the P3 Primary Cell 4D-Nucleofector X kit L (Lonza) according to the Amaxa 4D-Nucleofector protocol for mESCs. After transfection, cells were seeded onto 2 × 104 MEFs per square centimeter of tissue culture-grade plastic dishes containing glass cover slips. After 48 h, allowing for three to four divisions, cells were washed in PBS and mounted on glass slides for confocal microscopy.
Immunolocalization
The NRPD1 antibody (Pontier et al. 2005) was provided by T. Lagrange (Centre National de la Recherche Scientifique, University of Perpignan). Immunodetections were performed as described (Pillot et al. 2010; She et al. 2013). Briefly, pistils from stage 1-I to maturity were fixed overnight at 4°C in 2% paraformaldehyde, 1× PBS, and 2% Triton fixative; washed three times in 1× PBS; and dissected to isolate the ovules. The dissected ovules were embedded in acrylamide as described (Bass et al. 1997) in order to facilitate manipulation and maintain the three-dimensional architecture of the tissues. Samples were digested in an enzymatic solution (1% driselase, 0.5% cellulase, 1% pectolyase, 1% BSA [all from Sigma]) for 25 min to 1 h at 37°C depending on the developmental stage, subsequently rinsed three times in 1× PBS, and permeabilized for 2 h in 1× PBS and 2% Tween20. They were then incubated overnight at 4°C with primary antibodies used at the following dilutions: 1:200 for the GFP-Booster chromobody (Chromotek) and 1:200 for NRPD1. The slides were washed day long in 1× PBS and 0.2% Triton. For the GFP-Booster chromobody, which was conjugated to an Atto 488 fluorochrome, slides were then incubated with 1 µg/mL DAPI in 1× PBS for 1 h, washed for 2 h in 1× PBS, and mounted in Prolong medium (Molecular Probes). For NRPD1, slides were incubated overnight at 4°C with an Alexa fluor 488-conjugated secondary antibody (Molecular Probes) used at 1:400 dilution. After washing in 1× PBS and 0.2% Triton for a minimum of 6 h, the slides were incubated with 1 µg/mL DAPI in 1× PBS for 1 h, washed for 2 h in 1× PBS, and mounted in Prolong.
Sample preparation for microscopy
Imaging of reporter lines during male and female sporogenesis and female gametogenesis was obtained from freshly dissected anthers and carpels and isolated young embryos from freshly dissected seeds. All of the experiments were performed on T2 or T3 generations from at least three independent transgenic lines. To compare fluorescence intensity in the egg cells of mutants, a very potent mCHH-Venus line (H9-11) was introgressed into all of the tested mutants. Microscope settings were first set with the reference mCHH-YFP line (H9-11) and kept identical to study its fluorescent pattern in all of the mutant backgrounds. Image acquisition was performed on homozygous mutants homozygous for CHH-Venus. Fluorescence was imaged immediately after mounting in half Murashige and Skoog (MS) medium with 0.3% (w/v) Phytagel. For imaging roots, sterilized seeds were germinated under an agar block in glass-bottomed dishes (GWSt-5040, WillCoWells) (Larrieu et al. 2015). Roots growing against the glass bottom were selected to perform time-lapse imaging of mCG-Venus in mitotic cells.
Microscopy
Imaging of fluorescence for DYNAMETs and other reporter lines from freshly dissected tissues was performed with an inverted laser scanning confocal microscope (Zeiss LSM780) and a 40× oil immersion objective lens (plan apo, N.A = 1.3; Zeiss) with settings for detection of Venus (excitation, 514 nm; emission, band-pass 515–545 nm) or mCherry (excitation, 561 nm; emission, band-pass 561–621 nm). Fluorescence was acquired with a GaAsP detector with the pinhole set to one Airy unit and optimal sectioning as suggested by the software (Zen). For quantification of the immunostaining experiments, we acquired images using a Zeiss LSM780 inverted confocal scanning microscope with sequential acquisitions for each fluorochrome (excitation, 405 nm for DAPI and 488 nm for Atto 488 and Alexa 488). For each stack, the number of sections was calculated to ensure nonoverlapping contiguous sections in a fluorochrome-dependent manner. For each region of interest (ROI), signal intensity was measured for each individual section and each fluorochrome using the FIJI (Schindelin et al. 2012) measurement module and summed. The raw data were imported into R to calculate means, standard deviations, and statistical significance using R's implementation of the Student's test.
Time-lapse imaging
Time-lapse movies of mCG-Venus in dividing root cells were obtained using an inverted laser scanning confocal microscope (Zeiss, LSM780) with settings as described above and a 20× objective lens (plan apo 0.8; Zeiss). Time-series images were acquired every 2 min. Time-lapse movies of the mCHH-Venus reporter during male sporogenesis and gametogenesis were obtained with an upright multiphoton microscope (LSM880 Examiner Z1, Zeiss) fitted with a coherent laser (Chameleon Ultra II) and an automatable stage (Zen Extension NEce, Ingesym) commuting between the microscope and a vibratome (Microm HM650V, Thermo Scientific). Open flowers and siliques were removed with a dissecting needle from a primary inflorescence of the CHH-Venus line (line H9-11) to conserve only the young buds. The inflorescence was immersed into a homemade embedding cassette filled with an in vitro culture medium (Nitsch medium [Duchefa], 5% [w/v] trehalose, 0.05% [w/v] MES-KOH at pH 5.8, 1× Gamborg's vitamin solution [Sigma]) (Gooh et al. 2015) supplemented with 8% agarose (Sigma). The hardened block containing the inflorescence was placed on the commuting stage and sectioned with the vibratome until the desired stages of microsporogenesis or male gametogenesis were observed. Fluorescence was detected with a nondescanned detector (BiG.2) using the following parameters: 20× water immersion objective lens (plan apo, N.A = 1.0, Zeiss); excitation, 980 nm; and emission, BP500-550. Z-section stacks were performed through the whole depth of anthers with developing meiocytes or male gametophytes at 1- and 1.75-μm intervals, respectively. Time-series images were acquired every 30 min. The images and time-lapse movies were acquired using Zen (Zeiss). Digital image and movie processing was performed with Fiji and Adobe Photoshop/Illustrator software. Image registration for the time-lapse images was performed using Fiji's Trackem2 plug-in.
ChIP
Five-hundred micrograms of inflorescence tissues was collected for each sample and used for ChIP experiments using anti-GFP antibody (Chromotek, GFP-TRAP), which also recognizes YFP. Detailed ChIP protocols are in the Supplemental Material. Indexed libraries were generated using Illumina's TruSeq ChIP sample preparation kit and sequenced on either an Illumina HiSeq 2500 sequencer by Fasteris SA or a NextSeq machine at the Cold Spring Harbor Genomic Core Facility following the manufacturer's instructions. Two replicates were generated for each genotype.
Bioinformatics
Quality filtering and base calling were performed using the CASAVA pipeline. Reads were mapped to the Arabidopsis TAIR10 annotations using Bowtie2, allowing one mismatch. Binning and read counts/coverage relative to annotations were performed using BedTools2 intersectBed. ChIP peak detection was performed using MACS's call peak function, relative to input DNA used as control. Aggregate profiles for exon/intron coverage were performed using ngsplot. Aggregate profiles over TEs were calculated with VAP (Coulombe et al. 2014). Data sets for the Arabidopsis methylome and MeDIP-seq were downloaded from Gene Expression Omnibus (GEO; accession nos. GSM1499351 for methylome data and GSM1326797 for MeDIP data). To estimate correlation coefficients between the MeDIP and MBD-Venus data, reads were binned over 500-bp windows, and correlations were calculated using R. To estimate the correlation between ChIP enrichment and methylation levels, we classified TEs and genes having low, high, and mid methylation levels. Thresholds for methylation levels were adjusted differently for CG, CHG, and CHH to account for the difference in overall methylation between all three contexts and recover sufficient loci in each category for statistical analyses. All file manipulations were performed in R with custom scripts, which are available on request. The statistical significance data in Supplemental Figure S5 were determined in R using ANOVA.
Purification of mCG-Venus-6xHis protein
The sequence of mCG-Venus was amplified and cloned into pET28b expression vector (Novagen). Recombinant histidine-tagged mCG-Venus protein production was performed in BL21(DE3) Escherichia coli strain (New England Biolabs) after addition of 100 mM IPTG. After 4 h at 37°C, cells were pelleted, resuspended in a purification buffer (50 mM Tris at pH 8, 300 mM NaCl, 1 mM TCEP, 10 mM imidazole, protease inhibitor cocktail [Complete, Roche]), and disrupted by sonication (Branson sonicator). Immobilized nickel beads (Sigma) were added to the cleared soluble fraction and incubated for 30 min. Unbound proteins were washed by several cycles of centrifugation/resuspension in fresh extraction buffer. MBD-Venus-6xHis was released from beads by a final resuspension in elution buffer (50 mM Tris at pH 8, 300 mM NaCl, 1 mM TCEP, 250 mM imidazole). Finally, eluted recombinant protein was dialyzed overnight with a buffer containing 50 mM Tris (pH 8), 100 mM NaCl, 1 mM TCEP, and 10% glycerol.
Electrophoretic mobility shift assay (EMSA)
Oligonucleotides used for the EMSA experiments were synthesized as described (Zemach and Grafi 2003) and labeled at their 5′ ends with a 6FAM fluorescent dye (Eurogentec). Double-stranded oligonucleotides were generated after annealing of complementary oligonucleotides. DNA-binding assays were performed in 25 mM Tris (pH 7.5), 5% glycerol, 60 mM NaCl, 10 mM MgCl2, 2 mM DTT, and 2.5 μg/mL fish DNA as a nonspecific competitor. Recombinant mCG-Venus-6xHis (200 nM) was mixed with 15 nM double-stranded oligonucleotides unmethylated, hemimethylated, or fully methylated in the CG, CHG, or CHH context. Binding of recombinant mCG-Venus-6xHis was further characterized using 200 nM recombinant protein and hemimethylated oligonucleotides in the CG context with concentrations ranging from 5 nM to 200 nM. Interactions between labeled oligonucleotides and recombinant protein were resolved by electrophoresis in 8% acrylamide (19:1) gels containing 5% glycerol and Tris-borate-EDTA buffer and detected in a GE Healthcare Typhoon 9400 scanner.
Proteasome inhibition
The proteasome inhibition protocol was adapted from She et al. (2013). Briefly, 200 mg of whole inflorescences were harvested and incubated for 18 h in half MS medium with mock (water) or 5 µM syringolin A (Groll et al. 2008). After tissue grinding in liquid nitrogen, total proteins were extracted in 200 µL of lysis buffer (125 mM Tris at pH 8, 10 % glycerol, 1 % SDS, 10 mM DTT, protease inhibitor cocktail [Roche]). Samples were homogenized and centrifuged at 15,000 rpm for 10 min. The soluble fraction was then separated on a SDS-PAGE gel and blotted onto Hybond ECL nitrocellulose membrane (GE Healthcare). The DYNAMET fusion proteins were detected by using an anti-GFP antibody (Abcam, Ab6556).
Accession numbers
ChIP-seq data sets have been deposited in GEO under accession GSE70455.
Supplementary Material
Acknowledgments
We thank T. Harrop, F. Roudier, and M. Mirouze for critical reading of the manuscript; G Conejero, J. Cau, and M. Lartaud from the Montpellier RIO Imaging Facility for assistance in microscopy and image processing; S. Jacobsen for the quadruple mutant drm1 drm2 cmt2 cmt3; and T. Lagrange for the NRPD1 antibody. We acknowledge funding from the European Union H2020 program (REP-658900-1) to D.G.; the Agence Nationale de la Recherche (ANR-12-BSV2-0013) to D.G., C.M., B.S., and M.I.; and the Howard Hughes Medical Institute-Gordon and Betty Moore Foundation, the National Science Foundation (DBI-1025830), and the National Institutes of Health (5R01GM067014) to R.A.M.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.289397.116.
References
- Autran D, Baroux C, Raissig MT, Lenormand T, Wittig M, Grob S, Steimer A, Barann M, Klostermeier UC, Leblanc O, et al. 2011. Maternal epigenetic pathways control parental contributions to Arabidopsis early embryogenesis. Cell 145: 707–719. [DOI] [PubMed] [Google Scholar]
- Bass HW, Marshall WF, Sedat JW, Agard DA, Cande WZ. 1997. Telomeres cluster de novo before the initiation of synapsis: a three-dimensional spatial analysis of telomere positions before and during meiotic prophase. J Cell Biol 137: 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K, Weigel D. 2011. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480: 245–249. [DOI] [PubMed] [Google Scholar]
- Calarco JP, Borges F, Donoghue MT, Van Ex F, Jullien PE, Lopes T, Gardner R, Berger F, Feijo JA, Becker JD, et al. 2012. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151: 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Lee HJ, Smallwood SA, Kelsey G, Reik W. 2016. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol 17: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough SJ, Bent AF. 1998. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743. [DOI] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe C, Poitras C, Nordell-Markovits A, Brunelle M, Lavoie MA, Robert F, Jacques PE. 2014. VAP: a versatile aggregate profiler for efficient genome-wide data representation and discovery. Nucleic Acids Res 42: W485–W493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuerda-Gil D, Slotkin RK. 2016. Non-canonical RNA-directed DNA methylation. Nat Plants 2: 16163. [DOI] [PubMed] [Google Scholar]
- Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR. 2012. Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci 109: E2183–E2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Gruetzner R, Kandzia R, Marillonnet S. 2009. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One 4: e5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. 2010. Epigenetic reprogramming in plant and animal development. Science 330: 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald J, Luo M, Chaudhury A, Berger F. 2008. DNA methylation causes predominant maternal controls of plant embryo growth. PLoS One 3: e2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooh K, Ueda M, Aruga K, Park J, Arata H, Higashiyama T, Kurihara D. 2015. Live-cell imaging and optical manipulation of Arabidopsis early embryogenesis. Dev Cell 34: 242–251. [DOI] [PubMed] [Google Scholar]
- Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M, Dudler R. 2008. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 452: 755–758. [DOI] [PubMed] [Google Scholar]
- Hagmann J, Becker C, Muller J, Stegle O, Meyer RC, Wang G, Schneeberger K, Fitz J, Altmann T, Bergelson J, et al. 2015. Century-scale methylome stability in a recently diverged Arabidopsis thaliana lineage. PLoS Genet 11: e1004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P. 2011. Epigenetic reprogramming in the germline: towards the ground state of the epigenome. Philos Trans R Soc Lond B Biol Sci 366: 2266–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Martienssen RA. 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157: 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Jacobsen SE. 2007. Epigenetic inheritance in plants. Nature 447: 418–424. [DOI] [PubMed] [Google Scholar]
- Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL, Zilberman D. 2009. Genome-wide demethylation of Arabidopsis endosperm. Science 324: 1451–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra CA, Feng X, Schoft VK, Hsieh TF, Uzawa R, Rodrigues JA, Zemach A, Chumak N, Machlicova A, Nishimura T, et al. 2012. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337: 1360–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingouff M, Jullien PE, Berger F. 2006. The female gametophyte and the endosperm control cell proliferation and differentiation of the seed coat in Arabidopsis. Plant Cell 18: 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingouff M, Hamamura Y, Gourgues M, Higashiyama T, Berger F. 2007. Distinct dynamics of HISTONE3 variants between the two fertilization products in plants. Curr Biol 17: 1032–1037. [DOI] [PubMed] [Google Scholar]
- Ingouff M, Rademacher S, Holec S, Soljic L, Xin N, Readshaw A, Foo SH, Lahouze B, Sprunck S, Berger F. 2010. Zygotic resetting of the HISTONE 3 variant repertoire participates in epigenetic reprogramming in Arabidopsis. Curr Biol 20: 2137–2143. [DOI] [PubMed] [Google Scholar]
- Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V, Simon M, Agier N, Bulski A, Albuisson J, Heredia F, Audigier P, et al. 2009. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet 5: e1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, Callis J, Jacobsen SE. 2007. The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 17: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LM, Law JA, Khattar A, Henderson IR, Jacobsen SE. 2008. SRA-domain proteins required for DRM2-mediated de novo DNA methylation. PLoS Genet 4: e1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien PE, Susaki D, Yelagandula R, Higashiyama T, Berger F. 2012. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr Biol 22: 1825–1830. [DOI] [PubMed] [Google Scholar]
- Karimi M, Bleys A, Vanderhaeghen R, Hilson P. 2007. Building blocks for plant gene assembly. Plant Physiol 145: 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Berger F. 2014. Epigenetic reprogramming in plant sexual reproduction. Nat Rev Genet 15: 613–624. [DOI] [PubMed] [Google Scholar]
- Lampropoulos A, Sutikovic Z, Wenzl C, Maegele I, Lohmann JU, Forner J. 2013. GreenGate—a novel, versatile, and efficient cloning system for plant transgenesis. PLoS One 8: e83043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu A, Champion A, Legrand J, Lavenus J, Mast D, Brunoud G, Oh J, Guyomarc'h S, Pizot M, Farmer EE, et al. 2015. A fluorescent hormone biosensor reveals the dynamics of jasmonate signalling in plants. Nat Commun 6: 6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau OS, Bergmann DC. 2015. MOBE-ChIP: a large-scale chromatin immunoprecipitation assay for cell type-specific studies. Plant J 84: 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dai H, Martos SN, Xu B, Gao Y, Li T, Zhu G, Schones DE, Wang Z. 2015. Distinct roles of DNMT1-dependent and DNMT1-independent methylation patterns in the genome of mouse embryonic stem cells. Genome Biol 16: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et al. 2004. Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471–476. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. 2008. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen RA, Colot V. 2001. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293: 1070–1074. [DOI] [PubMed] [Google Scholar]
- Martinez G, Panda K, Kohler C, Slotkin RK. 2016. Silencing in sperm cells is directed by RNA movement from the surrounding nurse cell. Nat Plants 2: 16030. [DOI] [PubMed] [Google Scholar]
- Matzke MA, Kanno T, Matzke AJ. 2015. RNA-directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu Rev Plant Biol 66: 243–267. [DOI] [PubMed] [Google Scholar]
- Moreno-Romero J, Jiang H, Santos-Gonzalez J, Kohler C. 2016. Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm. EMBO J 35: 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamagata K, Hong K, Wakayama T, Zhang Y. 2010. A role for the elongator complex in zygotic paternal genome demethylation. Nature 463: 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Kim MY, Vickers M, Park JS, Hyun Y, Okamoto T, Zilberman D, Fischer RL, Feng X, Choi Y, et al. 2016. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc Natl Acad Sci 113: 15138–15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszkowski J, Grossniklaus U. 2011. Selected aspects of transgenerational epigenetic inheritance and resetting in plants. Curr Opin Plant Biol 14: 195–203. [DOI] [PubMed] [Google Scholar]
- Pillot M, Baroux C, Vazquez MA, Autran D, Leblanc O, Vielle-Calzada JP, Grossniklaus U, Grimanelli D. 2010. Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell 22: 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontier D, Yahubyan G, Vega D, Bulski A, Saez-Vasquez J, Hakimi MA, Lerbs-Mache S, Colot V, Lagrange T. 2005. Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev 19: 2030–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulski M, Lu Z, Kendall J, Donoghue MT, Reinders J, Llaca V, Deschamps S, Smith A, Levy D, McCombie WR, et al. 2013. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res 23: 1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders J, Wulff BB, Mirouze M, Mari-Ordonez A, Dapp M, Rozhon W, Bucher E, Theiler G, Paszkowski J. 2009. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev 23: 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Berard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al. 2011. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 30: 1928–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H, Mittelsten Scheid O, Paszkowski J. 2003. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34: 65–69. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Ecker JR. 2012. Epigenetic and epigenomic variation in Arabidopsis thaliana. Trends Plant Sci 17: 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Lewsey MG, O'Malley RC, Urich MA, Libiger O, Schork NJ, Ecker JR. 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 334: 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoft VK, Chumak N, Mosiolek M, Slusarz L, Komnenovic V, Brownfield L, Twell D, Kakutani T, Tamaru H. 2009. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep 10: 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secco D, Wang C, Shou H, Schultz MD, Chiarenza S, Nussaume L, Ecker JR, Whelan J, Lister R. 2015. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. Elife 4: e09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. 2013. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc Lond B Biol Sci 368: 20110330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She W, Grimanelli D, Rutowicz K, Whitehead MW, Puzio M, Kotlinski M, Jerzmanowski A, Baroux C. 2013. Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development 140: 4008–4019. [DOI] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. 2014. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira FK, Heredia F, Sarazin A, Roudier F, Boccara M, Ciaudo C, Cruaud C, Poulain J, Berdasco M, Fraga MF, et al. 2009. A role for RNAi in the selective correction of DNA methylation defects. Science 323: 1600–1604. [DOI] [PubMed] [Google Scholar]
- Vongs A, Kakutani T, Martienssen RA, Richards EJ. 1993. Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928. [DOI] [PubMed] [Google Scholar]
- Weigel D, Colot V. 2012. Epialleles in plant evolution. Genome Biol 13: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Custard KD, Brown RC, Lemmon BE, Harada JJ, Goldberg RB, Fischer RL. 2006. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 18: 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K. 2010. DNA methylation profiling using live-cell imaging. Methods 52: 259–266. [DOI] [PubMed] [Google Scholar]
- Yelagandula R, Stroud H, Holec S, Zhou K, Feng S, Zhong X, Muthurajan UM, Nie X, Kawashima T, Groth M, et al. 2014. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 158: 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Grafi G. 2003. Characterization of Arabidopsis thaliana methyl-CpG-binding domain (MBD) proteins. Plant J 34: 565–572. [DOI] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. 2013. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, et al. 2006. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126: 1189–1201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.