

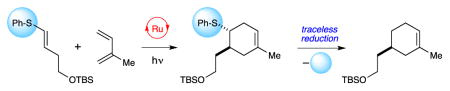
Abstract
The incorporation of an easily oxidized arylsulfide moiety facilitates the photocatalytic generation of alkene radical cations that undergo a variety of cycloaddition reactions with electron-rich reaction partners. The sulfide moiety can subsequently be reductively cleaved in a traceless fashion, affording products that are not otherwise directly accessible using photoredox catalysis. This approach constitutes a novel oxidative “redox auxiliary” strategy that offers a practical means to circumvent a fundamental thermodynamic limitation facing photoredox reactions.
Graphical Abstract
Alkene radical ions are open-shell reactive intermediates that can participate in a wide variety of organic transformations. Many features of these reactions are synthetically attractive: they often proceed with very low activation barriers, and their regiochemical outcomes generally complement those involving closed-shell neutral alkenes.1 In the past several years, there has been a renewed interest in the application of radical ion chemistry to synthesis, due in part to the recognition that photoredox catalysis offers a convenient means to access the distinctive reactivity of these odd-electron intermediates under relatively mild and convenient conditions.2 Recent reports of synthetic transformations involving photogenerated alkene radical cations have included a variety of cycloaddition reactions3 and anti-Markovnikov hydrofunctionalization reactions,4 each of which would be challenging to accomplish using alternate synthetic strategies.
One limitation common to all photoredox reactions is that the scope of a given method is dictated by the thermodynamic feasibility of the initial photoinduced electron-transfer step; substantially endergonic photoredox steps result in poor overall reactivity. On the one hand, the availability of a large number of structurally varied photocatalysts spanning a range of excited state redox potentials can be exploited to broaden the scope of these reactions.5 Nevertheless, a substrate’s redox potential remains a fundamental thermodynamic constraint on the success of photoredox methods. For instance, simple mono- and disubstituted aliphatic alkenes have proven too difficult to oxidize (> +2.5 V vs SCE)6 using even the most powerfully oxidizing photoredox catalysts in common usage and have not successfully been engaged in photooxidatively triggered transformations.
We recently described the concept of a “redox auxiliary,” which we defined as an easily removable moiety that can be temporarily installed on a substrate to enable its activation by single-electron transfer processes.7 Our initial demonstration of this concept was a radical anion [2+2] photoredox cycloaddition using 2-acylimidazoles as readily reducible analogues of enoate esters that would otherwise be resistant towards photoreductive activation. We wondered if an analogous redox auxiliary strategy might be applied to facilitate photooxidatively initiated organic transformations. We imagined that installation of an electron-rich redox auxiliary onto an otherwise unactivated alkene would facilitate its one-electron oxidation; the resulting radical cations could subsequently be induced to undergo a number of characteristic alkene radical cation reactions, including Diels–Alder3bfg and [2+2] cycloadditions.3ad Subsequent cleavage of the redox auxiliary group would afford the products of formal cycloadditions involving unactivated alkene substrates (Figure 1).8. 9
Figure 1.
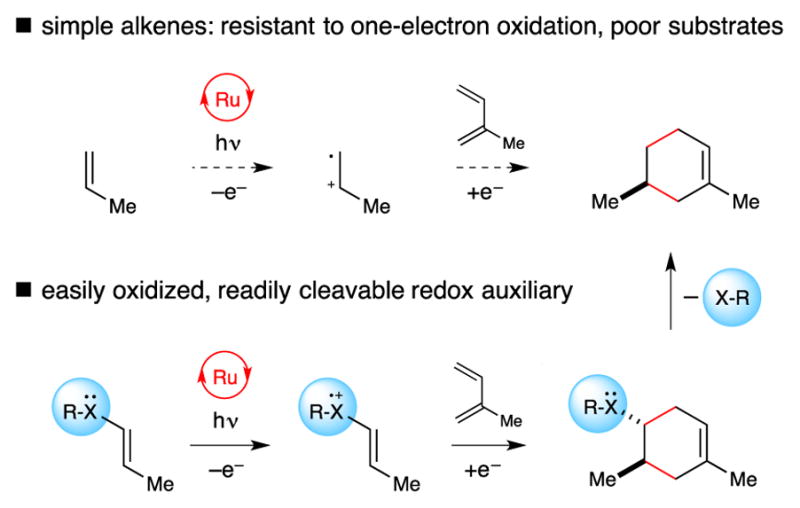
Traceless redox auxiliary strategy for radical cation reactions.
Very recently, Cooke and co-workers described an intriguing first step towards an alternate oxidative redox auxiliary strategy. They demonstrated that vinyl ferrocene is powerfully activated towards Diels–Alder and hydrothiolation reactions upon one-electron chemical oxidation.10 This study verified that the incorporation of a reversibly oxidizable moiety onto an alkene can indeed be used to facilitate redox-promoted transformations. However, this strategy involves a separate activation step using stoichiometric chemical oxidant rather than an in situ redox catalyst, and the requisite ferrocenyl moiety is not removable in a traceless fashion.
We imagined a different strategy utilizing a reversibly oxidizable sulfide moiety as a redox auxiliary (Figure 1). We were attracted to the use of vinyl sulfides in this context for a number of reasons. First, aryl vinyl sulfides generally possess oxidation potentials ranging from +1.1 V to +1.4 V vs SCE,11 which are readily accessible using the well-characterized Ru(II) polypyridyl photoredox catalysts that are increasingly being utilized in synthetic chemistry. Second, Bauld has studied radical cation Diels–Alder cycloadditions of aryl vinyl sulfides using triarylaminium salts as chemical oxidants or aromatic nitriles as PET sensitizers.11 This valuable precedent demonstrates that vinyl sulfide radical cations are indeed activated towards cycloaddition reactions. Finally, C–S bonds are relatively weak, and a variety of mild, operationally facile methods for their cleavage have been utilized in the synthesis of complex molecules.12 We imagined that successful development of this sequence would enable the preparation of formal cycloaddition products of simple alkenes that are not amenable to direct activation by photoredox catalysis and would also be challenging to engage in classical thermal cycloaddition methods.
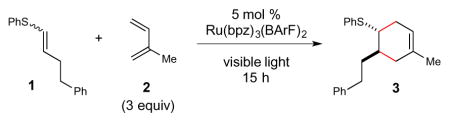
As a starting point for our investigations, we optimized conditions for the radical cation Diels–Alder cycloaddition of vinyl sulfide 1 with isoprene (2) (Table 1).13 We first examined conditions adapted from our previous study of photocatalytic radical cation Diels–Alder cycloadditions with styrenic dienophiles, utilizing Ru(bpz)3(BArF)2 as a strongly oxidizing, organic-soluble photocatalyst. The yield of this model cycloaddition, however, was disappointingly low in a variety of solvents (entries 1–4). A study of additives quickly revealed a sensitivity of the reaction to the presence of water (entry 5). This observation led us to investigate the effect of dessicants such as MgSO4, which resulted in a significant increase in the yield of the cycloaddition (entry 6), along with improved reproducibility. Intense blue LEDs are not required; the photocatalytic reaction proceeds using a less-intense CFL light bulb, albeit with slower rates (entry 7). Interestingly, although radical cation cycloaddition reactions of electron-rich styrenes can typically be conducted open to air, we observed diminished yields and poor mass balance when this Diels–Alder reaction was run under ambient atmosphere rather than in degassed solvent (entry 8), an observation we attribute to the sensitivity of sulfides towards reactive oxygen species.14 Finally, consistent with the photocatalytic nature of this reaction, control experiments indicated that no product is formed either in the absence of Ru(bpz)32+ or in the dark (entries 9 and 10).
Table 1.
Optimization studies for vinyl sulfide radical cation Diels–Alder cycloadditionsa
| ||||
|---|---|---|---|---|
| entry | solvent | light source | additive | yield (%) |
| 1 | MeCN | blue LED | -- | 40 |
| 2 | CH2Cl2 | blue LED | -- | 22 |
| 3 | DMF | blue LED | -- | 0 |
| 4 | DMSO | blue LED | -- | 0 |
| 5 | MeCN | blue LED | H2O (10 mol%) | 11 |
| 6 | MeCN | blue LED | MgSO4 | 84 |
| 7 | MeCN | 23 W CFL | MgSO4 | 44 |
| 8 | MeCN | blue LED | MgSO4, air | 24 |
| 9 | MeCN | -- | MgSO4 | 0 |
| 10b | MeCN | blue LED | MgSO4 | 0 |
Unless otherwise noted, reactions were conducted using 5 mol % Ru(bpz)3(BArF)2, 1 equiv 1, and 3 equiv 2 in solvent degassed using three freeze/pump/thaw cycles. A 15 W blue LED was used as the irradiation source.
Reaction conducted without Ru(bpz)3(BArF)2.
Studies examining the scope of the radical cation Diels–Alder cycloaddition of vinyl sulfides under optimized photocatalytic conditions are summarized in Scheme 1. A range of simple cyclic and acyclic dienes participate readily in this process (3–7), though sterically bulky dienes require longer reaction times (5), and electron-rich dienes provide somewhat lower yields (6). Simple cyclic dienes (7), however, work well in this reaction. The structure of the vinyl sulfide partner can also be modified. An examination of simple alkyl-substituted dienophiles reveals a sensitivity to steric bulk; larger vinyl substituents result in substantially slower Diels–Alder reactions (10 and 11), and β,β-disubstituted vinyl sulfides do not provide any observable cycloadducts. On the other hand, the reaction tolerates various functional groups including esters, silyl ethers, and phthalimides (12–14). These conditions were also found to be applicable to intramolecular cycloadditions (15).15
Scheme 1.

Scope studies for photocatalytic vinyl sulfide radical cation Diels–Alder cycloadditionsa
aReactions were conducted using 5 mol % Ru(bpz)3(BArF)2, 1 equiv vinyl sulfide, 3 equiv diene, and 2 weight equiv MgSO4 in degassed MeCN. A 15 W blue LED was used as the irradiation source.
These studies were premised on the hope that sulfide redox auxiliaries might be broadly applicable not only to Diels–Alder cycloadditions but also to a range of useful transformations involving alkene radical cations. As a first attempt to investigate the generality of this strategy, we studied the use of vinyl sulfides in intermolecular [2+2] cycloaddition reactions.3d We had previously investigated the factors controlling the crossed selectivity of such reactions involving electron-rich styrenes and were pleased to observe that similar considerations are applicable to [2+2] radical cation cycloadditions of vinyl sulfides (Scheme 2). Thus, when 1 is irradiated in the presence of electron rich monosubstituted alkenes, the corresponding unsymmetrical cyclobutanes are produced in good yield. Vinyl ethers were excellent reaction partners in this reaction, affording good yields and excellent selectivities regardless of the steric bulk of the ether substituent (16–20).16 An enamide also provided synthetically useful yields of the corresponding acetamide-substituted cyclobutane (21). Finally, although simple aliphatic olefins and vinyl esters did not participate in this reaction, styrenes are successful reaction partners (22 and 23), consistent with the stepwise radical mechanism expected for this cycloaddition.
Scheme 2.
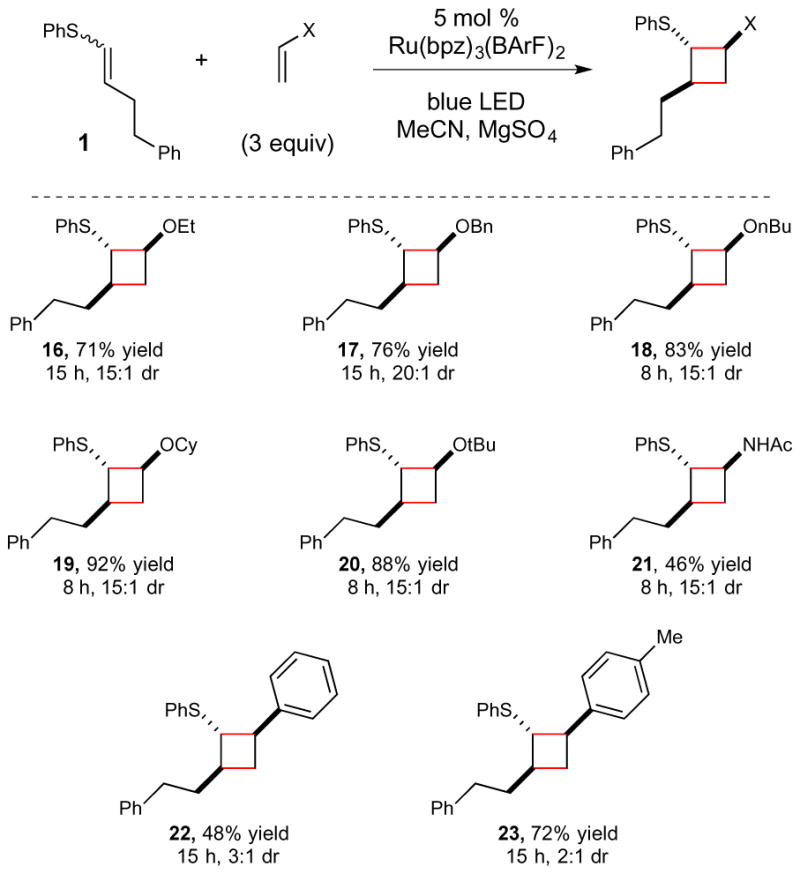
Scope studies for photocatalytic vinyl sulfide radical cation [2+2] cycloadditions.a
aUnless otherwise noted, reactions were conducted using 5 mol % Ru(bpz)3(BArF)2, 1 equiv vinyl sulfide, 3 equiv alkene reaction partner, and 2 weight equiv MgSO4 in degassed MeCN. A 15 W blue LED was used as the irradiation source. b Reaction conducted using 10 equiv of N-vinylacetamide.
The concluding step in this redox auxiliary strategy requires the cleavage of the sulfide moiety, which can readily be accomplished using various reductive protocols (Scheme 3). First, treatment of [4+2] cycloadduct 3 with freshly prepared lithium naphthalenide rapidly affords the corresponding desulfurized product without competitive reduction of the alkene moiety (eq 1). Importantly, the resulting cyclohexene 24 would not be directly accessible using alternate thermal or redox-promoted Diels–Alder methods. Similarly, cycloadduct 13 bearing a TBS-protected primary alcohol undergoes desulfurization to afford 25 without cleavage of the silyl protecting group (eq 2). The reduction of [2+2] cycloadduct 23 can readily be accomplished by treatment with Raney nickel (eq 3), and the diastereomer ratio of the resulting cyclobutane (26) is identical to that of the starting material, indicating that epimerization does not occur under these conditions. The desulfurization of functionalized cycloadduct 16 occurs without observable cleavage or elimination of the alkoxy substituent. Thus removal of these sulfide auxiliary groups can be accomplished under relatively mild conditions that tolerate a variety of common functional groups.
Scheme 3.
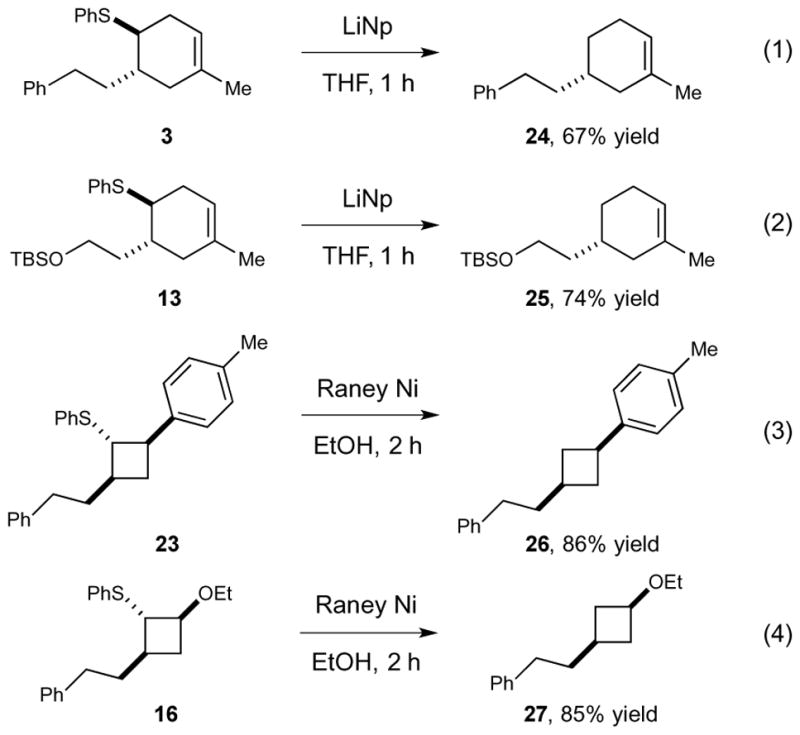
Reductive cleavage of the redox auxiliary group
In conclusion, vinyl sulfides are easily activated by visible light promoted photooxidation and subsequently undergo cycloaddition reactions characteristic of alkene radical cations. The activating sulfide moiety can be tracelessly removed after the photoredox reaction to afford cycloadducts that could not be directly synthesized by reactions of simple unfunctionalized alkenes. These results, along with the reductive redox auxiliary strategy our group reported several years ago,7 suggest that the use of redox auxiliary groups present a practical strategy to circumvent a fundamental limitation on the feasibility of photoredox reactions and could be used to significantly increase the scope of products that are available using this powerful mode of activation.
Supplementary Material
Acknowledgments
The authors are grateful to the NIH (GM098886) for financial support and to Sigma–Aldrich for a generous gift of RuCl3. Analytical instrumentation at UW–Madison is supported in part by the NIH (1S10 OD020022-1) and by a generous gift from the Paul J. Bender fund.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and spectroscopic data (PDF)
References
- 1.(a) Bauld NL. Tetrahedron. 1989;45:5307–5363. [Google Scholar]; (b) Schmittel M, Burghart A. Angew Chem Int Ed Engl. 1997;36:2550–2589. [Google Scholar]; (c) Mella M, Freccero M, Fasani E, Albini A. Chem Soc Rev. 1998;27:81–89. [Google Scholar]; (d) Staettel NJ, Oxgaard J, Wiest O. Eur J Org Chem. 2001:1429–1439. [Google Scholar]
- 2.(a) Ischay MA, Yoon TP. Eur J Org Chem. 2012:3359–3372. [Google Scholar]; (b) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Margrey KA, Nicewicz KA. Acc Chem Res. 2016;49:1997–2006. doi: 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lin S, Ischay MA, Fry CG, Yoon TP. J Am Chem Soc. 2011;133:19350–19353. doi: 10.1021/ja2093579. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Parrish JD, Ischay MA, Lu Z, Guo S, Peters NR, Yoon TP. Org Lett. 2012;14:1640–1643. doi: 10.1021/ol300428q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ischay MA, Ament MS, Yoon TP. Chem Sci. 2012;3:2807–2811. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Riener M, Nicewicz DA. Chem Sci. 2013;4:2625–2629. doi: 10.1039/C3SC50643F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Stephenson SM, Shores MP, Ferreira EM. Angew Chem Int Ed. 2015;54:6506–6510. doi: 10.1002/anie.201501220. [DOI] [PubMed] [Google Scholar]; (g) Higgins RF, Fatur SM, Shepard SG, Stevenson SM, Boston DB, Ferreira EM, Damrauer NH, Rappé AK, Shores MP. J Am Chem Soc. 2016;138:5451–5464. doi: 10.1021/jacs.6b02723. [DOI] [PubMed] [Google Scholar]
- 4.(a) Hamilton DS, Nicewicz DA. J Am Chem Soc. 2012;134:18577–18580. doi: 10.1021/ja309635w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grandjean J, Nicewicz DA. Angew Chem Int Ed. 2013;52:3968–3971. doi: 10.1002/anie.201210111. [DOI] [PubMed] [Google Scholar]; (c) Nguyen TM, Nicewicz DA. J Am Chem Soc. 2013;135:9588–9591. doi: 10.1021/ja4031616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Perkowski AJ, Nicewicz DA. J Am Chem Soc. 2013;135:10334–10337. doi: 10.1021/ja4057294. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nguyen TM, Manohar N, Nicewicz DA. Angew Chem Int Ed. 2014;53:6198–6201. doi: 10.1002/anie.201402443. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wilger DJ, Grandjean JM, Lammert T, Nicewicz DA. Nature Chem. 2014;6:720–726. doi: 10.1038/nchem.2000. [DOI] [PubMed] [Google Scholar]; (g) Morse PD, Nicewicz DA. Chem Sci. 2015;6:270–274. doi: 10.1039/c4sc02331e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gesmundo NJ, Grandjean JM, Nicewicz DA. Org Lett. 2015;17:1316–1319. doi: 10.1021/acs.orglett.5b00316. [DOI] [PubMed] [Google Scholar]; (i) Cavanaugh CL, Nicewicz DA. Org Lett. 2015;17:6082–6085. doi: 10.1021/acs.orglett.5b03113. [DOI] [PubMed] [Google Scholar]
- 5.Douglas JJ, Nguyen JD, Cole KP, Stephenson CRJ. Aldrichim Acta. 2014;47:15–25. [Google Scholar]
- 6.Roth HG, Romero NA, Nicewicz DA. Synlett. 2016;27:714–723. [Google Scholar]
- 7.Tyson EL, Farney EP, Yoon TP. Org Lett. 2012;14:1110–1113. doi: 10.1021/ol3000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida pioneered the use of “electroauxiliaries” that are analogous to the concept that we propose here. Electroauxiliaries are generally Si, Sn, or S substituents that similarly facilitate the oxidation of organic substrates that are otherwise resistant to one-electron oxidation. In Yoshida’s formualation, however, the electroauxiliary group is poised to undergo mesolytic cleavage upon oxidation, resulting in a cationic or radical reactive intermediate. In contrast, “redox auxiliaries,” as we define them, are moieties that remain covalently bound throughout the net transformation, enabling the radical cation reactivity of the substrate to be directly exploited.
- 9.For leading references on the use of electroauxiliary groups in synthesis, see: Yoshida J, Takada K, Ishichi Y, Isoe S. J Chem Soc, Chem Commun. 1994:2361–2362.Yoshida J, Sugawara M, Kise N. Tetrahedron Lett. 1996;37:3157–3160.Yoshida J, Nishiwaki K. J Chem Soc, Dalton Trans. 1998:2589–2596.Yoshida J, Sugawara M, Tatsumi M, Kise N. J Org Chem. 1998;63:5950–5961. doi: 10.1021/jo980601x.Shoji T, Kim S, Yamamoto K, Kawai T, Okada Y, Chiba K. Org Lett. 2014;16:6404–6407. doi: 10.1021/ol503198p.Nokami T, Isoda Y, Sasaki N, Takaiso A, Hayase S, Itoh T, Ryutaro H, Shimizu A, Yoshida J. Org Lett. 2015;17:1525–1528. doi: 10.1021/acs.orglett.5b00406.
- 10.Wiles AA, Zhang X, Fitzpatrick B, Long DL, Macgregor SA, Cooke G. Dalton Trans. 2016;45:7220–7225. doi: 10.1039/c6dt00875e. [DOI] [PubMed] [Google Scholar]
- 11.(a) Pabon RA, Bellville DJ, Bauld NL. J Am Chem Soc. 1984;106:2730–2731. [Google Scholar]; (b) Harirchian B, Bauld NL. J Am Chem Soc. 1989;111:1826–1828. [Google Scholar]; (c) Aplin JT, Bauld NL. J Chem Soc, Perkin Trans. 2;1997:853–855. [Google Scholar]
- 12.(a) Lee JH, Zhang Y, Danishefsky SJ. J Am Chem Soc. 2010;132:14330–14333. doi: 10.1021/ja1073855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Toribio G, Marjanet G, Alibés R, de March P, Font J, Bayón P, Figueredo M. Eur J Org Chem. 2011:1534–1543. doi: 10.1021/jo800107h. [DOI] [PubMed] [Google Scholar]
- 13.Both (E) and (Z) linear vinyl sulfides provide identical reaction rates and diastereomer ratios. Thus the studies described herein were conducted using a mixture of geometric isomers arising from the synthesis of the substrates via photoinduced radical thiol-ene addition. See the Supporting Information for details.
- 14.(a) Schenck GO, Krauch CH. Angew Chem. 1962;74:510–510. [Google Scholar]; (b) Foote CS, Peters JW. J Am Chem Soc. 1971;93:3795–3796. [Google Scholar]
- 15.(a) Harirchian B, Bauld NL. Tetrahedron Lett. 1987;28:927–930. [Google Scholar]; (b) Lin SS, Padilla CE, Ischay MA, Yoon TP. Tetrahedron Lett. 2012;53:3073–3076. doi: 10.1016/j.tetlet.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.We observe no evidence for competitive photooxiation of the vinyl ethers, presumably because their redox potentials (ca. +1.67 V vs. SCE) are outside of the reasonable working range of the Ru*(bpz)32+ photocatalyst (+1.45 V vs. SCE). See: Koch D, Schäfer H, Steckhan E. Chem Ber. 1974;107:3640–3657.Crutchley RJ, Lever ABP. J Am Chem Soc. 1980;102:7128–7129.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.