

ABSTRACT
CarD is an essential RNA polymerase (RNAP) interacting protein in Mycobacterium tuberculosis that stimulates formation of RNAP-promoter open complexes. CarD plays a complex role in M. tuberculosis growth and virulence that is not fully understood. Therefore, to gain further insight into the role of CarD in M. tuberculosis growth and virulence, we determined the effect of increasing the affinity of CarD for RNAP. Using site-directed mutagenesis guided by crystal structures of CarD bound to RNAP, we identified amino acid substitutions that increase the affinity of CarD for RNAP. Using these substitutions, we show that increasing the affinity of CarD for RNAP increases the stability of the CarD protein in M. tuberculosis. In addition, we show that increasing the affinity of CarD for RNAP increases the growth rate in M. tuberculosis without affecting 16S rRNA levels. We further show that increasing the affinity of CarD for RNAP reduces M. tuberculosis virulence in a mouse model of infection despite the improved growth rate in vitro. Our findings suggest that the CarD-RNAP interaction protects CarD from proteolytic degradation in M. tuberculosis, establish that growth rate and rRNA levels can be uncoupled in M. tuberculosis and demonstrate that the strength of the CarD-RNAP interaction has been finely tuned to optimize virulence.
IMPORTANCE Mycobacterium tuberculosis, the causative agent of tuberculosis, remains a major global health problem. In order to develop new strategies to battle this pathogen, we must gain a better understanding of the molecular processes involved in its survival and pathogenesis. We have previously identified CarD as an essential transcriptional regulator in mycobacteria. In this study, we detail the effects of increasing the affinity of CarD for RNAP on transcriptional regulation, CarD protein stability, and virulence. These studies expand our understanding of the global transcription regulator CarD, provide insight into how CarD activity is regulated, and broaden our understanding of prokaryotic transcription.
KEYWORDS: Mycobacterium tuberculosis, RNA polymerase, protein stability, protein-protein interactions, rRNA, transcription initiation factor
INTRODUCTION
Mycobacterium tuberculosis remains a major global health problem, resulting in 9.6 million new cases of tuberculosis (TB) in 2014 and 1.5 million TB-related deaths that year (1). Control of the M. tuberculosis epidemic is hampered by the increasing prevalence of multidrug-resistant strains, which resulted in 480,000 patients developing multidrug-resistant TB in 2014. In order to develop new strategies to battle this pathogen, we must gain a better understanding of the molecular processes involved in its survival and pathogenesis. Recent studies have identified numerous aspects of mycobacterial physiology that differ from what has been identified in model organisms and highlight the importance of research directly in mycobacteria to understand their lineage specific physiology. We have identified CarD as an essential transcriptional regulator in mycobacteria that is not conserved in the model organism Escherichia coli, which has traditionally been used to study mechanisms of transcription (2). carD is conserved in all mycobacteria and numerous other bacteria (2–5) but is not found in eukaryotes. Therefore, studying CarD will broaden our understanding of prokaryotic transcription while also characterizing a promising potential target for desperately needed new therapeutic strategies for TB.
Chromatin immunoprecipitation-sequencing (ChIP-seq) experiments in mycobacteria demonstrated that CarD is localized with RNA polymerase (RNAP) holoenzyme at promoters throughout the genome, indicating that CarD is a global regulator of transcription initiation (4). CarD interacts directly with the β1 lobe of the RNAP-β subunit through its N-terminal RNAP interaction domain (RID) (2, 5, 6) and with DNA just upstream of the −10 element of the promoter through a conserved basic patch in its C-terminal domain (3, 4, 7). Within the basic patch, studies have identified a highly conserved tryptophan residue that is proposed to wedge into the minor groove at the upstream edge of the transcription bubble (3, 4). Thus far, CarD's activity has primarily been studied on rRNA promoters. Using in vitro transcription assays, we have shown that CarD stabilizes RNAP-promoter open complexes at rRNA promoters during transcription initiation and that CarD requires interactions with both RNAP and DNA, as well as the activity of the conserved tryptophan to do so (4, 6–8). Recently, a bulk fluorescence assay was used to measure the effect of CarD on the kinetics of transcription initiation at the M. tuberculosis rRNA promoter rrnAP3. These studies revealed that, compared to the Escherichia coli RNAP, the Mycobacterium bovis RNAP forms a significantly less stable open complex at rrnAP3 in the absence of CarD. Addition of CarD stabilizes M. bovis RNAP-rrnAP3 open complexes by binding to the RNAP-promoter open complex with high affinity and preventing collapse to closed complex. With lower affinity, CarD also binds the RNAP-promoter closed complex and promotes melting of the DNA to form open complex. Importantly, the cellular concentration of CarD in wild-type (WT) M. tuberculosis is sufficient for both of these activities to be physiologically relevant (8). Mycobacterial strains expressing mutants of CarD with weakened affinity for the RNAP-β subunit, weakened affinity for DNA, or that are mutated at the conserved tryptophan all express lower levels of rRNA exhibit lower rRNA promoter activation in promoter-lacZ fusion experiments, grow slower, and are more sensitive to multiple stresses and antibiotics compared to strains expressing WT CarD (4, 6, 7).
In this study, we use CarD mutants with increased affinity for RNAP to dissect the role of CarD binding to RNAP in vitro and in vivo. We detail the distinct effects of increasing the affinity of CarD for RNAP on open complex stability, transcriptional regulation, CarD protein stability, and the effects of these changes on growth and virulence. These studies expand our understanding of the global transcription regulator CarD and the impact of CarD activity on the physiology of M. tuberculosis, while also providing insight into how CarD activity is regulated.
RESULTS
Identification of amino acid substitutions in the N terminus of CarD that increase the affinity of CarD for RNAP-β.
To identify substitutions that strengthen the M. tuberculosis CarD/RNAP-β β1 lobe interaction, we used site-directed mutagenesis guided by a comparison between our X-ray structure of Thermus thermophilus CarD in complex with the β1 lobe of T. thermophilus RNAP (Protein Data Bank [PDB] accession no. 4XAX) (3) (Fig. 1A), solved to 2.4-Å resolution, and the structure of M. tuberculosis CarD in complex with the RNAP-β β1 and β2 lobes of M. tuberculosis RNAP (PDB accession no. 4KBM) (9), solved to 2.11 Å. The β1 lobes from each structure were aligned over 919 atoms in PyMOL (version 1.8; Schrödinger, LLC) to give a root mean square (RMS) of 1.095 Å, and the interactions between the β1 lobe and the CarD RID were compared. Many of the interactions were conserved between the T. thermophilus and M. tuberculosis structures (Fig. 1B and D), but a significant difference was noted in a region that included a loop at the tip of the RID (residues 26 to 29 in M. tuberculosis) that was not modeled in the M. tuberculosis structure, presumably due to a lack of density. Cartoon rendering by B-factors (PyMOL) of both structures revealed that this loop was highly disordered in M. tuberculosis but well ordered in T. thermophilus, presumably due to its interaction with the β1 lobe (Fig. 1C). In particular, the T. thermophilus structure revealed a nonpolar interaction between loop residue V28 from CarD (I27 in M. tuberculosis) and I101 and I108 from the β1 lobe (I140 and I147 in M. tuberculosis) that was not visible in the M. tuberculosis structure due to disorder (Fig. 1B). Based on the T. thermophilus structure of the β1 lobe in complex with CarD, we predicted that the isoleucine side chain at position 27 of M. tuberculosis CarD likely contributes to an interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe that may not have been observed in the crystal structure due to crystal packing constraints. Furthermore, structural modeling predicts that removal of the isoleucine side chain at position 27 of the M. tuberculosis CarD RID would weaken the interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe, whereas substitutions of position 27 with residues containing side chains that are more hydrophobic than isoleucine might strengthen the interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe.
FIG 1.

Single point mutations in CarD specifically increase CarD's affinity for RNAP-β. (A) Crystal structure of T. thermophilus CarD in complex with the β1 lobe of T. thermophilus RNAP reveals molecular interactions between the two proteins at high-resolution (2.4-Å) detail. CTD, C-terminal domain. (B) Structural alignment of the β1 lobes of T. thermophilus CarD:β1-lobe and M. tuberculosis CarD:β1-lobe structures to compare the interactions between the CarD RID and RNAP β1 in each organism. (C) Atom displacement was rendered visually using PyMOL B-factor rendering. The spectrum scale above illustrates a range of low B-factors (highly ordered, blue indicates under 30%) to high B-factors (disordered, red indicates 90% or higher). (D) Alignment of T. thermophilus (top) and M. tuberculosis (bottom) CarD RIDs comparing interactions with their cognate RNAP β1 lobe. Black shading indicates residues that are identical and yellow shading indicates residues that are homologous. The following groups are considered homologous: R and K; E and D; V, I, L, M, and A; S and T; and Q and N. Bullets are defined in the figure by interactions unique to each organism or shared by both. The red box designates the region that is observed in the T. thermophilus complex but not the M. tuberculosis complex. (E) Results of β-Gal assays (6, 15) examining the effects of amino acid substitutions at position 27 of the M. tuberculosis CarD RID on the interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe. The bar graphs show β-Gal activity in Miller units observed in assays done using cells containing the indicated M. tuberculosis CarD RID derivative and the M. tuberculosis β1 lobe, as well as assays performed using cells containing only the M. tuberculosis β1 lobe. Each graph shows the means ± the SEM of data from five replicates. Statistical significance was analyzed by analysis of variance (ANOVA) and Tukey's multiple-comparison test (n.s., not significant; **, P ≤ 0.01; ****, P ≤ 0.0001). (F) Immunoprecipitation experiments with a monoclonal antibody specific for HA in the M. smegmatis ΔcarD attB::tet-carD strain expressing M. tuberculosis CarDWT-HA (lane 1), CarDR25E-HA (lane 2), CarDI27F-HA (lane 3), or CarDI27W-HA (lane 4). Immunoprecipitated eluates were analyzed by Western blotting with monoclonal antibodies specific for either RNAP-β or CarD.
To test these predictions, we used an E. coli-based bacterial two-hybrid assay that detects the interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe (6). In this assay, the interaction between the WT M. tuberculosis CarD RID (amino acids 1 to 66) and the M. tuberculosis β1 lobe results in a 3-fold increase in β-galactosidase (β-Gal) activity from the two-hybrid test promoter (Fig. 1E). Substitution of I27 of the CarD RID with alanine or glycine reduced the β-Gal activity to near background levels, while substitution of I27 of the CarD RID with the bulkier hydrophobic residues phenylalanine and tryptophan resulted in 7- and 9-fold increases, respectively, in β-Gal activity relative to that observed with the WT M. tuberculosis CarD RID (Fig. 1E). The results of the two hybrid assays indicate that I27A and I27G substitutions weaken the interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe whereas I27F and I27W substitutions strengthen the interaction between the M. tuberculosis CarD RID and the M. tuberculosis β1 lobe, a finding consistent with the predictions from the structural modeling.
To determine whether the CarD I27F and I27W mutations increase the affinity of the interaction between CarD and RNAP-β subunit in vivo, we constructed Mycobacterium smegmatis strains expressing hemagglutinin (HA)-tagged wild-type CarD (CarDWT), CarD with an R-to-E change at position 25 (CarDR25E), CarDI27F, or CarDI27W. Coimmunoprecipitation experiments in M. smegmatis demonstrated that CarDI27W and CarDI27F mutants coprecipitated more RNAP-β than CarDWT (Fig. 1F). As previously reported, the CarDR25E mutant, which has lower affinity for RNAP-β than CarDWT, coprecipitated less RNAP-β than CarDWT (6). The CarDI27W mutant associated with more RNAP-β than CarDI27F, indicating a higher-affinity interaction.
CarDI27F and CarDI27W mutants stabilize open complexes at lower concentrations than CarDWT.
CarD regulates transcription initiation by binding to and stabilizing RNAP-promoter open complexes (3, 4, 7, 8, 10). We have previously developed a fluorescence assay for CarD activity that reports on open complex formation in real-time. In this assay, a Cy3 label, which exhibits a 2-fold fluorescence enhancement in open complex, is incorporated at the +2 position on the nontemplate strand of the M. tuberculosis rrnAP3 promoter within a linear fragment of M. tuberculosis genomic DNA containing nucleotides 1470151 to 1470300 (8). M. bovis RNAP-σA holoenzyme with or without M. tuberculosis CarD is then mixed with the labeled DNA fragment via stopped-flow spectrophotometry, and the fluorescence is monitored for 20 min. The Cy3 label increases fluorescence in the open complex conformation, and thus the rate of open complex formation can be monitored as a change in fluorescence. The amplitude of the fluorescence intensity curve correlates to the equilibrium amount of open complex in a given condition (8). To monitor the effect of increasing the affinity of CarD for RNAP on the formation and stability of open complexes, we performed this assay with concentrations of CarDI27F and CarDI27W ranging for 0 to 1800 nM and fixed concentrations of RNAP (225 nM) and promoter DNA (10 nM) (Fig. 2A). We found that the CarD concentration necessary to reach half of the maximum level of open complex at saturation (half-maximal concentration) was lower for CarDI27F (17 ± 2 nM) and CarDI27W (23 ± 3 nM) than for CarDWT (59 ± 10 nM), which is consistent with a higher affinity of the CarD I27 mutants for RNAP in initiation complexes compared to CarDWT (Fig. 2A).
FIG 2.

Effects of increasing the affinity of CarD for RNAP on open complex stability. (A) Equilibrium fluorescence fold change normalized to the fold change at high CarD concentrations (saturation) for each mutant of CarD with 225 nM M. bovis RNAP-σA and 10 nM Cy3-labeled M. tuberculosis rrnAP3 promoter. Both M. tuberculosis CarDI27F (red) and CarDI27W (blue) mutants achieve a half-maximal effect (dashed line) at lower concentrations (17 ± 2 nM for CarDI27F and 23 ± 3 nM for CarDI27W) than M. tuberculosis CarDWT (black; 59 ± 10 nM). (B) kobs3 values, calculated using ProData Viewer software from Applied Photophysics, of open complex formation as a function of CarD concentration for M. tuberculosis CarDWT (black), CarDI27F (red), and CarDI27W (blue). (C) In vitro transcription assay results showing a representative gel and a graph of the ratio of the amount of 3-nt initial transcript formed by 200 nM M. bovis RNAP-σA from 10 nM a linear DNA fragment in the presence of 2 μM CarD versus in the absence of CarD for reaction mixtures containing no CarD, M. tuberculosis CarDWT, CarDR25E, CarDI27F, or CarDI27W. The graph shows the means ± the SEM of data from at least four replicates. Statistical significance was analyzed by ANOVA and Tukey's multiple-comparison test (***, P ≤ 0.001; ****, P ≤ 0.0001).
CarDI27F and CarDI27W mutants accelerate promoter opening at lower concentrations than CarDWT.
We have previously reported modeling based on the trends of the slowest observed rate in the real-time fluorescent traces (kobs3) that suggests that CarD stabilizes open complex through a two-tiered concentration-dependent mechanism where CarD associates with both open and closed complexes with different affinities (8). More specifically, the model predicts that at low concentrations (i.e., <100 nM), CarDWT binds to open complex and prevents bubble collapse, resulting in more open complex and a slower observed rate. The model further predicts that at higher concentrations, CarDWT binds to the closed complex and accelerates the rate of opening, resulting in still more open complex and an acceleration in the observed rate. Analysis of kobs3 as a function of CarD concentration was performed for the CarD I27 mutants as previously described (8) (Fig. 2B). We found that the I27 mutants begin to accelerate kobs3 at lower concentrations (<50 nM) than WT CarD (100 nM). In fact, for CarDI27W we did not observe a deceleration in kobs3 even at the lowest concentration tested (16 nM). The two-tiered kinetic model predicts that acceleration in kobs3 arises from CarD binding to closed complex, which increases the rate of promoter opening. Thus, our working model predicts that, even at 16 nM, CarDI27W is associating with closed complex and accelerating opening. The acceleration of the observed rate at lower concentrations coupled with the lower half-maximal concentration for open complex stabilization is consistent with a model where the CarD I27 mutants have higher affinities to both closed and open RNAP-promoter complexes compared to CarDWT.
Increasing the affinity of CarD for RNAP increases formation of initial RNA products.
To investigate whether increasing the affinity of CarD for RNAP affects RNA synthesis, we monitored initial product formation in a multiround in vitro transcription assay (10). M. bovis RNAP-σA holoenzyme was incubated with the promoter template used in the fluorescence assays, but without the Cy3 label, in the presence or absence of WT or mutant M. tuberculosis CarD. GpU dinucleotide was used as the initiating nucleotide, and radiolabeled UTP was added as the extending nucleotide. Reaction mixtures were incubated for 20 min, after which the amounts of initial trinucleotide RNA products (GpUpU) were quantified. Addition of CarDWT protein increases the amount of initial RNA product formation ∼6-fold compared to the amount of product produced in the absence of CarD (Fig. 2C), whereas CarDR25E, which has decreased affinity for RNAP, only increases the amount of product formed ∼2-fold. In contrast, CarDI27F and CarDI27W increase the amount of product formed ∼9-fold. These data demonstrate that increasing the affinity of CarD for the RNAP results in an increase in initial RNA product formation. We infer that the effect of strengthening the interaction between CarD and RNAP on initial RNA product formation is a consequence of an increase in the formation of RNAP-promoter complexes (Fig. 2).
Increasing the affinity of CarD for RNAP stabilizes the CarD protein in M. tuberculosis.
In previous work, we constructed a strain of M. tuberculosis in which a copy of carD is expressed from a constitutive promoter at the attB site, a nonendogenous chromosomal locus, and the endogenous copy of carD is deleted (2). We have previously shown that a strain of M. tuberculosis that expresses carDR47E, which encodes a CarD protein that has an R-to-E change at position 47 and weakened affinity for the RNAP, from the attB site grows more slowly than a strain expressing wild-type carD (carDWT) from the attB site (6). To determine the effects of increasing the affinity of CarD for RNAP in the bacteria, we constructed M. tuberculosis strains expressing carDI27F or carDI27W from the attB site. We first determined the expression levels of the carD alleles in each strain by measuring carD transcript and protein levels. We found the CarDWT, CarDI27F, CarDI27W, and CarDR47E strains all produce equal levels of carD transcript, as expected given that the strains express the carD gene from the same promoter (Fig. 3A). However, when we examined the protein content in these strains we found that the CarDI27F and CarDI27W strains reproducibly had more CarD protein than the CarDWT strain (Fig. 3B, RNAP β levels also shown as a loading control). In addition, this effect is proportional to the magnitude of the change in affinity, since the CarDI27W strain reproducibly has more CarD protein than the CarDI27F strain and the CarDWT strain has more CarD protein than CarDR47E. Given the equal amount of transcript being produced, the different levels of protein indicate posttranslational differences in the stability of CarD protein in these strains. CarD has previously been identified as a Clp protease substrate in M. tuberculosis (11). Taken together, our data can be explained in the context of a model where CarD is protected from proteolysis when associated with RNAP.
FIG 3.

Increasing CarD's affinity for RNAP results in more stable CarD protein and faster growth without a change in rRNA levels. (A) The ratio of carD transcript levels in exponential cultures of the ΔcarD attB::tet-carD strain expressing CarDI27F, CarDI27W, or CarDR47E to levels in the CarDWT strain when grown in Sauton's medium. Transcript levels were determined using qRT-PCR and normalized to sigA. (B) Western blot analysis results of two biological replicates of lysates from the same cultures as used in panel A. Membranes were blotted with a monoclonal antibody against RNAP-β (top) or a polyclonal antibody against CarD (bottom). RNAP-β was used as a loading control for each sample. (C) Representative growth curves of the ΔcarD attB::tet-carD strain expressing CarDWT, CarDI27F, or CarDI27W in Sauton's medium. (D) Doubling times of the ΔcarD attB::tet-carD strain expressing CarDWT, CarDI27F, or CarDI27W in Sauton's medium. Doubling times were calculated for each of five biological replicate growth curves using the exponential growth equation and excluding values after the optical density at 600 nm (OD600) reached 1 to focus on the growth rate in the exponential growth phase. (E) Ratio of 16S rRNA levels in exponential cultures of the ΔcarD attB::tet-carD strain expressing CarDI27F or CarDI27W to levels in the CarDWT strain when grown in Sauton's medium. Transcript levels were determined using qRT-PCR and normalized to sigA. Each graph shows the means ± the SEM of data from at least three replicates. Statistical significance was analyzed by ANOVA and Tukey's multiple-comparison test (n.s., not significant; *, P ≤ 0.05).
Increasing CarD's affinity for RNAP increases the growth rate in M. tuberculosis.
We have previously reported that decreasing CarD's affinity for RNAP decreases growth rate in M. tuberculosis strain (6). To determine the effect of increasing CarD's affinity for RNAP on growth rate, we back-diluted cultures in the exponential growth phase (to avoid an initial lag phase) into fresh liquid Sauton's medium and monitored growth. We found that the CarDI27F and CarDI27W strains had a faster doubling time than the CarDWT stain (26.31-, 25.75-, and 30.74-h doubling times, respectively) (Fig. 3C and D). These results, as well as the growth defect previously reported in a strain expressing a CarD mutant with lower affinity for RNAP (6), demonstrate that the affinity of CarD for the RNAP can impact growth rate in M. tuberculosis.
Increasing the affinity of CarD for RNAP does not result in differences in rRNA transcript levels in M. tuberculosis, despite the effects on growth rate.
We have previously shown that decreasing the affinity of CarD for RNAP results in decreased rRNA levels and slower growth (6, 7). Because rRNA levels are often correlated to growth rate (12–14), the decreased rRNA levels in the CarD mutants with lower affinity for RNAP could contribute to the growth defect associated with these mutations. To determine the effect of increasing the affinity of the interaction between CarD and RNAP on rRNA levels in the bacterium, we isolated RNA from exponential cultures of CarDWT, CarDI27F, and CarDI27W strains in Sauton's medium and performed quantitative real-time PCR (qRT-PCR) analysis for the levels of stable 16S rRNA using primers internal to the 16S rRNA transcript (Fig. 3E). The CarD I27 mutants that have higher affinity for RNAP express the same amount of rRNA as the CarDWT strain, indicating that the faster growth in these strains is not a result of increasing rRNA expression. Therefore, the rRNA levels and growth rate are uncoupled in strains expressing CarD proteins with various affinities to RNAP. Thus, the effects of altering CarD's affinity for RNAP on growth rate can occur independently of effects on rRNA levels. To the best of our knowledge, this is also the first report of rRNA levels and growth rate being uncoupled in M. tuberculosis.
Increasing the affinity of CarD for RNAP attenuates survival of M. tuberculosis during infection of mice.
We have previously shown that weakening the interaction of CarD with RNAP results in decreased bacterial survival by 35 days postinfection (dpi) in mice (6). To determine the effect of increasing the affinity of CarD for RNAP on virulence, we infected C57BL/6J mice with the M. tuberculosis CarDWT, CarDI27F, or CarDI27W strains and measured the bacterial burden in the lungs at 21 and 35 dpi (Fig. 4). Both the CarDI27F and the CarDI27W mutants were attenuated for survival in the mice and displayed significantly lower bacterial burden in the lungs (Fig. 4). Specifically, the CarDI27W strain displayed significantly lower bacterial burden than the CarDWT strain at 21 and 35 dpi, whereas the CarDI27F strain had lower bacterial burdens than CarDWT at 35 dpi. The severity of the virulence defect in the CarD mutants mirrors the effect of these mutations on the affinity of CarD for RNAP, with CarDI27W exhibiting both a higher affinity and a more pronounced virulence defect than CarDI27F. These data show that although increasing the affinity of CarD for RNAP increases the rate of formation of transcription competent RNAP-promoter complexes and increases growth rates in culture, it results in a loss of virulence. The degree of attenuation at 35 dpi for the CarD I27 mutants is similar to that previously reported for M. tuberculosis strains expressing a mutant CarD protein with lower affinity for the RNAP (6). Thus, either decreasing or increasing the affinity of CarD for RNAP has a detrimental effect on the virulence of M. tuberculosis. We therefore propose that this indicates that the affinity of CarD for RNAP is finely tuned to the physiology of this obligate pathogen to optimize virulence.
FIG 4.
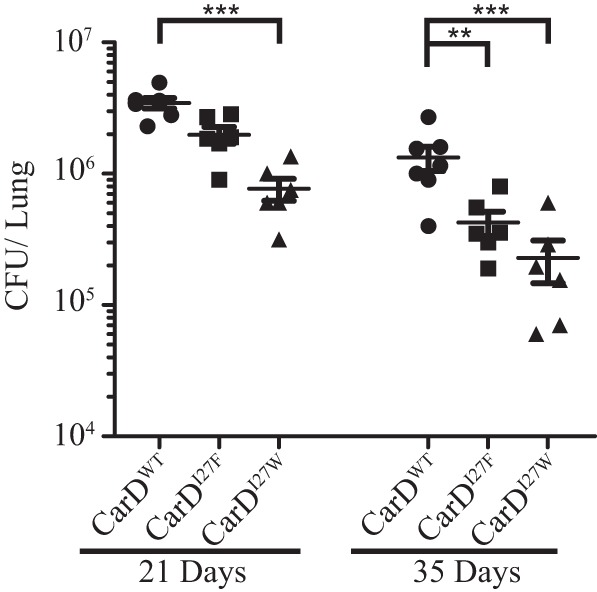
Increasing CarD's affinity for RNAP attenuates M. tuberculosis in a mouse model of infection. Dot plot of CFU in the lungs of C57BL/6J mice infected with the ΔcarD attB::tet-carD strain expressing either CarDWT (●), CarDI27F (■), or CarDI27W (▲). Each time point is the means ± the SEM of data from at least six mice per strain. Statistical significance was analyzed by ANOVA and Tukey's multiple-comparison test (**, P ≤ 0.01; ***; P ≤ 0.001). All comparisons were tested, and only significantly different comparisons are shown.
DISCUSSION
In this study, we investigated both the biochemical and the physiological effects of increasing the affinity of CarD for RNAP. Our findings reveal new aspects of CarD regulation and the effect of increasing the affinity of CarD for RNAP-β on growth rate and virulence.
Using a real-time fluorescence assay, we found that increasing the affinity of CarD for RNAP leads to a decrease in the concentration of CarD required for half-maximal open complex formation and stability (Fig. 2A). Our data further show that the higher-affinity CarD mutants result in increased stability of an RNAP-promoter complex and an increase in the formation of initial RNA products (Fig. 2C). However, despite these positive effects of the CarD I27 mutants on the formation of transcription competent RNAP-promoter complexes at rrnAP3 in vitro, M. tuberculosis strains expressing CarDI27F or CarDI27W did not contain higher levels of rRNA than the CarDWT strain (Fig. 3E). This observation may be explained by the presence of other factors in vivo that impact whether rRNA levels respond to changes in the affinity of CarD for the RNAP. In addition, increasing the affinity of CarD for RNAP-β may affect other stages of transcription after open complex formation (i.e., promoter escape) that ultimately impact full transcript production. These possibilities will be explored in future investigations.
We found that increasing the affinity of CarD for RNAP results in an increase in growth rate without changing the levels of rRNA (Fig. 3C and D). This suggests that in WT strains rRNA and ribosomes would be in excess of the amounts required to accommodate a higher growth rate. We also found that increasing the affinity of CarD for RNAP in M. tuberculosis results in attenuated virulence in mice (Fig. 4). Together, these findings demonstrate that the differences in growth rate and virulence of the CarD I27 mutant strains are not a result of altered rRNA content but are instead another consequence of varied levels or activity of CarD proteins, thus uncoupling the role of CarD in regulating rRNA content from its effect on growth rate. Therefore, the essential function of CarD is unlikely to be limited to regulation of rRNA levels. These data also indicate that there exist non-rRNA genes that are more sensitive to changes in the affinity of CarD for RNAP than the rRNA genes. We have previously shown that in the absence of CarD, the mycobacterial RNAP is only weakly able to form open complexes at rrnAP3 (3, 8, 10). In addition, CarD is localized to promoter-bound RNAP holoenzymes throughout the genome (4). These studies suggest that CarD may in fact be a general component of the transcription initiation machinery in mycobacteria and that its essential role is enabling the mycobacterial RNAP to efficiently form open complexes at all promoters in the genome. In particular, changes in the affinity of CarD for the RNAP could result in a global dysregulation of gene expression. Alternatively, phenotypes of CarD mutants may result from the deregulation of a specific subset of genes differentially regulated by CarD. It should also be noted that, despite the presence at all promoters, at this point CarD activity has only been evaluated at mycobacterial rRNA promoters and consensus promoters from E. coli. Therefore, a priority of future work will be to clarify the role of CarD in global transcription regulation.
Using multiple CarD mutants, we have also revealed a role for the CarD-RNAP interaction in regulating CarD protein levels. Increasing the affinity of CarD for RNAP increases the cellular concentration of CarD without affecting carD transcript levels (Fig. 3A and B). The effect on CarD protein concentration is proportional to the affinity for RNAP and, of the strains expressing carD from the attB site, CarDI27W has the highest affinity for RNAP and the highest concentration of CarD protein, while CarDR47E has the lowest affinity for CarD and the lowest CarD protein levels (Fig. 3A and B). These data suggest that CarD is protected from proteolytic degradation while associated with RNAP. Since CarD is a known target of the Clp protease in mycobacteria (11), one possibility is that Clp-mediated degradation of CarD is more efficient when CarD is not associated with RNAP. Thus, our findings provide a mechanistic explanation for our previous observation that mycobacteria contain a similar concentration of CarD and the housekeeping sigma factor (8) and suggest that there may be a detrimental effect of excess, free cellular CarD.
MATERIALS AND METHODS
Bacterial two-hybrid assays.
The bacterial two-hybrid assay used in this study is based upon the demonstration that contact between a protein domain fused to the α subunit of RNA polymerase and a partner protein fused to the bacteriophage λCI protein activates transcription of a lacZ reporter gene under the control of a test promoter bearing an upstream λ operator (15). Assays were performed as described using E. coli FW102 F′ OL2-62 reporter strain cells, which contain the test promoter placOL2-62 driving the expression of a linked lacZ gene on an F′ episome (15). Plasmids used in these assays included pBRα-Mt CarD (1 to 66), which encodes residues 1 to 248 of the E. coli RNAP α subunit fused to residues 1 to 66 of M. tuberculosis CarD (6); plasmid pBRα, which encodes the WT α subunit; and plasmid pACλCI-Mt β1, which encodes residues 1 to 236 of the bacteriophage λCI protein fused to residues 52 to 178 of the β subunit of M. tuberculosis RNAP fused to residues 379 to 440 of the β subunit of M. tuberculosis RNAP via two glycine residues (6). Plasmids carrying amino acid substitutions at residue I27 of M. tuberculosis CarD were introduced into pBRα-CarD (1 to 66) by PCR.
FW102 F′ OL2-62 were cotransformed with either the indicated pBRα-Mt CarD (1 to 66) derivative and pACλCI-Mt β1 or pBRα and pACλCI-Mt β1. Individual transformants were selected and grown in Luria-Bertani (LB) medium supplemented with carbenicillin (100 μg/ml), chloramphenicol (25 μg/ml), and kanamycin (50 μg/ml). No IPTG (isopropyl-β-d-thiogalactopyranoside) was present in the growth medium. β-Galactosidase assays were performed as described previously (6). Graphs in Fig. 1E depict the β-Gal activity in cells containing the indicated pBRα-Mt CarD (1 to 66) derivative and pACλCI-Mt β1. The bar labeled “No CarD” in Fig. 1E depicts the β-Gal activity observed in cells containing pBRα and pACλCI-Mt β1.
Bacterial strains and growth conditions. (i) M. tuberculosis.
All M. tuberculosis strains were derived from the Erdman strain and were grown at 37°C in Sauton's broth medium (0.5 g liter−1 KH2PO4, 0.5 g liter−1 MgSO4, 4 g liter−1 l-asparagine, 60 ml of glycerol, 0.05 g liter−1 ferric ammonium citrate, 2.0 g liter−1 citric acid, 0.1 ml liter−1 1% ZnSO4, 0.05% Tween 80 [pH 7.0]) or 7H10 agar medium (Difco) supplemented with 60 μl liter−1 oleic acid, 5 g liter−1 bovine serum albumin (BSA), 2 g liter−1 dextrose, 0.003 g liter−1 catalase (OADC), and 0.5% glycerol. Gene switching was used to construct strains of mycobacteria expressing different carD alleles and to test for their viability (6, 16). Specifically, the M. tuberculosis ΔcarD attB::tet-carD strain (described previously in reference 2) was transformed with pMSG430Rv3583cI27F, pMSG430Rv3583cI27W, or pMSG430Rv3583cR47E (expresses M. tuberculosis CarDWT, CarDI27F, CarDI27W, or CarDR47E, respectively, from a constitutive Pmyc1-tetO promoter, kanamycin resistant) or pDB19Rv3583c (expresses CarDWT from a constitutive Pmyc1-tetO promoter, zeocin resistant). The transformants were selected on 20 μg ml−1 kanamycin or 1.25 μg ml−1 zeocin. The carD gene from each transformant was sequenced to confirm the presence of the correct sequence. The M. tuberculosis ΔcarD attB::tet-carD strains transformed with pDB19Rv3583cWT, pMSG430Rv3583cI27F, pMSG430Rv3583cI27W, or pMSG430Rv3583cR47E were named mgm3080, csm230, csm231 and csm195, respectively.
(ii) M. smegmatis.
All M. smegmatis strains were derived from mc2155 and were grown at 37°C in LB medium supplemented with 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 (broth). M. smegmatis strains expressing HA-tagged CarDWT, CarDR25E, CarDI27F, or CarDI27W were engineered as described for the analogous M. tuberculosis strains using pMSG430 expression plasmids and the M. smegmatis ΔcarD attB::tet-carD strain (described previously in reference 2). The M. smegmatis ΔcarD attB::tet-carD strains expressing C-terminal HA-tagged versions of M. tuberculosis CarDWT, M. tuberculosis CarDR25E, M. tuberculosis CarDI27F, or M. tuberculosis CarDI27W from a constitutive Pmyc1-tetO promoter at the attB site of M. smegmatis ΔcarD attB::tet-carD were named mgm3090, mgm3091, csm228, and csm229, respectively.
Cell lysate.
Next, 20-ml portions of exponential cultures of M. tuberculosis were lysed in 500 μl of NP-40 buffer (10 mM sodium phosphate [pH 8.0], 150 mM NaCl, and 0.25% NP-40) by bead beating three times.
Immunoprecipitation.
Lysate from M. smegmatis strains expressing HA-tagged alleles of CarD was bound to monoclonal anti-HA–agarose (Sigma), and the complexes were eluted as previously described (6, 7).
Western blot analysis.
Protein samples were mixed with an SDS-PAGE loading buffer and run on a 4 to 12% Bis-Tris protein gel (Invitrogen). HA-tagged CarD in the immunoprecipitation experiment was detected using a mouse monoclonal antibody (clone 10F05; Memorial Sloan-Kettering Cancer Center Monoclonal Antibody Core Facility). To determine protein concentrations in cell lysate, CarD was detected with a rabbit polyclonal antibody (2). RNAP-β was detected with a mouse monoclonal antibody in both experiments (clone 8RB13; Neoclone, Madison, WI). Horseradish peroxidase-conjugated secondary antibodies against mouse or rabbit (Perkin-Elmer) were used for detection.
Quantitative real-time PCR.
RNA was prepared from 20 to 30 ml of M. tuberculosis Erdman (WT M. tuberculosis), mgm3080 (CarDWT), csm195 (CarDR47E), csm230 (CarDI27F), and csm231 (CarDI27W) during the exponential growth phase. 16S rRNA and carD levels were measured and normalized to sigA transcript levels as previously described (2).
Mouse infections.
Infection of mice with exponentially replicating M. tuberculosis Erdman strain (WT M. tuberculosis), csm3080 (CarDWT), csm230 (CarDI27F), and csm231 (CarDI27W) strains and the determination of bacterial loads were performed as previously described (6, 7). All procedures involving animals were conducted according to the National Institutes of Health (NIH) guidelines for the housing and care of laboratory animals, and all procedures were performed in accordance with institutional regulations after protocol review and approval by the Institutional Animal Care and Use Committee of the Washington University in St. Louis School of Medicine (protocol 20130156, analysis of mycobacterial pathogenesis). Washington University is registered as a research facility with the U.S. Department of Agriculture and is fully accredited by the American Association of Accreditation of Laboratory Animal Care. The Animal Welfare Assurance documentation is on file with the Office for Protection from Research Risks of the NIH. All animals used in these experiments were subjected to no or minimal discomfort. All mice were euthanized by CO2 asphyxiation, which is approved by the American Veterinary Medical Association Panel on Euthanasia.
Protein preparation for biochemical assays.
Recombinant Mycobacterium bovis core RNAP was purified from E. coli using a system kindly supplied by Robert Landick (17, 18) as previously described (7). Recombinant M. bovis σA, which is identical to M. tuberculosis σA, was purified from E. coli and added to the core RNAP to reconstitute the RNAP holoenzyme.
Preparation of fluorescent promoter DNA fragments.
The DNA template contains nucleotides 1470151 to 1470300 of the M. tuberculosis Erdman genomic DNA, which includes the rrnAP3 promoter. Cy3-labeled promoter DNA was prepared as previously described (8).
Stopped-flow fluorescence assay.
Stopped-flow fluorescence experiments were performed as previously described (8). All experiments were performed using a final M. bovis RNAP-σA holoenzyme concentration of 225 nM and a final promoter DNA concentration of 10 nM. Experiments were performed at 25°C under the following final solution conditions: 14 mM Tris (pH 8.0), 120 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 0.1 mg/ml BSA, and 10% glycerol by volume. Equal volume mixing of protein with promoter DNA was performed in a stopped-flow apparatus (Applied Photophysics SX-20, total shot volume 150 μl, dead time < 2 ms), so the initial protein and DNA solutions were each diluted by half in order to reach their final reaction concentrations. Due to slight hardware variations in different stopped-flow instruments, excitation light was provided by either a 510 nm fixed-wavelength LED light source or a 515-nm light source from an arc lamp passed through a monochromator. The difference in excitation light had no effect on the data. In all cases emission was collected at 570+ nm using a long-pass filter. The fluorescence was monitored for 20 min.
At least two traces were collected and averaged per condition. Traces were plotted as fold change over DNA according to the formula (F − F0)/F0, where F0 is the buffer-subtracted signal for DNA alone, and F is the buffer-subtracted signal for DNA mixed with protein. The final point of each trace was used as a measure of equilibrium fluorescence. The fold change traces were fit to a triple exponential from 0.1 to 1200 s using the ProData Viewer software from Applied Photophysics. The slowest observed rate dominated the fractional amplitude of the fits and was used as a measurement of kobs3. Conditions that were repeated multiple times on different days were used to estimate standard errors of the mean (SEM). An average SEM was used to estimate uncertainty for specific conditions that were only repeated multiple times on the same day.
In vitro transcription assays.
CarD proteins used in in vitro transcription assays were diluted into 1× dialysis buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1 mM 2-mercaptoethanol). For the aborted 3-nucleotide (nt) transcript assay (10), a linear fragment of dsDNA M. tuberculosis Erdman strain genomic DNA containing nucleotides 1470151 to 1470300, which includes the M. tuberculosis rrnAP3 promoter, was prepared by annealing and extending primers overlapping 85-bp primers (Integrated DNA Technologies, Coralville, IA). Reaction conditions were as follows: 200 nM M. bovis core RNAP, 2 μM M. bovis σA, 2 μM CarD or equivalent volume of buffer, 10 nM linear DNA template, 210 μM GpU dinucleotide, 21 μM UTP, 0.1 μl [α32-P]UTP, 13.25 mM Tris (pH 8.0), 59 mM NaCl, 10.12 mM MgCl2, 5% (vol/vol) glycerol, 1 mM DTT, and 0.1 mg/ml BSA (NEB) in a total volume of 20 μl. M. bovis core and σA were incubated for 10 min at 37°C, followed by the addition of CarD and 10 more minutes of incubation at 37°C. The DNA template was added, and the reactions were diluted to 17.5 μl, followed by incubation at 37°C for 10 min. Reaction mixtures were initiated with addition of a 2.5 μl of mixture containing GpU, UTP, and the radiolabeled UTP, followed by incubation at 37°C. After 20 min, the reactions were stopped with 2× formamide buffer (98% [vol/vol] formamide, 5 mM EDTA) and run on a 22% urea-PAGE gel. Transcripts were quantified using phosphorimaging and analyzed using Image Gauge software (FujiFilm).
ACKNOWLEDGMENTS
We thank Tim Lohman for the use of his stopped-flow spectrophotometer, Tom Ellenberger for the use of the high-pressure and fast-performance liquid chromatography in his laboratory, and Yocheved Gensler for assistance with cloning. We are grateful to Katherine Mann and Jerome Prusa for their thoughtful review of the manuscript.
We declare that we have no conflicts of interest with the contents of this article.
C.L.S. and E.A.G. are supported by grant GM107544 from the National Institutes of Health. B.E.N. is supported by grant GM118059 from the National Institutes of Health. E.A.C. is supported by grant GM114450 from the National Institutes of Health. A.L.G. is supported by the NIGMS Cell and Molecular Biology Training grant GM007067 and the Stephen I. Morse Graduate Fellowship. J.R. is supported by a Sigma-Aldrich Predoctoral Fellowship. J.P.H. is supported by National Science Foundation Graduate Research Fellowship DGE-1143954.
A.L.G., B.E.N., E.A.C., S.A.D., and C.L.S. conceived the project, designed experiments, analyzed results, and wrote the manuscript. A.L.G., J.R., J.P.H., L.A.W., L.M.O., B.B., J.C., D.J., and A.R.M. performed experiments and analyzed the results. E.A.G. analyzed results and edited the manuscript.
REFERENCES
- 1.WHO. 2015. Global tuberculosis report. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/en/. [Google Scholar]
- 2.Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. 2009. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae B, Chen J, Davis E, Leon K, Darst SA, Campbell EA. 2015. CarD uses a minor groove wedge mechanism to stabilize the RNA polymerase open promoter complex. eLife 4:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava DB, Leon K, Osmundson J, Garner AL, Weiss LA, Westblade LF, Glickman MS, Landick R, Darst SA, Stallings CL, Campbell EA. 2013. Structure and function of CarD, an essential mycobacterial transcription factor. Proc Natl Acad Sci U S A 110:12619–12624. doi: 10.1073/pnas.1308270110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-Moreno D, Abellón-Ruiz J, García-Heras F, Murillo FJ, Padmanabhan S, Elías-Arnanz M. 2010. CdnL, a member of the large CarD-like family of bacterial proteins, is vital for Myxococcus xanthus and differs functionally from the global transcriptional regulator CarD. Nucleic Acids Res 38:4586–4598. doi: 10.1093/nar/gkq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss LA, Harrison PG, Nickels BE, Glickman MS, Campbell EA, Darst SA, Stallings CL. 2012. Interaction of CarD with RNA polymerase mediates Mycobacterium tuberculosis viability, rifampin resistance, and pathogenesis. J Bacteriol 194:5621–5631. doi: 10.1128/JB.00879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garner AL, Weiss LA, Manzano AR, Galburt EA, Stallings CL. 2014. CarD integrates three functional modules to promote efficient transcription, antibiotic tolerance, and pathogenesis in mycobacteria. Mol Microbiol 93:682–697. doi: 10.1111/mmi.12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rammohan J, Ruiz Manzano A, Garner AL, Stallings CL, Galburt EA. 2015. CarD stabilizes mycobacterial open complexes via a two-tiered kinetic mechanism. Nucleic Acids Res 43:3272–3285. doi: 10.1093/nar/gkv078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gulten G, Sacchettini JC. 2013. Structure of the M. tuberculosis CarD/RNAP β-lobes complex reveals the molecular basis of interaction and presents a distinct DNA-binding domain for M. tuberculosis CarD. Structure 21:1859–1869. doi: 10.1016/j.str.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis E, Chen J, Leon K, Darst SA, Campbell EA. 2015. Mycobacterial RNA polymerase forms unstable open promoter complexes that are stabilized by CarD. Nucleic Acids Res 43:433–445. doi: 10.1093/nar/gku1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raju RM, Unnikrishnan M, Rubin DHF, Krishnamoorthy V, Kandror O, Akopian TN, Goldberg AL, Rubin EJ. 2012. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog 8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bremer H, Dennis PP. 1996. Modulation of chemical composition and other parameters of the cell by growth rate, p 1553–1569. In Escherichia coli and Salmonella Typhimurium: cellular and molecular biology. ASM Press, Washington, DC. [Google Scholar]
- 13.Schaechter M, Maaloe O, Kjeldgaard NO. 1958. Dependency on medium and temperature of cell size and chemical composition during balanced growth of Salmonella typhimurium. J Gen Microbiol 19:592–606. doi: 10.1099/00221287-19-3-592. [DOI] [PubMed] [Google Scholar]
- 14.Binder BJ, Liu YC. 1998. Growth rate regulation of rRNA content of a marine Synechococcus (cyanobacterium) strain. Appl Environ Microbiol 64:3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nickels BE. 2009. Genetic assays to define and characterize protein-protein interactions involved in gene regulation. Methods 47:53–62. doi: 10.1016/j.ymeth.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 16.Pashley CA, Parish T. 2003. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol Lett 229:211–215. doi: 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- 17.Huff J, Czyz A, Landick R, Niederweis M. 2010. Taking phage integration to the next level as a genetic tool for mycobacteria. Gene 468:8–19. doi: 10.1016/j.gene.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Czyz A, Mooney RA, Iaconi A, Landick R. 2014. Mycobacterial RNA polymerase requires a U-tract at intrinsic terminators and is aided by NusG at suboptimal terminators. mBio 5:1–10. doi: 10.3391/mbi.2014.5.1.01. [DOI] [PMC free article] [PubMed] [Google Scholar]