

Abstract
APOBEC3A/B cytidine deaminase is responsible for the majority of cancerous mutations in a large fraction of cancer samples. However, its role in heritable mutagenesis remains very poorly understood. Recent studies have demonstrated that both in yeast and in human cancerous cells, most APOBEC3A/B-induced mutations occur on the lagging strand during replication and on the nontemplate strand of transcribed regions. Here, we use data on rare human polymorphisms, interspecies divergence, and de novo mutations to study germline mutagenesis and to analyze mutations at nucleotide contexts prone to attack by APOBEC3A/B. We show that such mutations occur preferentially on the lagging strand and on nontemplate strands of transcribed regions. Moreover, we demonstrate that APOBEC3A/B-like mutations tend to produce strand-coordinated clusters, which are also biased toward the lagging strand. Finally, we show that the mutation rate is increased 3′ of C→G mutations to a greater extent than 3′ of C→T mutations, suggesting pervasive trans-lesion bypass of the APOBEC3A/B-induced damage. Our study demonstrates that 20% of C→T and C→G mutations in the TpCpW context—where W denotes A or T, segregating as polymorphisms in human population—or 1.4% of all heritable mutations are attributable to APOBEC3A/B activity.
Understanding the processes responsible for heritable mutations is important for a broad range of evolutionary, population genetics, and medical questions (Shendure and Akey 2015). Recent studies have shown that different numbers of de novo mutations are inherited from the father and from the mother, with the contribution of paternal mutations being two to four times higher (Kong et al. 2012; Francioli et al. 2015; Wong et al. 2016; Yuen et al. 2016). Additionally, the number of de novo mutations strongly depends on the father's age at conception (Kong et al. 2012; Francioli et al. 2015; Wong et al. 2016; Yuen et al. 2016) and, to a lesser extent, on the mother's age at conception (Goldmann et al. 2016; Wong et al. 2016). At the molecular level, the understanding of mechanisms of heritable mutagenesis is very limited. Only a few of the mutation types can be attributed to specific molecular processes. Most prominently, the CpG→TpG substitutions are known to result from spontaneous cytosine deamination of methyl-cytosine in the CpG context together with poor efficiency of subsequent base excision repair (Pfeifer 2006; Chen et al. 2014); some mutations in the CpCpC motif arise due to activity of the APOBEC3G protein, which is normally involved in protection against viruses and retroelements (Knisbacher and Levanon 2016; Pinto et al. 2016), and small insertions and deletions result from polymerase slippage on homonucleotide tracts and tandem repeats (Montgomery et al. 2013).
The sources of somatic mutations, in particular those in cancers, are better understood. The rates of such mutations were related to age-dependent cytosine deamination, to activity of APOBEC3A/B/G and AID, to deficiencies in systems responsible for the fidelity of DNA repair and replication, and to exposure to external and internal mutagens (Alexandrov et al. 2013; Lawrence et al. 2013).
APOBEC3A/B-induced mutations were described for many cancer types (Alexandrov et al. 2013; Burns et al. 2013b; Roberts et al. 2013). Mutations produced by APOBEC3A/B have known properties confirmed both in yeast and in human cancers: (1) They are C→D mutations in the TpCpN context (the more specific APOBEC3A/B signature is TpCpW→K, where D denotes A, T, or G; W denotes A or T; and K denotes G or T) (Burns et al. 2013a; Taylor et al. 2013; Chan et al. 2015; Seplyarskiy et al. 2016); (2) they often form strand-coordinated clusters (Nik-Zainal et al. 2012; Roberts et al. 2013; Taylor et al. 2013); (3) they are strongly biased toward the lagging strand during replication (Haradhvala et al. 2016; Hoopes et al. 2016; Morganella et al. 2016; Nik-Zainal et al. 2016; Seplyarskiy et al. 2016); (4) they are biased toward the nontranscribed strand, at least in breast and bladder cancer (Nordentoft et al. 2014; Morganella et al. 2016); and (5) cytosines deaminated to uracils by APOBEC frequently result in C→G substitutions. According to the current models, these mutations arise due to incomplete repair of U-G mismatches resulting in abasic sites. In turn, abasic sites are bypassed by REV1, which inserts exactly 1 nucleotide (nt) opposite to the site, and this single-nucleotide primer is then extended by the low-fidelity polymerase ζ (Nik-Zainal et al. 2012; Chan et al. 2013; Seplyarskiy et al. 2015). Additionally, as we show here, (6) strand-coordinated clusters in cancers are very strongly biased toward the lagging strand.
We previously demonstrated that fork polarity (fp) calculated as the derivative of replication timing allows to discriminate between the strands replicated as leading versus lagging (Seplyarskiy et al. 2016) and that accumulation of APOBEC3A/B mutations in human cancers is strongly strand specific. Here, we asked if this specificity, as well as other properties of APOBEC3A/B-induced mutagenesis, is also manifested in heritable mutations.
Results
Heritable mutations in the APOBEC3A/B context are 20% more frequent on the lagging strand
Rare polymorphisms tend to be young (Mathieson and McVean 2014). Therefore, their mutational spectra and genomic distribution much better reflect the spectra of de novo mutations compared with the spectra and distribution of substitutions between species that are affected by nonmutational processes such as selection or biased gene conversion operating over the lifetime of a mutation as it spreads to fixation (Rahbari et al. 2016; Terekhanova et al. 2016). To study heritable mutations, we thus mainly focused on rare polymorphisms (The 1000 Genomes Project Consortium 2015), i.e., those with the frequency of the derived allele in the human population <1%; we excluded singletons from the main analyses as they are enriched in cell line artefacts (Mathieson and Reich 2016).
We measured the ratio of the frequencies of C→K mutations to those of complementary G→M (where M denotes A or C) mutations in different contexts and asked how this ratio depends on whether the analyzed strand is preferentially replicated as leading or lagging. The rates of both provisionally APOBEC3A/B-induced mutation types (to T and to G) in both APOBEC3A/B contexts (TpCpT and TpCpA) were 10%–20% higher on the lagging strand (Fig. 1), while a weak bias was observed in only one of the comparisons for the corresponding mutations in the non-APOBEC3A/B VpCpW context (where V denotes A, C, or G) (Fig. 1A). Among the 24 possible mutations in NpCpW contexts, the TpCpW→K mutations are the most asymmetric. These estimates hold for all seven cell types with measured replication timing (Table 1; Supplemental Table S1).
Figure 1.

Mutations in different APOBEC3A/B contexts are more frequent on the lagging strand (A–D). Horizontal axis indicates the propensity of the region of the DNA strand to be replicated as lagging or leading; vertical axis, ratio of the frequencies of the two complementary mutation types on the strand in this category. Vertical bars represent 95% confidence intervals. V corresponds to A, C, or G.
Table 1.
APOBEC3A/B-driven replication asymmetry is concordantly observed in seven cell types with known RT
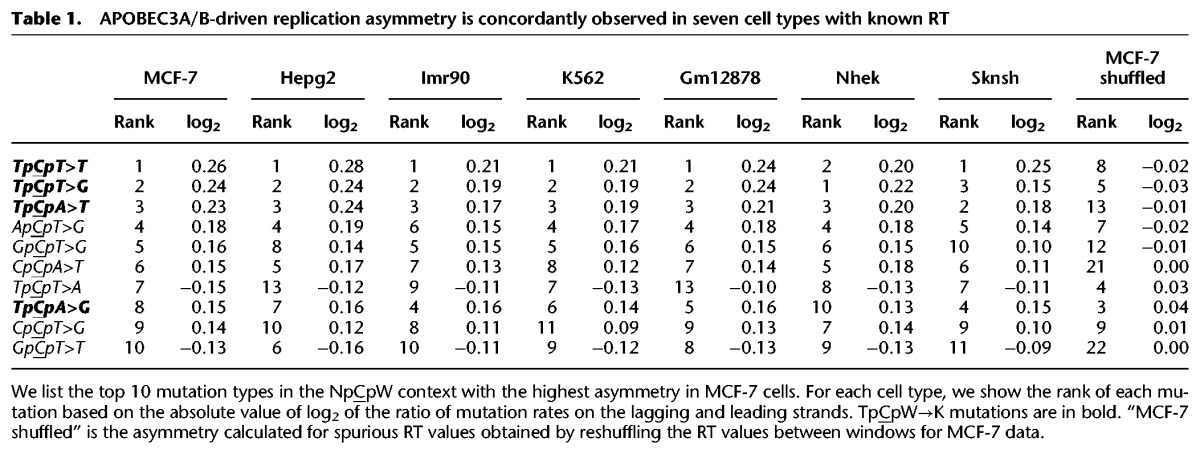
The context specificity of the asymmetry, together with the known lagging strand preferences of APOBEC3A/B-induced mutations, suggests that the observed bias is caused by an excess of APOBEC3A/B-induced mutations on the lagging strand. If so, we can estimate the contribution of APOBEC3A/B to mutagenesis, assuming that it is the sole reason for these biases (for details, see Methods). Under these assumptions, we infer that ∼15%–30% of TpCpW→K substitutions resulted from APOBEC3A/B deamination, with a lower fraction for the TpCpA→G mutations (∼15%). The differences in strand bias between different APOBEC3A/B contexts are in line with lower frequencies of TpCpA→G among APOBEC3A/B-induced mutations in cancers (Alexandrov et al. 2013) and among APOBEC3B-induced mutations in human cells lines (Akre et al. 2016). Similar biases were observed for SNPs at all frequencies (Supplemental Table S2) and for interspecies differences accumulated in the human lineage since divergence from the last common ancestor with the chimpanzee, albeit they are slightly less obvious for divergence (Supplemental Table S2; Supplemental Fig. S1). Possibly, slightly weaker asymmetry observed in divergence reflect minor changes in RT, as it was shown that RT varies even within the human population (Koren et al. 2014).
APOBEC3A- and APOBEC3B-induced mutations preferentially occur when, respectively, Y (T or C) or R (A or G) is observed 2 nt upstream of the mutated C (Taylor et al. 2013; Chan et al. 2015). Therefore, by analyzing this extended context, we are able to discriminate between these two enzymes. The strand bias is stronger for YpTpCpW contexts than for RpTpCpW contexts (P < 0.0024 for all comparisons) (Supplemental Fig. S2), suggesting that similarly to cancers with a high burden of APOBEC-induced mutations (Chan et al. 2015), APOBEC3A likely contributes more to the observed mutations than APOBEC3B.
In cancers most affected by APOBEC3A/B-induced mutagenesis, the mutation rate in the TpCpW context is increased by up to 40-fold compared with the VpCpW context (Seplyarskiy et al. 2016). From strand asymmetry, we estimate that only 15%–30% of heritable TpCpW→K mutations are induced by APOBEC3A/B. We asked whether this APOBEC3A/B-induced mutagenesis is also manifested in an increased genome-wide mutation rate in the corresponding nucleotide context. However, the frequencies of TpCpW→K were not uniformly higher than the frequencies of VpCpW→K mutations (Supplemental Table S3). This is probably because mechanisms other than those associated with APOBEC3A/B, with their own context specificities (Supplemental Fig. S3; Aggarwala and Voight 2016), contribute more to polymorphism data compared with cancer samples where APOBEC3A/B virtually monopolizes the mutation process.
Heritable mutations in the APOBEC3A/B context are more frequent on the nontemplate strands of transcribed regions
To understand other genomic features that could be associated with APOBEC3A/B activity, we analyzed the density of rare polymorphisms in the APOBEC3A/B context in genomic regions that may be prone to deamination by APOBEC3A/B: transcribed regions, recombination hotspots, and expressed transposons. No APOBEC3A/B-specific differences from the background rate were observed in recombination hotspots or in transposons (Supplemental Fig. S4).
For transcribed regions, we saw evidence for APOBEC3A/B activity at the nontemplate strand. Here, the rate of TpCpW→K mutations was increased compared with intergenic regions (Fig. 2A). No such increase was observed for the template strand (Fig. 2B). As a result, TpCpW→K mutations were strongly biased toward the nontemplate strand. Indeed, at the nontemplate strand, the TpCpT→G mutations had the strongest mutational asymmetry among all 24 mutations in the NpCpW contexts, and other TpCpW→K mutations were also biased toward the nontranscribed strand (Fig. 2C). These observations are in line with the transcriptional asymmetry of APOBEC3A/B-induced mutations in bladder cancer and breast cancer (Nordentoft et al. 2014; Morganella et al. 2016) and likely reflect the accessibility of the nontranscribed strand to APOBEC3A/B, because the nontemplate strand is prone to single-strandedness (Skourti-Stathaki and Proudfoot 2014). The replication asymmetry of TpCpW→K mutations is observed both in transcribed and nontranscribed regions, implying that it is independent of the effect of transcription, although the extent of this asymmetry differs slightly between the template and the nontemplate strands because of contribution of chain-specific mutations (Fig. 2; Supplemental Table S4).
Figure 2.

APOBEC3A/B-induced mutations occur preferentially on the nontemplate strand. Color-coded log2 of the ratio of densities of rare polymorphisms on the nontemplate strand (A) or the template strand (B) and intergenic regions, or of the ratio of densities of rare polymorphisms on the nontemplate and template strands (C). Mutations in the TpCpW→K context that may be associated with APOBEC3A/B activity are in green boxes.
Heritable mutations in APOBEC3A/B context tend to form strand-coordinated clusters
As APOBEC family enzymes deaminate single-stranded DNA, they tend to produce strand-coordinated clusters. To study such clusters, we focused on pairs of rare single-nucleotide polymorphisms (SNPs) in strong linkage at distances of up to 5000 nt from each other (for details, see Methods). Mutations at sites closer than 10 nt to each other occur due to activity of low fidelity polymerases or other mechanisms not related to APOBEC3A/B (Terekhanova et al. 2013; Harris and Nielsen 2014; Zhu et al. 2015); therefore, we excluded such pairs from our analysis. Over half of the remaining pairs of TpCpW→K mutations were strand coordinated, and for three out of the four considered mutation types, the fraction of strand-coordinated pairs was significantly higher than for the same mutations in the VpCpW context used as a control (Fig. 3A–D). Similar results were obtained for interspecies divergence data (Supplemental Table S5). Conversely, strand-coordinated clusters composed of many (more than six) C→K mutations were enriched in mutations in an APOBEC3A/B-prone context. Indeed, in such clusters, the fraction of TpCpW→K mutations was about three times higher than in noncoordinated clusters with similar properties (Fig. 3E). Moreover, many of the strand-coordinated clusters included only TpCpN→K mutations (Supplemental Table S6) and likely represented pure traces of APOBEC3A/B activity.
Figure 3.
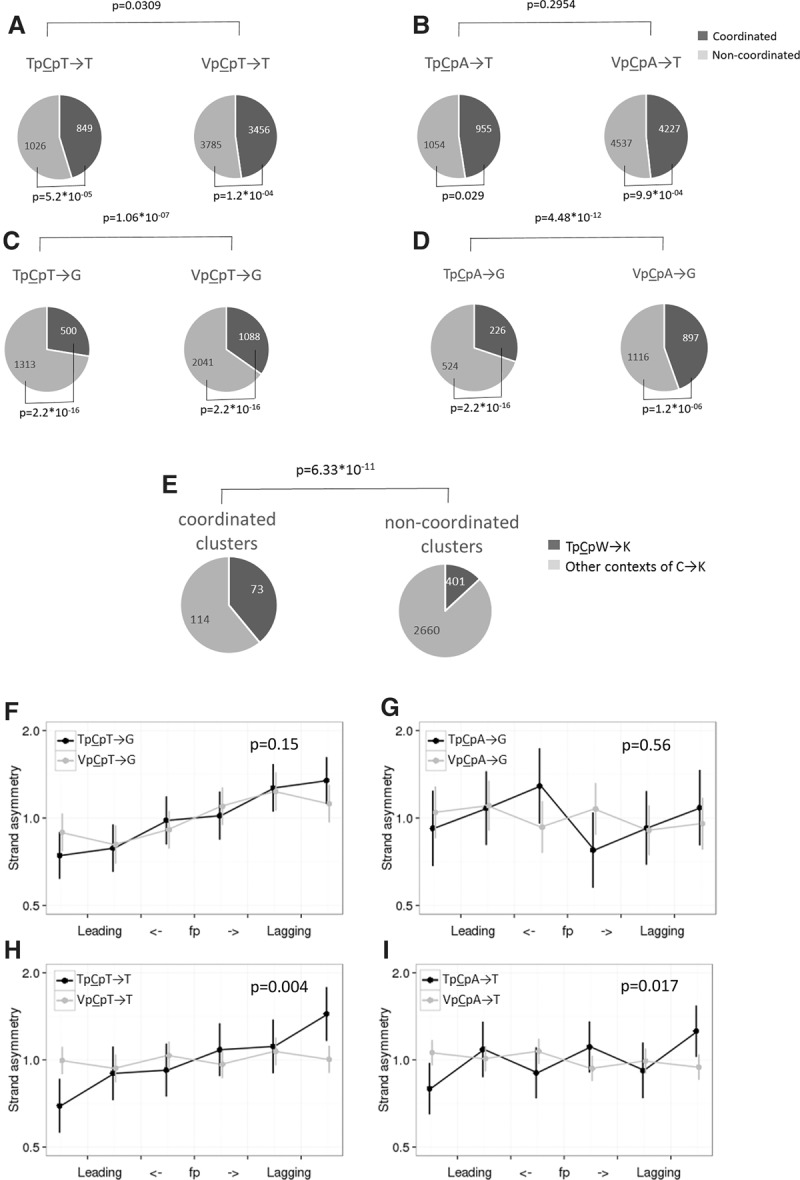
Clustered mutations in the TpCpW→K context are enriched in strand-coordinated clusters and biased toward the lagging strand. (A–D) Pairs of linked SNPs in contexts prone to APOBEC3A/B-induced mutations tend to be strand coordinated. (E) Strand-coordinated clusters that are composed exclusively of C→K mutations and contain at least six mutations are approximately threefold enriched in mutations in the TpCpW context, compared with noncoordinated clusters. (F–I) Strand-coordinated pairs are biased toward the lagging strand. The axes and notation are as in Figure 1. The P-value is calculated for the differences between the asymmetries of TpCpW→K and VpCpW→K mutations in the regions with the highest fork polarity (i.e., the first and the last bin).
APOBEC3A/B-induced mutations are biased toward the lagging strand in cancer and in yeasts. We asked whether this pattern is also observed for strand-coordinated clusters. In cancers with a strong prevalence of APOBEC3A/B-related mutagenesis, strand-coordinated clusters occur sixfold more frequently on the lagging strand, demonstrating the highest level of replicative strand asymmetry reported for mammalian cells (Supplemental Fig. S5). Therefore, we would expect an excess of strand-coordinated pairs of heritable TpCpW→K mutations on the lagging strand if APOBEC3A/B plays a role in their formation. Indeed, clustered mutations preferentially occur on the lagging strand (Fig. 3F–I), and the level of strand asymmetry for them is higher than that for dispersed mutations (Supplemental Fig. S6), reflecting an enrichment of APOBEC3A/B-induced clusters among strand-coordinated clusters.
An unknown mechanism produces clusters of heritable NpCpT→G mutations
Notably, a tendency to form strand-coordinated clusters was observed not only for the TpCpW→K mutations but also, to a lesser extent, for the VpCpW→K mutations (Fig. 3A–D). To better understand this, we focused on the VpCpT→G mutations for which this trend is particularly strong (Fig. 3C) and which are also biased toward the lagging strand (Fig. 1A). Notably, C→G is among the most frequent mutations in de novo mutational clusters (Francioli et al. 2015). We investigated C→G mutations in more detail in two recently published whole-genome data sets on human trios (Francioli et al. 2015; Wong et al. 2016) and found that within 23 pairs of C→G mutations at distances of up to 20 kb from each other, 18 were strand coordinated, which is more than expected if they were independent (P = 0.01) (Fig. 4A). The context preferences of the clustered C→G mutations differed from those of nonclustered mutations (P = 0.019), with the clustered C→G mutations enriched in the NpCpT→G context (P = 0.04) (Fig. 4C). Therefore, while the mechanism behind the strand-coordinated NpCpT→G mutations remains unclear, they are observed in all data types.
Figure 4.

Properties of C→G mutations involved in clusters of de novo mutations. (A) Pairs of C→G de novo mutations tend to form strand-coordinated clusters. (B,C) Fractions of different nucleotides at adjacent sites 5′ (B) and 3′ (C) of the mutated position calculated for clustered and dispersed C→G de novo mutations. Vertical bars represent 95% confidence intervals. (*) P < 0.05 (χ2 test).
Density of SNPs is increased at 3′ from C→G mutation
In cancers, up to half of APOBEC3A/B-induced mutations are C→G. These mutations arise due to bypass of abasic sites that originate from unfinished repair of cytosines deaminated by APOBEC3A/B. These and other DNA damages may be bypassed by low fidelity polymerase ζ. It introduces C>G mutations and is recruited to bypass different DNA damages (Diaz et al. 2003; Helleday et al. 2014) by replicating a stretch of DNA downstream from the damage (Kochenova et al. 2015). If similar processes affect heritable mutations, we expect to observe a higher mutation rate at 3′ of the C→G mutations that mark polymerase ζ–dependent DNA synthesis. We compared the mutation rates at distances of up to 5 kb 5′ and 3′ of TpCpW→K mutations. The mutation rates were increased ∼1–2 kb 3′ of C→G mutations in the APOBEC3A/B contexts both on the leading and the lagging strand (Fig. 5A,C,E,G). A higher mutation rate 3′ of TpCpW→G SNPs on both replicative strands at distances of up to 2 kb is in agreement with the key role of polymerase ζ in generation of these mutations and with its known low (∼1 kb) processivity (Kochenova et al. 2015). However, an increase in the mutation rate is also observed 3′ of VpCpW→G mutations (Supplemental Fig. S7) and on both chains in the case of APOBEC3A/B-prone context, implying that the role of polymerase ζ in the accumulation of heritable C→G mutations is not limited to bypassing of APOBEC3A/B-induced abasic sites.
Figure 5.
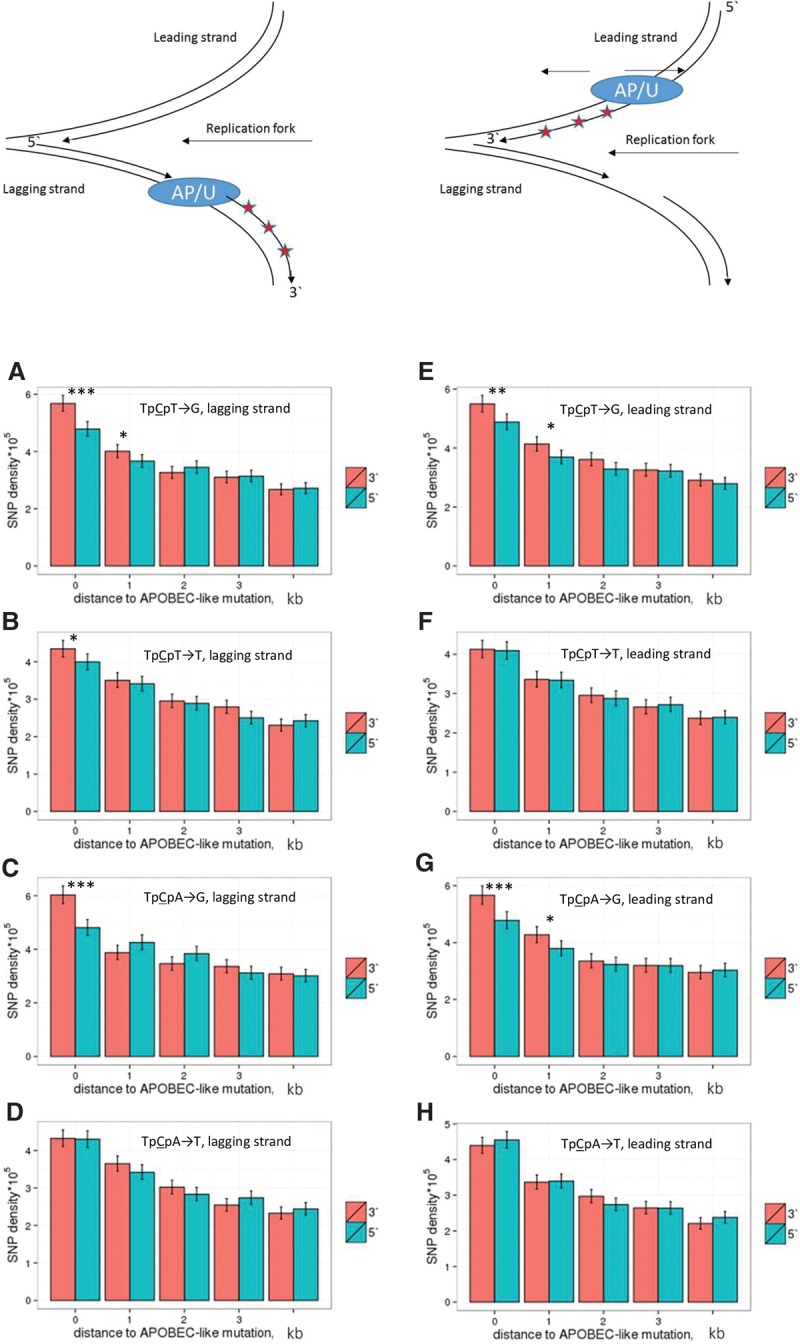
Density of linked SNPs 5′ and 3′ of a TpCpW→K mutation. In the schematic depiction of a replication fork at the top of the figure, red asterisks correspond to de novo mutations, and AP/U is the position of the TpCpW→G mutation interpreted as the position of the abasic site or the position of the TpCpW→T mutation interpreted as the position of the uracil. The rate of mutations on the lagging (A–D) or the leading (E–H) strand is measured in five nonoverlapping 1-kb windows at increasing distance from the TpCpW→K SNPs. (***) P < 0.001; (**) P < 0.01; (*) P < 0.05 (χ2 test).
A weaker effect was observed in one of the four comparisons for the C→T mutation (Fig. 5B): a mutation rate asymmetry in the vicinity of TpCpT→T mutations on the lagging strand. This effect is particularly strong in the 400 nt nearest to the C→T mutation (Supplemental Fig. S8). The TpCpT→T mutations demonstrate the strongest bias toward the lagging strand compared with the control mutation types (Fig. 1), suggesting that APOBEC3A/B causes a high fraction of these mutations on the lagging strand. Therefore, our observations suggest that the mutation rate is increased 3′ of APOBEC3A/B-induced mutations. The difference between the mutation rates 5′ and 3′ of the mutation of interest is observed only for linked SNPs (Supplemental Fig. S9), implying that this association is due to the co-occurrence of mutations in a single mutational event rather than some underlying properties of the corresponding genomic regions.
The rate of all mutations is also strongly increased in the vicinity of a SNP in the same haplotype (Fig. 5), in line with previous results (Schrider et al. 2011; Terekhanova et al. 2013; Harris and Nielsen 2014; Zhu et al. 2015). This effect is weaker and decays more gradually with distance between the mutations not linked with each other compared with linked mutations (cf. Supplemental Fig. S9 and Fig. 5).
Discussion
Recent studies have linked most somatic mutations, that is, mutations associated with cancers or with dedifferentiation of induced pluripotent stem cells, with specific processes (Alexandrov et al. 2013; Lawrence et al. 2013; Rouhani et al. 2016). In contrast, only a minority of heritable mutations are attributable to known mechanisms. Analyses of clustered mutations and L1 transposons have revealed a major role of low fidelity polymerase ζ and APOBEC3G in the generation of heritable mutations (Harris and Nielsen 2014; Seplyarskiy et al. 2015; Zhu et al. 2015; Knisbacher and Levanon 2016; Pinto et al. 2016). Direct experiments have also uncovered the role of recombination in mutagenesis (Arbeithuber et al. 2015; Yang et al. 2015). Still, the causes of many described and pervasive patterns observed in heritable mutations such as the cryptic variation of the site-specific mutation rate and heterogeneity of mutational spectra (Hodgkinson et al. 2009; Johnson and Hellmann 2011; Seplyarskiy et al. 2012) remain unknown.
Among mutation types, clusters of adjacent or nearby mutations can be attributed to specific mutational mechanisms most reliably, as chance occurrence of such clusters by conventional mechanisms is unlikely. However, the majority of mutations are dispersed, and few methods to study their origin are available. A type of mutation can be relatively easily linked to a specific mechanism only if its rate is unusually high, as is the case for CpG→T mutations.
Here, we show that APOBEC3A/B contributes substantially to heritable mutagenesis. We show that TpCpW→K mutations in human polymorphism and divergence exhibit all the properties of APOBEC3A/B-induced mutations, unlike non-APOBEC-induced VpCpW→K mutations. We estimate that ∼20% of heritable TpCpW→K mutations, or 1.4% of all heritable mutations, are linked to APOBEC3A/B activity, corresponding to 970,000 of mutations from the 1000 Genomes Project Consortium (2015).
Replication asymmetry of TpCpW→K mutations is not due to asymmetry in coreplicative repair or error rate of replicative polymerases
Many mutations are associated with replication and preferentially occur at segments of DNA strands replicated as leading or lagging. A computational approach has been developed to discriminate, by determining the prevalent direction of the replication fork at each genomic region, between the leading and the lagging DNA strands (Chen et al. 2011; Baker et al. 2012; Haradhvala et al. 2016; Seplyarskiy et al. 2016), providing a powerful tool to investigate strand-biased mechanisms that give rise even to dispersed mutations (Chen et al. 2011; Baker et al. 2012; Haradhvala et al. 2016; Morganella et al. 2016; Nik-Zainal et al. 2016; Seplyarskiy et al. 2016). APOBEC3A/B-induced mutations, as well as mutations in mismatch repair (MMR)-deficient cells, are strongly biased toward the lagging strand (Lujan et al. 2012; Andrianova et al. 2016; Haradhvala et al. 2016; Hoopes et al. 2016; Seplyarskiy et al. 2016). Here, we show that such biases in the germline shape the mutational lanscape that gives rise to human variation.
Conceivably, the observed replication asymmetry could reflect accumulation of nonrepaired mismatches during DNA doubling. Indeed, mutations accumulated in cells with a deficiency in MMR or decreased fidelity of major replicative leading or lagging strand polymerases have a strong replicative asymmetry (Lujan et al. 2012, 2014; Andrianova et al. 2016; Haradhvala et al. 2016). However, the direction of the observed replicative asymmetry is inconsistent with this. Indeed, in cancer cells prone to mutations caused by wild-type polymerases or by polymerase ε or polymerase δ without exonuclease activity, C→T mutations are accumulated on the leading strand (Andrianova et al. 2016). In contrast, heritable C→T mutations in the TpCpW context preferentially occurred on the lagging strand (Fig. 1). Therefore, a specific context-dependent coreplicative mutational process needs to be invoked.
APOBEC3A and/or APOBEC3B proteins are most plausible causes of the replicative asymmetry
The asymmetry is most pronounced for cytosines in the TpCpW context, is associated with DNA replication, and produces strand-coordinated clusters, strongly suggesting that a protein from the APOBEC family plays a key role. While deamination in the TpCpW context excludes some APOBECs from consideration, APOBEC1, APOBEC3A, APOBEC3B, APOBEC3F, and APOBEC3H (Taylor et al. 2013; Kim et al. 2014; Saraconi et al. 2014) all are plausible suspects. Overexpression of APOBEC1, APOBEC3A, and APOBEC3B is associated with increased mutation rate in vertebrate cell lines, with mutations distributed all over the genome (Saraconi et al. 2014; Akre et al. 2016; Green et al. 2016). Notably, however, APOBEC1 causes C→A mutations in experimental systems (Saraconi et al. 2014), while the heritable replication asymmetry is largely restricted to C→T and C→G mutations (Table 1). Recently, one of the alleles of APOBEC3H has been linked with lung cancer (Starrett et al. 2016), suggesting this protein as another possible candidate. However, this APOBEC3H allele is barely stable and has only a weak effect even in specific essays (Starrett et al. 2016), arguing against the role of APOBEC3H in the patterns observed in germline. For APOBEC3A and APOBEC3B, mutations are known to accumulate on the lagging strand (Haradhvala et al. 2016; Hoopes et al. 2016; Seplyarskiy et al. 2016), and mutagenesis is associated with replication (Green et al. 2016). Finally, APOBEC3A and APOBEC3B are major mutators in a broad range of cancer types (Alexandrov et al. 2013; Burns et al. 2013b; Roberts et al. 2013). Therefore, although we cannot indisputably exclude other APOBECs, indirect evidence concordantly suggests APOBEC3A and APOBEC3B as the most probable candidates. Our analysis of extended contexts make APOBEC3A a somewhat better explanation than APOBEC3B (Supplemental Fig. S2), although both proteins may contribute.
Leading vs. lagging strand bias indicates that 20% of heritable mutations are induced by APOBEC3A/B
For all types of heritable single-nucleotide substitutions, the asymmetry between the leading and the lagging strand is weak (Chen et al. 2011; Andrianova et al. 2016). Therefore, even a small admixture of mutations that are two to three times more prevalent on one of the strands can be detected (Fig. 1), and the amount of such admixture can be estimated from the level of asymmetry. De novo TpCpW→K mutations obtained from Francioli et al. (2015) were inferred to be 1.38 times more frequent on the lagging strand than on the leading strand (Haradhvala et al. 2016). In our analyses of the same data set, the corresponding ratio is 1.23 but is not significantly different from 1.0 (P > 0.05). The difference between the results is likely due to differences in how fp is measured. The only assumption made by the method we use is that replication fork velocity is nearly constant throughout the genome (Guilbaud et al. 2011; Baker et al. 2012), and this assumption has been confirmed experimentally (Guilbaud et al. 2011; Baker et al. 2012). Still, the strand asymmetry of TpCpW→K de novo mutations in a larger joined data set (Francioli et al. 2015; Wong et al. 2016) yielded a significant 1.17-fold difference between strands (P-value = 0.0174). This is in line with the approximately 1.15-fold asymmetry observed for rare SNPs. The observed ∼15% asymmetry corresponds to a ∼20% admixture of APOBEC3A/B-induced mutations. Still, the effect of adjacent nucleotides on the mutation rate appears to mask this contribution of APOBEC3A/B, so that we do not observe any tendency of C→K mutations to occur in the TpCpW context among de novo mutations or in polymorphism data (Supplemental Tables S3, S7), in line with Francioli et al. (2015).
Our results show that knowledge of replication fork direction may be used to estimate the contribution of a specific process to heritable mutagenesis. We suggest that a similar set of approaches can be also used to quantitatively estimate the fraction of APOBEC3G-induced mutations, especially among dispersed mutations, because recent experiments on bacteria showed that these mutations, too, are strand asymmetric (Bhagwat et al. 2016).
TpCpW→K mutations demonstrate transcriptional asymmetry
Some mutation types are known to be transcriptionally asymmetric in human germline or soma (Green et al. 2003; Polak and Arndt 2008; Mugal et al. 2009; Pleasance et al. 2010; Nordentoft et al. 2014; Haradhvala et al. 2016). Here, we describe context-specific transcriptional asymmetry for C→T and C→G mutations in the germline (Fig. 2C). This asymmetry is elevated in TpCpW contexts. Transcription-coupled repair is the main source of transcriptional asymmetry in cancers (Pleasance et al. 2010; Haradhvala et al. 2016). However, if transcription-coupled repair was the main cause of the observed transcriptional asymmetry in the germline, we would not expect the mutation rate to be elevated at either of the strands compared with the intergenic regions. In contrast, we found that TpCpW→K SNPs were more frequent on the nontemplate strand compared with intergenic regions (Fig. 2A). Genes tend to be located in regions of early RT (Farkash-Amar et al. 2008) and experience high levels of background selection (Mu et al. 2011); these factors could also reduce the number of SNPs but should not increase it, especially in a strand-specific manner. Therefore, a specific mutational mechanism needs to be invoked to explain the bias of TpCpW→K mutations toward the nontemplate strand. A similar asymmetry in the same context has been described for APOBEC3A/B-induced mutations in breast and bladder cancer (Nordentoft et al. 2014; Morganella et al. 2016), suggesting that the transcriptional asymmetry likely reflects APOBEC3A/B-induced heritable mutations on the nontemplate strand. As APOBEC3A/B-induced mutagenesis affects single-stranded DNA, we suggest that, alongside the lagging strand during replication, it may also produce mutations on the nontemplate strand in transcribed regions, due to single strangeness of this strand during R loop formation (Skourti-Stathaki and Proudfoot 2014). The asymmetry observed in the CpCpW context may be related to the APOBEC3G transcription–associated activity that has been shown recently (Pinto et al. 2016).
Fraction of APOBEC3A/B-induced mutations is high in strand-coordinated clusters
In cancer, APOBEC3A/B gives rise to mutational clusters spanning ∼10 kb (Nik-Zainal et al. 2012; Roberts et al. 2013). Linked SNPs, especially young ones, may be used to study such events, as mutational clusters observed in them have not yet been disrupted by recombination and are still detectible at distances of a few kb (Harris and Nielsen 2014). Clustered mutations in the TpCpW context are enriched in strand-coordinated clusters by a factor of up to two. Moreover, for strand-coordinated clusters, we observed a stronger bias toward the lagging strand than for dispersed mutations. Thus, the orthogonal approach based on strand coordination also detects the prevalence of APOBEC3A/B-induced mutations.
Contribution of APOBEC3A/B-induced mutations to the mutation load in humans
APOBEC3A/B-induced mutations fuel cancer development, representing the second most prevalent mutational signature in it. Therefore, the cancer-related activity of APOBEC3A/B should be slightly deleterious, although selection against it may be weak (Martincorena and Campbell 2015). Here, we have shown that APOBEC3A/B also causes heritable mutations, thus increasing the mutation load. The fact that it is conserved by negative selection implies that its positive role in protection against retroelements or viruses outweighs its deleterious mutability. Thus, the maintenance of APOBEC3A/B represents a tradeoff between its advantageous function and deleterious mutagenesis.
Methods
Mutational data
The main set of results was obtained using polymorphism data from the 1000 Genomes Project Consortium (2015). We excluded exons and 10 nt adjacent to each exon to reduce the contribution of selection, and we only considered nonsingleton variants with low (<1%) derived allele frequency, using the ancestral variant determined by the 1000 Genomes Project Consortium (2015). Clusters were defined as pair or more of SNPs of a particular type at distances between 10 and 5000 nt from each other, such that at least half of the genotypes carrying the derived allele for one of the SNPs also carry the derived allele for any other SNP within cluster and vice versa. The same criteria were used to subdivide SNPs as linked (Fig. 5; Supplemental Figs. S8, S9) or unlinked (Supplemental Fig. S7) in analyses of SNP densities 5′ or 3′ of the considered SNP. For analyses of interspecies divergence, we used the human–chimpanzee–orangutan multiple alignment from the UCSC Genome Browser (https://genome.ucsc.edu/). We inferred substitutions in the human lineage after its divergence from the chimpanzee by maximum parsimony, using the orangutan as the outgroup. Somatic mutations in cancers for whole-genome sequences were obtained from Alexandrov et al. (2013) and the TCGA consortium (Hoadley et al. 2014; https://tcga-data.nci.nih.gov/tcga/). Mutational clusters in cancers were determined as described by Seplyarskiy et al. (2016) and mutational clusters of de novo heritable mutations obtained from trio data as described by Francioli et al. (2015). Cancers with a strong prevalence of APOBEC3A/B signature were defined on the basis of an increased rate of the TpCpW→K mutation; this matches the definition of APOrich cancers in the work by Seplyarskiy et al. (2016). Mutation rates in all analyses were calculated as the number of events divided on the number of corresponding sites.
We have written Perl scripts (Supplemental perl scripts) and R scripts (Supplemental R scripts) to analyze the data.
Genomic annotations
Information about genomic coordinates and which strand is the template for transcribed genes and expressed transposons was downloaded from the UCSC Genome Browser, files knownGene and ucscRetroAli5, correspondingly. Hotspots of meiotic double-strand breaks, also called recombination hotspots, were obtained from Pratto et al. (2014). We use all hotspots observed in at least one individual (file 1256442_DatafileS1.txt, column 17).
Leading vs. lagging strand asymmetry
The derivative of the replication timing at the position of the mutation was used as a proxy for the probability that the reference strand is replicated as leading or lagging in the current position, as previously described (Seplyarskiy et al. 2016). The genome was categorized by these values into six (Fig. 3F–I) or 20 (Fig. 1) equal bins, with low value of the derivative corresponding to the propensity of the DNA segment to be replicated as lagging; high value, as leading. For each bin, the numbers of substitutions and target sites were calculated. Each substitution was counted twice: (1) as a substitution on the reference strand with the corresponding derivative of the replication timing and (2) as a complementary substitution with the inverse derivative. Thus, each plot of substitution asymmetry (Figs. 1, 3F–I) is symmetric with respect to zero. Confidence intervals were obtained for the relative risk in a 2 × 2 table. As a measure of the asymmetry, we used the ratio of the frequencies of complementary mutations between strands in the most extreme (first or last) bin, where the determination of the fp was the most confident. Since tissue-matched replication timing for heritable mutations is unavailable, MCF-7 replication timing was used for the main analysis. Results replicated for all seven cell types with available RT data (Supplemental data fp). To reshuffle MCF-7 RTs, we randomly picked a value of RT for each 1-kb segment from the distribution of MCF-7 RT values.
Estimation of the admixture of APOBEC3A/B mutations from the leading vs. lagging strand asymmetry
Previously, using non-tissue-specific replication timing data as in the current analysis of heritable mutations, we found that about two-thirds of APOBEC3A/B mutations in cancer occurred on the lagging strand (Seplyarskiy et al. 2016). By using tissue-matched data on replication timing for analyses of asymmetry in cancers (MCF-7 for breast cancers, and IMR90 for lung cancers), we increase the observed level of asymmetry (Supplemental Fig. S10) compared with our previous study and studies by other groups (Haradhvala et al. 2016; Morganella et al. 2016; Seplyarskiy et al. 2016). Therefore, in our non-tissue-matched data, the observed ratio of mutational frequencies on the lagging and on the leading strands (strand bias) s is
where x is the fraction of APOBEC3A/B-induced mutations among all mutations; 0.67x and 0.33x are the fractions of APOBEC3A/B-induced mutations on the lagging and on the leading strand, respectively, with the coefficients 0.67 and 0.33 estimated from cancer data; and the coefficient 0.5 corresponds to the equal distribution of non-APOBEC3A/B-induced mutations between leading and lagging strands. Therefore,
so that s = 1.15 estimated for heritable mutations implies an admixture of APOBEC3A/B-induced mutations of 21%.
Supplementary Material
Acknowledgments
We thank Wendy Wong, Shamil Sunyaev, Ruslan Soldatov, and Nadezhda Terekhanova for useful discussion and Wendy Wong for help with data retrieving from the study by Wong et al. (2016). This work was performed in IITP RAS and supported by the Russian Science Foundation (grant no. 14-50-00150).
Author contributions: V.B.S. conceived the study and performed the analyses; all authors contributed to study design, intensively discussed the study, and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.210336.116.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwala V, Voight BF. 2016. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat Genet 48: 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akre MK, Starrett GJ, Quist JS, Temiz NA, Carpenter MA, Tutt ANJ, Grigoriadis A, Harris RS. 2016. Mutation processes in 293-based clones overexpressing the DNA cytosine deaminase APOBEC3B. PLoS One 11: e0155391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianova M, Bazykin GA, Nikolaev S, Seplyarskiy V. 2016. Human mismatch repair system corrects errors produced during lagging strand replication more effectively. bioRxiv 45278 https://doi.org/10.1101/045278. [Google Scholar]
- Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I. 2015. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc Natl Acad Sci 112: 2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A, Audit B, Chen C-L, Moindrot B, Leleu A, Guilbaud G, Rappailles A, Vaillant C, Goldar A, Mongelard F, et al. 2012. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput Biol 8: e1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat AS, Hao W, Townes JP, Lee H, Tang H, Foster PL. 2016. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc Natl Acad Sci 113: 2176–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. 2013a. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494: 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, Harris RS. 2013b. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 45: 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Resnick MA, Gordenin DA. 2013. The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair 12: 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PA, et al. 2015. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet 47: 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-L, Duquenne L, Audit B, Guilbaud G, Rappailles A, Baker A, Huvet M, d'Aubenton-Carafa Y, Hyrien O, Arneodo A, et al. 2011. Replication-associated mutational asymmetry in the human genome. Mol Biol Evol 28: 2327–2337. [DOI] [PubMed] [Google Scholar]
- Chen J, Miller BF, Furano AV. 2014. Repair of naturally occurring mismatches can induce mutations in flanking DNA. eLife 3: e02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz M, Watson NB, Turkington G, Verkoczy LK, Klinman NR, McGregor WG. 2003. Decreased frequency and highly aberrant spectrum of ultraviolet-induced mutations in the hprt gene of mouse fibroblasts expressing antisense RNA to DNA polymerase ζ. Mol Cancer Res 1: 836–847. [PubMed] [Google Scholar]
- Farkash-Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, Simon I. 2008. Global organization of replication time zones of the mouse genome. Genome Res 18: 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, Genome of the Netherlands Consortium, van Duijn CM, Swertz M, Wijmenga C, et al. 2015. Genome-wide patterns and properties of de novo mutations in humans. Nat Genet 47: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann JM, Wong WSW, Pinelli M, Farrah T, Bodian D, Stittrich AB, Glusman G, Vissers LELM, Hoischen A, Roach JC, et al. 2016. Parent-of-origin-specific signatures of de novo mutations. Nat Genet 48: 935–939. [DOI] [PubMed] [Google Scholar]
- Green P, Ewing B, Miller W, Thomas PJ, NISC Comparative Sequencing Program, Green ED. 2003. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet 33: 514–517. [DOI] [PubMed] [Google Scholar]
- Green AM, Landry S, Budagyan K, Avgousti DC, Shalhout S, Bhagwat AS, Weitzman MD. 2016. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle 15: 998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G, Rappailles A, Baker A, Chen C-L, Arneodo A, Goldar A, d'Aubenton-Carafa Y, Thermes C, Audit B, Hyrien O. 2011. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput Biol 7: e1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. 2016. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 164: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K, Nielsen R. 2014. Error-prone polymerase activity causes multinucleotide mutations in humans. Genome Res 24: 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. 2014. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 15: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, et al. 2014. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158: 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, Ladoukakis E, Eyre-Walker A. 2009. Cryptic variation in the human mutation rate. PLoS Biol 7: e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA. 2016. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep 14: 1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PLF, Hellmann I. 2011. Mutation rate distribution inferred from coincident SNPs and coincident substitutions. Genome Biol Evol 3: 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E-Y, Lorenzo-Redondo R, Little SJ, Chung Y-S, Phalora PK, Maljkovic Berry I, Archer J, Penugonda S, Fischer W, Richman DD, et al. 2014. Human APOBEC3 induced mutation of human immunodeficiency virus type-1 contributes to adaptation and evolution in natural infection. PLoS Pathog 10: e1004281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knisbacher BA, Levanon EY. 2016. DNA editing of LTR retrotransposons reveals the impact of APOBECs on vertebrate genomes. Mol Biol Evol 33: 554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenova OV, Daee DL, Mertz TM, Shcherbakova PV. 2015. DNA polymerase ζ-dependent lesion bypass in Saccharomyces cerevisiae is accompanied by error-prone copying of long stretches of adjacent DNA. PLoS Genet 11: e1005110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Handsaker RE, Kamitaki N, Karlić R, Ghosh S, Polak P, Eggan K, McCarroll SA. 2014. Genetic variation in human DNA replication timing. Cell 159: 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Pursell ZF, Abdulovic-Cui AA, Clark AB, Nick McElhinny SA, Kunkel TA. 2012. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet 8: e1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Clausen AR, Clark AB, MacAlpine HK, MacAlpine DM, Malc EP, Mieczkowski PA, Burkholder AB, Fargo DC, Gordenin DA, et al. 2014. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res 24: 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Campbell PJ. 2015. Somatic mutation in cancer and normal cells. Science 349: 1483–1489. [DOI] [PubMed] [Google Scholar]
- Mathieson I, McVean G. 2014. Demography and the age of rare variants. PLoS Genet 10: e1004528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, Reich DE. 2016. Variation in mutation rates among human populations. bioRxiv 63578 https://doi.org/10.1101/063578. [Google Scholar]
- Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith KS, et al. 2013. The origin, evolution, and functional impact of short insertion–deletion variants identified in 179 human genomes. Genome Res 23: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, et al. 2016. The topography of mutational processes in breast cancer genomes. Nat Commun 7: 11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu XJ, Lu ZJ, Kong Y, Lam HYK, Gerstein MB. 2011. Analysis of genomic variation in non-coding elements using population-scale sequencing data from the 1000 Genomes Project. Nucleic Acids Res 39: 7058–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, von Grünberg H-H, Peifer M. 2009. Transcription-induced mutational strand bias and its effect on substitution rates in human genes. Mol Biol Evol 26: 131–142. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordentoft I, Lamy P, Birkenkamp-Demtröder K, Shumansky K, Vang S, Hornshøj H, Juul M, Villesen P, Hedegaard J, Roth A, et al. 2014. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep 7: 1649–1663. [DOI] [PubMed] [Google Scholar]
- Pfeifer GP. 2006. Mutagenesis at methylated CpG sequences. Curr Top Microbiol Immunol 301: 259–281. [DOI] [PubMed] [Google Scholar]
- Pinto Y, Gabay O, Arbiza L, Sams AJ, Keinan A, Levanon EY. 2016. Clustered mutations in hominid genome evolution are consistent with APOBEC3G enzymatic activity. Genome Res 26: 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin M-L, Ordóñez GR, Bignell GR, et al. 2010. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463: 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Arndt PF. 2008. Transcription induces strand-specific mutations at the 5′ end of human genes. Genome Res 18: 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratto F, Brick K, Khil P, Smagulova F, Petukhova GV, Camerini-Otero RD. 2014. DNA recombination. Recombination initiation maps of individual human genomes. Science 346: 1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, et al. 2016. Timing, rates and spectra of human germline mutation. Nat Genet 48: 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. 2013. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45: 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhani FJ, Nik-Zainal S, Wuster A, Li Y, Conte N, Koike-Yusa H, Kumasaka N, Vallier L, Yusa K, Bradley A. 2016. Mutational history of a human cell lineage from somatic to induced pluripotent stem cells. PLoS Genet 12: e1005932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraconi G, Severi F, Sala C, Mattiuz G, Conticello SG. 2014. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome Biol 15: 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hourmozdi JN, Hahn MW. 2011. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol 21: 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy VB, Kharchenko P, Kondrashov AS, Bazykin GA. 2012. Heterogeneity of the transition/transversion ratio in Drosophila and Hominidae genomes. Mol Biol Evol 29: 1943–1955. [DOI] [PubMed] [Google Scholar]
- Seplyarskiy VB, Bazykin GA, Soldatov RA. 2015. Polymerase ζ activity is linked to replication timing in humans: evidence from mutational signatures. Mol Biol Evol 32: 3158–3172. [DOI] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. 2016. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res 26: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Akey JM. 2015. The origins, determinants, and consequences of human mutations. Science 349: 1478–1483. [DOI] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ. 2014. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28: 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, et al. 2016. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun 7: 12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. 2013. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2: e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terekhanova NV, Bazykin GA, Neverov A, Kondrashov AS, Seplyarskiy VB. 2013. Prevalence of multinucleotide replacements in evolution of primates and Drosophila. Mol Biol Evol 30: 1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terekhanova NV, Seplyarskiy V, Soldatov RA, Bazykin GA. 2016. Evolution of local mutation rate and its determinants. bioRxiv 54825 https://doi.org/10.1101/054825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WSW, Solomon BD, Bodian DL, Kothiyal P, Eley G, Huddleston KC, Baker R, Thach DC, Iyer RK, Vockley JG, et al. 2016. New observations on maternal age effect on germline de novo mutations. Nat Commun 7: 10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang L, Huang J, Zhang X, Yuan Y, Chen J-Q, Hurst LD, Tian D. 2015. Parent–progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523: 463–467. [DOI] [PubMed] [Google Scholar]
- Yuen RK, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, Tong X, Sun Y, Cao D, Zhang T, et al. 2016. Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med 1: 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Cooper DN, Zhao Q, Wang Y, Liu R, Li Q, Férec C, Wang Y, Chen J-M. 2015. Concurrent nucleotide substitution mutations in the human genome are characterized by a significantly decreased transition/transversion ratio. Hum Mutat 36: 333–341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.