

Abstract
Background
Tobacco smoke and alcohol drinking are the major risk factors for squamous cell carcinoma of the head and neck (SCCHN). Smoking and drinking cause DNA damage leading to apoptosis, and insufficient apoptotic capacity may favor development of cancer because of the dysfunction of removing damaged cells. In the present study, we investigated the association between camptothecin (CPT)-induced apoptotic capacity and risk of SCCHN in a North American population.
Methods
In a case-control study of 708 SCCHN patients and 685 matched cancer-free controls, we measured apoptotic capacity in cultured peripheral blood lymphocytes (PBLs) in response to in vitro exposure to CPT by using the flow cytometry-based method.
Results
We found that the mean level of apoptotic capacity in the cases (45.9±23.3%) was significantly lower than that in the controls (49.0±23.1%) (P = 0.002). When we used the median level of apoptotic capacity in the controls as the cutoff value for calculating adjusted odds ratios (ORs), subjects with a reduced apoptotic capacity had an increased risk (adjusted OR = 1.42, 95% confidence interval [CI] = 1.13–1.78, P = 0.002), especially for those who were age ≥57 (1.73, 1.25–2.38, 0.0009), men (1.76, 1.36–2.27, < 0.0001) and ever drinkers (1.67, 1.27–2.21, 0.0003), and these variables significantly interacted with apoptotic capacity (Pinteraction = 0.015, 0.005 and 0.009, respectively). A further fitted prediction model suggested that the inclusion of apoptotic capacity significantly improved in the prediction of SCCHN risk.
Conclusion
Individuals with a reduced CPT-induced apoptotic capacity may be at an increased risk of developing SCCHN, and apoptotic capacity may be a biomarker for susceptibility to SCCHN.
Keywords: biomarker; Apoptosis; CPT, apoptotic capacity; head and neck cancer
1. Introduction
Head and neck cancer is a heterogeneous group of tumors that involve multiple sites and cellular origins within the head and neck region. Squamous cell carcinoma of the head and neck (SCCHN), including cancers of the oral cavity, oropharynx, hypopharynx, and larynx, is the most common histological type (i.e., 90% of cases) and one of the six most common cancers worldwide [1]. In the United States, approximately 60,000 new cases are diagnosed annually and 12,000 die of this disease each year [2]. It is well known that tobacco smoke and alcohol use as well as prior human papilloma virus (HPV) infection (particularly for the oropharynx subsite) are the major risk factors that play an important role in the etiology of SCCHN. However, only a small fraction of smokers and/or drinkers will develop SCCHN, and a small proportion of those exposed to HPV will develop oropharyngeal cancer (OPC), suggesting that there is genetic susceptibility to this disease in the general population [3–5]. Indeed, tobacco smoke and consumption of alcoholic beverages contain several carcinogens that can cause different types of DNA damage in the target cells [6, 7]. Apoptosis functions to eliminate damaged cells and thus is a critical factor in the protection against tobacco and alcohol induced cancers, including SCCHN [8].
Apoptosis, also called programmed cell death, is a critical mechanism to maintain the balance of cell survival and death by removing irreparable damaged cells [9–11]. Cells have two main apoptosis signaling pathways, an extrinsic (death receptor) and an intrinsic (mitochondrial) pathway [12–14]. The extrinsic pathway, or the death receptor pathway, is initiated from outside of the cell, usually when conditions in the extracellular environment determine cell death. This pathway can be induced through the activation of death receptors, such as death receptor 3 (DR3), death receptor 4 (DR4), death receptor 5 (DR5), FAS and tumor necrosis factor alpha receptor (TNFαR), by their respective ligands. The intrinsic pathway, or the mitochondrial pathway, begins when DNA damage occurs within the cell. It involves a diverse array of non-receptor-mediated stimuli that produce intracellular signals that act directly on targets within the cell. In both pathways, the signaling leads to activation of the caspase family proteases responsible for dismantling and removing the dying cell [15–17]. It is well known that apoptosis contributes to a wide variety of physiological and pathological processes, and dysregulation of apoptosis can disrupt the delicate balance between cell proliferation and cell death, leading to diseases including cancer [9, 18]. Increasing evidence has demonstrated that insufficient apoptotic capacity can promote development of cancer [19, 20]. Therefore, it is important to detect apoptotic levels and their association with cancer risk through epidemiologic research.
There are many different experimental methods that can induce apoptosis in vitro, including radiation, benzo[a]pyrene-7,8–9,10-diol epoxide (BPDE), thapsigargin and camptothecin (CPT), which have somewhat different underlying mechanisms [21–26]. For example, CPT is an effective chemotherapeutic drug for treatment of patients with cancer [27]. Although the mechanisms underlying CPT-mediated responses in cancer cells are not fully understood, it has been identified that CPT induces apoptosis by inhibiting topoisomerase I, resulting in high levels of internally accumulated DNA double-strand breaks in the cell, ultimately leading to cell death [28–30]. CPT is believed to possess promising anticancer effect against a broad spectrum of cancer cell lines, such as those of the breast, colon, lung, and ovarian cancers, because many cancer cells exhibit downregulation of the topoisomerase I activity [30, 31]. To date, most experimental studies of CPT-induced apoptosis were conducted with some cancer cell lines and animal models [22, 25, 27, 32], and there are no reported assays that measure the CPT-induced apoptotic capacity in humans, such as using peripheral blood lymphocytes (PBLs) by flow cytometry in epidemiologic research. In the present study, by measuring the CPT-induced apoptotic capacity in PBLs with a flow cytometry-based Terminal Transferase dUTP Nick End Labeling (TUNEL) assay as previously described [33], we tested the hypothesis that suboptimal apoptotic capacity measured in PBLs is associated with risk of SCCHN in a case-controls study of 703 cases and 685 cancer-free controls of a North American population.
2. Materials and Methods
2.1. Study subjects
The analysis included a total 703 cases who were patients with newly diagnosed, untreated primary tumors of the oral cavity (n = 214; 30.4%), oropharynx (n = 367; 52.4%), hypopharynx (n = 20; 2.8%), larynx (n = 101; 14.4%), and unknown primaries (n = 1; 0.1%). These cases were histologically confirmed and recruited at The University of Texas M.D. Anderson Cancer Center during the period between November 2005 and November 2012. Patients with second SCCHN primary tumors, primary tumors of the nasopharynx or sinonasal tract, or any histopathologic diagnosis other than SCCHN were excluded. By frequency matching on age (±5 years), sex, and ethnicity, we also identified an additional 685 cancer-free control subjects from among hospital visitors at M.D. Anderson Cancer Center during the same time period. Having provided a written informed consent, each of cases and controls completed a self-administered life-style questionnaire that collected information about risk factors, such as tobacco smoking and alcohol use, and donated a one-time sample of 30 ml of blood for biomarker tests. The University of Texas M.D. Anderson Cancer Center Institutional Review Board approved the research protocol.
2.2. Human peripheral blood lymphocytes (PBLs) culture and camptothecin (CPT) treatment
The method of PBL culture and CPT treatment have been previously reported [33]. Briefly, the PBLs were isolated from the whole blood by using Ficoll (Pharmacia Biotech Inc., Piscataway, NJ) gradient centrifugation and then cultured in RPMI 1640 supplemented with 15% fetal calf serum (GIBCO BRL) and 56.25 μg/ml phytohemagglutinin (Murex Diagnostics, Norcross, GA) for 48 hours at 37°C in an incubator with 5% CO2. We used CPT to selectively induce apoptosis of the PBLs. The baseline or spontaneous apoptosis index was obtained from the same individual’s samples that were not incubated in parallel with those treated by CPT. The dose of 250 nmol/L CPT (Cat# C9911; Sigma-Aldrich, Inc.) for the in vitro treatment of the cells for 24 hours was determined according to a previous report [22]. At the indicated time points, the aliquotted samples of cultured cells were harvested and fixed with 1% paraformaldehyde, washed with 1x PBS and finally stored in 70% ethanol at −20°C until used for the apoptotic detection by a flow cytometer.
2.3. Apoptotic analysis with flow cytometry
For the measurement of CPT-induced apoptotic capacity, we used the TUNEL assay as previously described [33]. Briefly, we detected apoptotic cells with an APO-Brdu kit (Phoenix Flow Systems, San Diego, CA), which contained apoptosis-positive and -negative cells as the assay controls, and we used a two-color staining method for labeling DNA breaks and total cellular DNA. The kit included washing, reaction, and rinsing buffers for processing each step of the assay; terminal deoxynucleotidyl transferase enzyme, bromodeoxyuridine triphosphate, and fluorescein-labeled anti-bromodeoxyuridine antibody for labeling DNA; and propidium iodide/RNase A solution for counterstaining the total DNA. The ratio of apoptotic cells (1 × 105) was calculated based on the measurements by flow cytometry (with the Epics Profile II Flow Cytometer, Beckman Coulter, Inc.) according to the instructions of the manufacturer.
2.4. Statistical analysis
The differences in selected demographic variables, smoking, and alcohol use between SCCHN cases and controls were evaluated by using the χ2-test. The two-sided Student’s t test was used to test the difference between cases and controls for baseline and CPT- induced apoptotic capacity levels. The apoptotic capacity was determined by the formula [(Percent experimental apoptosis – percent spontaneous apoptosis)/(100 – percent spontaneous apoptosis)] × 100% [34]. The median apoptotic capacity (%) of control subjects was used as the cutoff value: values greater than or equal to the median were considered high levels of apoptotic capacity, and values below the median were considered low/suboptimal levels of apoptotic capacity. The quartiles of apoptotic capacity in controls subjects were used as the cut-off values to evaluate the apoptotic capacity dose-response effect on SCCHN risk. We also dichotomized apoptotic capacity by the median of controls to evaluate its effect on risk of SCCHN by computing odds ratios (ORs) and their 95% confidence intervals (CIs) from both univariate and multivariate logistic regression models in the case-control analysis. These analyses were performed with or without adjustment for age (in years), sex, smoking and drinking status. Subjects who had smoked <100 cigarettes in their lifetimes were defined as never smokers; all others were defined as ever smokers. Among ever smokers, those who had quit and had not smoked for >1 year before the interview were defined as former smokers and the others were defined as current smokers. Similarly, subjects who had reported drinking alcoholic beverages at least once a week and longer than 1 year prior to diagnosis or interview were defined as ever drinkers and the remaining as never drinkers. Among ever smokers, those who had quit drinking for longer than 1 year prior to diagnosis or interview were defined as former drinkers and the others as current drinkers.
We also performed a stratified analysis followed by an assessment of interactions between the quartiled apoptotic capacity and selected variables, such as age, sex, ethnicity, smoking and drinking status, tumor sites and stages as previously described [35, 36]. The multiplicative interaction was estimated by including the interaction term (i.e., the quartiled apoptotic capacity × risk factor) in one multivariate logistic regression model, which also included the main effect of the quartiled apoptotic capacity and other covariates. To assess the effect of apoptotic capacity on SCCHN risk prediction, receiver operating characteristic (ROC) curves were plotted from the logistic regression models, and the area under the curve (AUC) were used to assess the classification performance of the models. Statistical significance of the improvement in AUC after adding apoptotic capacity to the model with existing covariates (e.g., age, sex, ethnicity, smoking and drinking status) was calculated by Delong’s test as previously described [36, 37]. All tests were two sided, and all statistical analyses were performed with the SAS software (version 9.1.3; SAS Institute, Inc., Cary, NC).
3. Results
3.1. Characteristics of the study population
As shown in Table 1, there were no significant differences in the distributions of age and sex between 703 cases and 685 cancer-free controls, with a similar age distribution (P = 0.745) with mean ages of 56.9 (±10.7) and 56.8 (±11.1) years, respectively, but the cases were more likely than the controls to be ever smokers (64.4% versus 46.9%) or ever drinkers (73.4% vs. 57.0%). Furthermore, the differences in tobacco smoke and alcohol consumption between cases and controls were statistically significant (both P <0.001). However, all these variables were further adjusted for any confounding effect in the multivariate logistic regression models later. Overall disease stage distribution of SCCHN patients was 13.8%, 10.3%, 14.4%, and 61.5% for stages I–IV, respectively (Table 1).
Table 1.
Characteristics of squamous cell carcinoma of the head and neck (SCCHN) cases and cancer-free controls in a North American population.
| Variables | Cases (n = 703) | Controls (n = 685) | P a | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Age (years) | 0.745 | ||||
| <57 | 352 | 50.1 | 337 | 49.2 | |
| ≥ 57 | 351 | 49.9 | 348 | 50.8 | |
| Sex | 0.882 | ||||
| Female | 170 | 24.2 | 168 | 24.5 | |
| Male | 533 | 75.8 | 517 | 75.5 | |
| Race | |||||
| Caucasians | 647 | 92.0 | 647 | 94.5 | 0.066 |
| Others | 56 | 8.0 | 38 | 5.5 | |
| Asian | 5 | 0.7 | 1 | 0.1 | |
| Black | 30 | 4.3 | 16 | 2.3 | |
| Hispanic | 19 | 2.7 | 21 | 3.1 | |
| American Indian | 2 | 0.3 | 0 | 0.0 | |
| Smoking statusb | < 0.001 | ||||
| Never | 250 | 35.6 | 363 | 53.2 | |
| Former | 223 | 31.8 | 255 | 37.3 | |
| Current | 229 | 32.6 | 65 | 9.5 | |
| Ever | 452 | 64.4 | 320 | 46.9 | |
| Alcohol useb | <0.0001 | ||||
| Never | 187 | 26.6 | 294 | 43.1 | |
| Former | 181 | 25.8 | 137 | 20.1 | |
| Current | 334 | 47.6 | 252 | 36.9 | |
| Ever | 515 | 73.4 | 389 | 57.0 | |
| Tumor siteb | |||||
| Oropharynx | 367 | 52.3 | |||
| Non-oropharynxc | 335 | 47.7 | |||
| T staged | |||||
| T1 | 201 | 28.7 | |||
| T2 | 260 | 37.0 | |||
| T3 | 123 | 17.5 | |||
| T4 | 118 | 16.8 | |||
| N staged | |||||
| N0 | 232 | 33.0 | |||
| N1 | 77 | 11.0 | |||
| N2 | 381 | 54.3 | |||
| N3 | 12 | 1.7 | |||
| Overall staged | |||||
| I | 97 | 13.8 | |||
| II | 72 | 10.3 | |||
| III | 101 | 14.4 | |||
| IV | 432 | 61.5 | |||
Two-sided χ2 test.
Missed 1 patients’ and 2 controls’ information.
Included oral cavity (n=214), hypopharyngeal (n=20) and larynx (n=101) cancer cases.
3.2. Association between apoptotic capacity and risk of SCCHN
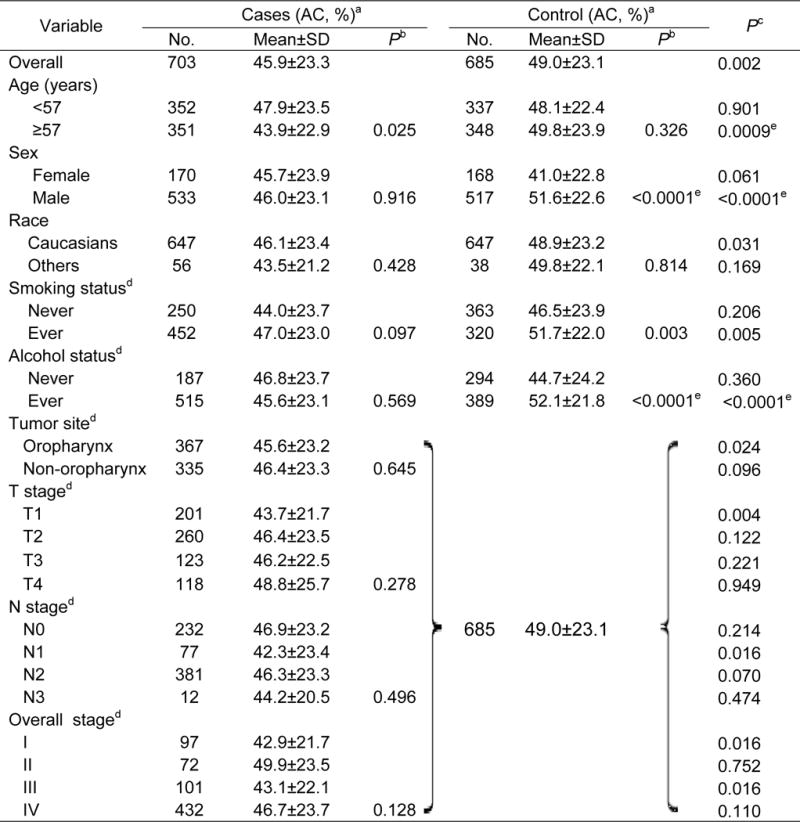
When apoptotic capacity (%) was analyzed as continuous variable, the mean apoptotic capacity in PBLs was significantly lower in the cases than in the controls (45.9+23.3% vs. 49.0+23.1%; P < 0.002; Table 2; Supplementary Figure 1). As shown in Table 2, in the controls, men, ever smokers and drinkers had significantly higher apoptotic capacity than did women, never smokers and drinkers (P < 0.0001, 0.003 and <0.0001, respectively); in the cases, the younger subjects (<57 years) had significantly higher apoptotic capacity than did older subjects (≥57 years, P = 0.025). There were no other significant differences in apoptotic capacity by subgroups of selected variables for both cases and controls. In the case-control analysis, we found that the cases were more likely to have a lower apoptotic capacity than the controls, particularly in the older subjects (≥57 years, P = 0.0009), men (P = <0.0001), Caucasians (P = 0.031), ever smokers (P = 0.005), ever drinkers (P < 0.0001), and OPC (P = 0.024) (Table 2; Supplementary Figure 1). We also controlled for multiple testing with the Bonferroni method, and found the results of older subjects, males and ever drinkers passed the Bonferroni threshold (0.05/38 tests = 0.0013) (Table 2).
Table 2.
Apoptotic capacity (AC, %) between squamous cell carcinoma of the head and neck (SCCHN) patients and healthy control subjects in a North American population.
|
AC= [100 × (experimental apoptosis − spontaneous apoptosis)/(100 − spontaneous apoptosis)].
P values for the differences between subgroups were determined by the Student t test.
P values for the difference between cases and controls.
Missed 1 patients’ and 2 controls’ information.
Tests that could pass the Bonferroni threshold (0.05/38 tests =0.0013) for multiple testing correction.
When apoptotic capacity values were categorized by the median or quartile of the controls, the decreased apoptotic capacity was consistently associated with an increased risk of SCCHN, as assessed in the logistic regression models. As shown in Table 3, when the dichotomized apoptotic capacity was used, we found that individuals with a low apoptotic capacity had a significantly increased risk (i.e., OR with adjustment for age, sex, ethnicity, smoking and drinking status) of SCCHN (adjusted OR = 1.42; 95% confidence interval [CI] = 1.13–1.78, P = 0.002), compared with those with a high apoptotic capacity. Similarly, compared with the highest apoptotic capacity quartile (≥67.9%) of the controls, this increased risk was dose dependent [the second highest quartile (57.3–67.8): adjusted OR, 1.18; 95% CI, 0.85–1.65; the third quartile (31.4%–57.2%): adjusted OR, 1.49; 95% CI, 1.08–2.06; and the fourth quartile (<31.4%): adjusted OR, 1.61; 95% CI, 1.17–2.22, respectively; P = 0.015]. This trend of increasing risk with decreasing apoptotic capacity was statistically significant (Ptrend = 0.002; Table 3, Figure 1).
Table 3.
Logistic regression analysis of categorized apoptosis capacity (AC, %) in patients with SCCHN and cancer-free controls in a North American population.
| Variable | Cases (n = 703) |
Controls (n = 685) |
Crude OR (95% CI) |
Adjusted OR (95% CI)a |
Pa |
|---|---|---|---|---|---|
| By median | |||||
| ≥51.3 | 301 (42.8) | 342 (49.9) | 1.00 | 1.00 | 0.002 |
| <51.3 | 402 (57.2) | 343 (50.1) | 1.33 (1.08–1.65) | 1.42 (1.13–1.78) | |
| By quartile | |||||
| ≥67.9 | 142 (20.2) | 172 (25.1) | 1.00 | 1.00 | 0.015 |
| 57.3–67.8 | 159 (22.6) | 170 (24.8) | 1.13 (0.83–1.55) | 1.18 (0.85–1.65) | |
| 31.4–57.2 | 197 (28.0) | 171 (25.0) | 1.39 (1.03–1.88) | 1.49 (1.08–2.06) | |
| <31.4 | 205 (29.2) | 172 (25.1) | 1.43 (1.06–1.93) | 1.61 (1.17–2.22) | |
| Ptrend | 0.007 | 0.002 |
Obtained from the logistic regression model with adjustment for age, sex, race, smoking and drinking status.
Figure 1.
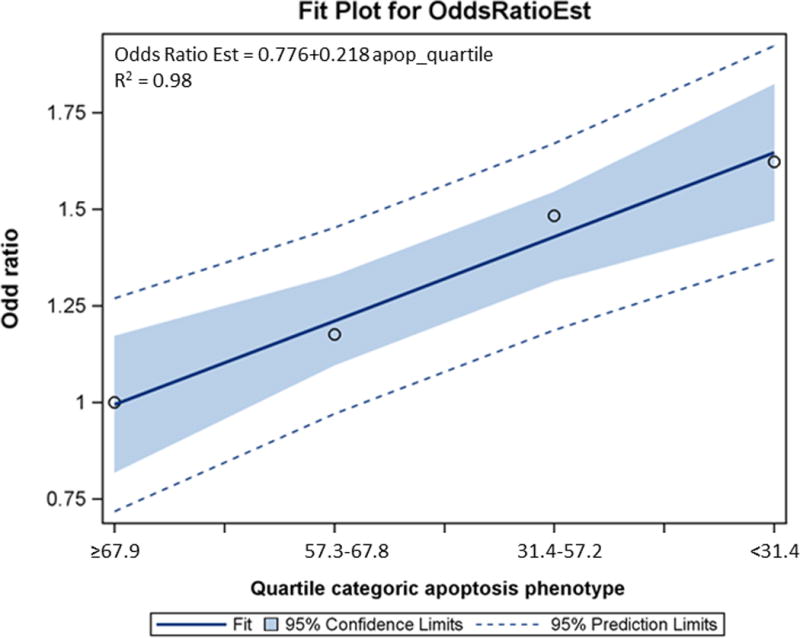
3.3. Stratified analysis of apoptotic capacity by selected variables
We assessed possible interactions on multiplicative scales, between apoptotic capacity and the related variables listed in Table 4. We found a significant interaction of apoptotic capacity with age and sex as well as drinking status (Pinteraction = 0.015, 0.005 and 0.009, respectively) on SCCHN risk, which suggested that older subjects (≥57 years), men and ever drinkers with low apoptotic capacity had the highest risk of SCCHN, as compared with their corresponding baseline group (Figure 2).
Table 4.
Stratified analysis for association between apoptosis capacity (AC, %) and risk of squamous cell carcinoma of the head and neck (SCCHN) in a North American population
| Variable | Cases (n = 703)a |
Controls (n = 685)a |
Crude OR (95% CI) |
Adjusted OR (95% CI)b |
Pc | P interationd |
|---|---|---|---|---|---|---|
| All subjects: | 402/301 | 343/342 | 1.33 (1.08–1.65) | 1.42 (1.13–1.78) | 0.002 | |
| Age | ||||||
| <57 (years) | 186/166 | 171/166 | 1.08 (0.80–1.46) | 1.17 (0.85–1.61) | 0.323 | |
| ≥57 (years) | 216/135 | 172/176 | 1.62 (1.20–2.19) | 1.73 (1.25–2.38) | 0.0009 | 0.015 |
| Sex | ||||||
| Female | 95/75 | 110/58 | 0.65 (0.42–1.00) | 0.66 (0.40–1.06) | 0.087 | |
| Male | 307/226 | 233/284 | 1.65 (1.29–2.11) | 1.76 (1.36–2.27) | <0.0001 | 0.005 |
| Race | ||||||
| Caucasians | 369/278 | 324/323 | 1.32 (1.06–1.65) | 1.41 (1.12–1.78) | 0.004 | |
| Others | 33/23 | 19/19 | 1.24 (0.53–2.90) | 1.98 (0.72–5.47) | 0.187 | 0.697 |
| Smoking status | ||||||
| Never | 153/97 | 195/168 | 1.36 (0.98–1.89) | 1.36 (0.97–1.90) | 0.074 | |
| Ever | 248/204 | 148/172 | 1.41 (1.06–1.88) | 1.48 (1.10–1.99) | 0.009 | 0.566 |
| Alcohol status | ||||||
| Never | 109/78 | 168/126 | 1.05 (0.72–1.52) | 1.02 (0.70–1.51) | 0.905 | |
| Ever | 292/223 | 175/212 | 1.60 (1.23–2.09) | 1.67 (1.27–2.21) | 0.0003 | 0.009 |
| Tumor site | ||||||
| Oropharynx | 215/152 | 343/342 | 1.40 (1.09–1.81) | 1.59 (1.21–2.07) | 0.0008 | |
| Non-oropharynx | 186/149 | 343/342 | 1.23 (0.95–1.60) | 1.26 (0.94–1.68) | 0.121 | |
| T stage | ||||||
| T1 | 120/81 | 343/342 | 1.47 (1.07–2.02) | 1.55 (1.12–2.16) | 0.009 | |
| T2 | 148/112 | 343/342 | 1.31 (0.98–1.75) | 1.46 (1.08–1.97) | 0.014 | |
| T3 | 71/52 | 343/342 | 1.33 (0.90–1.97) | 1.39 (0.91–2.10) | 0.126 | |
| T4 | 62/56 | 343/342 | 1.10 (0.74–1.62) | 1.37 (0.89–2.12) | 0.158 | |
| T1/T2 | 268/193 | 343/342 | 1.38 (1.09–1.75) | 1.47 (1.15–1.89) | 0.002 | |
| T3/T4 | 133/108 | 343/342 | 1.21 (0.90–1.63) | 1.34 (0.97–1.87) | 0.081 | |
| N stage | ||||||
| N0 | 130/102 | 343/342 | 1.26 (0.94–1.71) | 1.32 (0.96–1.82) | 0.092 | |
| N1 | 48/29 | 343/342 | 1.61 (0.99–2.61) | 1.77 (1.07–2.94) | 0.027 | |
| N2 | 217/164 | 343/342 | 1.31 (1.02–1.69) | 1.50 (1.15–1.97) | 0.003 | |
| N3 | 6/6 | 343/342 | 0.99 (0.32–3.10) | 1.19 (0.36–3.91) | 0.774 | |
| N0/N1 | 178/131 | 343/342 | 1.34 (1.02–1.76) | 1.41 (1.05–1.88) | 0.021 | |
| N2/N3 | 223/170 | 343/342 | 1.30 (1.01–1.67) | 1.49 (1.14–1.94) | 0.003 | |
| Overall stage | ||||||
| I | 60/37 | 343/342 | 1.61 (1.04–2.49) | 1.60 (1.02–2.50) | 0.041 | |
| II | 35/37 | 343/342 | 0.94 (0.58–1.52) | 1.01 (0.61–1.69) | 0.956 | |
| III | 65/36 | 343/342 | 1.76 (1.14–2.72) | 1.89 (1.20–2.99) | 0.006 | |
| IV | 241/191 | 343/342 | 1.25 (0.98–1.59) | 1.47 (1.13–1.90) | 0.004 | |
| I/II | 95/74 | 343/342 | 1.27 (0.91–1.79) | 1.28 (0.90–1.83) | 0.167 | |
| III/IV | 306/227 | 343/342 | 1.33 (1.06–1.67) | 1.51 (1.18–1.93) | 0.001 |
Low AC group (<51.3)/high AC group (≥51.3) using the median apoptotic capacity in controls as the cutoff point value.
Adjusted for age, sex and smoking status in logistic regression models when appropriate.
Obtained from the logistic regression model with adjustment for age, sex, race, smoking and alcohol status.
Interaction P values for quartile apoptotic capacity using logistic regression model with adjustment for age, sex, race, smoking and alcohol status.
Figure 2.
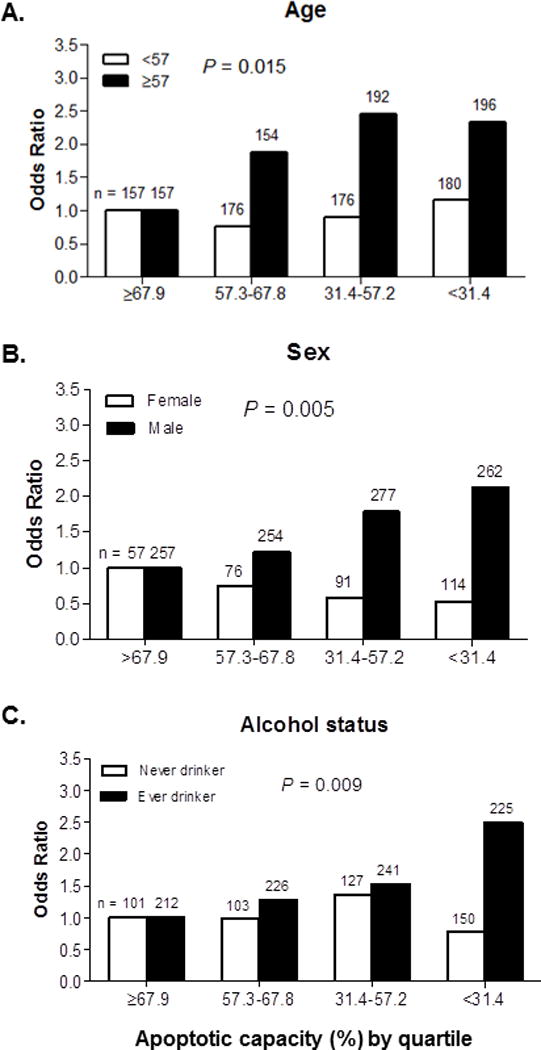
We further performed stratified analyses to evaluate the effects of apoptotic capacity on the risk of SCCHN in subgroups of age, sex, ethnicity, smoking status, alcohol consumption, tumor sites and tumor stages using the median value in controls for calculating ORs. As shown in Table 4, the stratified analysis showed that the inverse association of apoptotic capacity with SCCHN risk remained significant among subjects with the following characteristics: cases were more likely than the controls to have reduced apoptotic capacity: 1.73 times for the older subjects (adjusted OR = 1.73; 95% CI = 1.25–2.38, P = 0.0009); 1.76 times for male (adjusted OR = 1.76; 95% CI = 1.36–2.27, P < 0.0001); 1.48 times for ever smokers (adjusted OR = 1.48; 95% CI = 1.10–1.99, P = 0.009); 1.67 times for ever drinkers (adjusted OR = 1.67; 95% CI = 1.27–2.21, P = 0.0003); 1.59 times for OPC (adjusted OR = 1.59; 95% CI = 1.21–2.07, P = 0.0008); 1.47 times for T1/T2 stages (adjusted OR = 1.47; 95% CI = 1.15–1.89, P = 0.002); 1.49 times for N2/N3 stages (adjusted OR = 1.49; 95% CI = 1.14–1.94, P = 0.003); and 1.51 times for stages III/IV (adjusted OR = 1.51; 95% CI = 1.18–1.93, P = 0.001).
3.4. Prediction of the SCCHN risk by apoptotic capacity
To evaluate the performance of apoptotic capacity in SCCHN risk prediction models, we calculated the AUC using multivariate logistic regression and ROC. As shown in Figure 3, the model included age, sex, race, smoking and drinking status as classifiers, it had an AUC of 63.2%; with the addition of apoptotic capacity, the AUC was significantly improved to 64.4% (P = 0.029, by the DeLong’s test). With age, sex, ethnicity, smoking and drinking status as classifiers, the model specific for OPC had an AUC of 63.9%; with the addition of apoptotic capacity, the AUC was significantly improved to 66.0% (P = 0.010, by the DeLong’s test).
Figure 3.
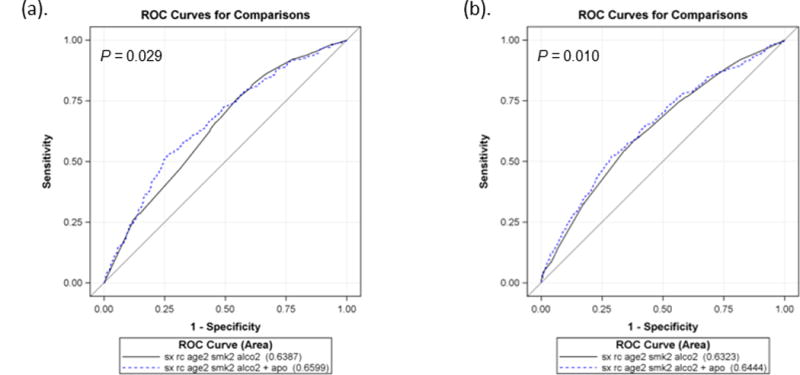
4. Discussion
In this hospital-based case-control study, we observed that SCCHN patients had a significantly lower CPT-induced apoptotic capacity in their PBLs than that of the controls. When using the median level of apoptotic capacity in the controls as a cutoff, individuals with a lower apoptotic capacity had a 1.42 times greater risk for having SCCHN than those with a higher apoptotic capacity, which suggests that reduced apoptotic capacity may play a role in the etiology of SCCHN. Stratified analysis further showed that this association was more evident in subgroups of older subjects (≥57 years), men, ever smokers, ever drinkers, OPC, tumor stages T1/T2, N2/N3, and overall stages III/IV. These findings are consistent with the notion that apoptosis capacity may be involved in cancer risk associated with exposure to tobacco smoke and alcohol drink, because old subjects, men and ever smokers and drinkers are more likely to have a higher level of exposure to tobacco smoke and alcoholic beverage [5, 38]. Our additional analyses of the AUC further suggested that apoptotic capacity could significantly improve the prediction of SCCHN risk.
Apoptosis plays a crucial role in many biological and cellular processes, while dysfunction of apoptosis may lead to oncogenesis and development of cancer [18, 39]; thus, evasion of apoptosis is considered one of the hallmarks of human cancers [40]. In fact, pro-apoptotic genes might act as tumor suppressors, whereas oncogenes might fulfill anti-apoptotic functions [40]. Apoptosis defects are now considered an important complement of proto-oncogene activation [41], and establishing targeted pro-apoptotic therapeutic protocols and developing apoptosis-inducing drugs have been under way since the 1990s, which have provided novel approaches to the successful treatment of cancers [42].
Apoptotic capacity has been suggested as a biomarker for risk of several types of cancer, including SCCHN, but results from epidemiologic studies have been inconclusive [19, 21, 43–45]. For example, Zhao et al. found that among 30 lung cancer patients and 22 controls, apoptosis levels were significantly lower in the cases than in controls [43]. In a pilot case-control study of 29 patients with neoplasms of the salivary and thyroid glands and 29 cancer-free control subjects, we observed that the low apoptotic capacity was associated with a 10 or four-fold increased risk of salivary and thyroid carcinoma [46]. In a study of 68 lung cancer patients and 74 cancer-free controls, we measured the apoptotic capacity in their cultured PBLs in response to in vitro exposure to an ultimate tobacco carcinogen, benzo[a]pyrene diol epoxide (BPDE), by using the terminal dUTP nucleotide end labeling (TUNEL) and flow cytometry, we observed a significantly lower apoptotic capacity in the cases than in the controls [19]. Similarly, in a 243 female breast cancer patients and 75 cancer-free controls after their PBLs were irradiated or mock-treated, breast cancer patients shown significantly reduced apoptotic response, compared with normal controls [44]. While in a study of 211 breast cancer patients and 170 cancer-free controls, Docherty et al. investigated the apoptotic response to ionizing radiation in PBLs, but the authors failed to reveal any difference in DNA damage-induced apoptosis between the cases and the controls [47].
In the present study that included 703 SCCHN cases and 685 matched cancer-free controls, we found an association between apoptotic capacity and risk of SCCHN, a finding consistent with previous studies that found that a low apoptotic capacity in PBLs was associated with an increased risk of several types of cancers, such as lung cancer [19, 43], breast cancer [44], and salivary and thyroid carcinoma [46]. Our finding also supports a previous study in which the Bcl-2 protein, an inhibitor of apoptosis, played a role in the regulation of T lymphocyte apoptosis [48]. In one relatively small study of 23 laryngeal carcinoma cases and 20 healthy controls in a Poland population, the expression of the Bcl-2 protein in T lymphocytes in CD4+ and CD8+ T lymphocytes measured by flow cytometry was significantly higher in laryngeal carcinoma patients than in the controls [48]. More recently, in another study of 43 SCCHN cases’ tumor tissues and 10 normal mucosa samples in a Chinese population, a higher expression of survivin (also called BIRC5, an inhibitor of apoptosis-related protein) in the nuclei of tumor cells than in those of normal mucosa cells [49], and one study identified that survivin inhibited apoptosis through inhibiting both Bax and Fas-induced apoptotic pathways [50]. In contrast with our observations, another study assessed the apoptosis levels using the TUNEL assay by flow cytometry in peripheral blood mononuclear cells from subjects of 22 head and neck cancer patients after treatment by surgery, or surgery and radiation or surgery and chemoradiotherapy and 16 healthy controls and found that cancer patients showed higher apoptotic response than healthy subjects at the time of blood draw (0 time) and after 24 hours incubation [51]. Such a small study is likely to generate unstable estimates.
In the stratified analysis, we also observed that OPC patients had a significantly lower apoptotic capacity in PBLs than that of the controls. It is well known that HPV infection has been implicated as the most important risk for the development of OPC. Among the known HPV types, high-risk HPV-16 is the most common, accounting for approximately 90% or more of HPV-positive OPC cases [52]. There is an increasing molecular evidence that HPV causes human cancers by expressing E6 oncogenic protein, which not only binds to p53 and inhibits its activity, resulting in reduced protein function, but also inhibits p53-independent apoptotic pathways, promoted by different stimuli [53–55]. It has been reported that HPV-16 E6 inhibited drug-induced apoptosis in cells lacking p53 [56]. In transgenic mice expressing HPV-16 E6 in the ocular lens, it has been identified that HPV E6 prevented apoptosis in both wild-type and p53-null animals [57]. Our finding in the present study provided additional epidemiologic evidence that low apoptotic capacity is associated with an increased risk of HPV-related OPC.
To the best of our knowledge, the present study is the first larger population study to epidemiologically show the association of reduced apoptotic capacity with an increased risk of SCCHN. However, the present study also had several limitations. First, this is a hospital-based case-control study of the CPT-induced apoptotic capacity assay with some inherited selection bias, although we had applied a stringent frequency-matching strategy in recruiting cases and controls to control for potentially confounding variables. Second, no information of genetic susceptibility was involved in the present study, although such as apoptotic gene mutations and single nucleotide polymorphisms (SNPs) may also influence apoptotic capacity in individuals [19, 58], but the phenotypic measurement of apoptosis may have reflected the sum of all possible genetic variants. Third, although reduced apoptotic capacity has been found to be associated with increased SCCHN risk, we cannot exclude the influence of other potential confounder (e.g., HPV infection status) that was not controlled in the present study, and the retrospective nature of the present study may not exclude the possibility that the disease status may have some effect on the apoptotic capacity. Fourth, the apoptotic capacity was measured in PBLs that were used as a surrogate for head and neck epithelial cells; however, it has been reported that lymphocytes are a good surrogate for estimating cancer risk associated with apoptosis phenotypes as markers for genetic susceptibility [19, 41]. Finally, variations in apoptotic capacity may be due to difference in the interval between blood sample collection and the cell-based experiments, because no repeated blood samples were collected in the study design.
In conclusion, the present study offers the first evidence that reduced CPT-induced apoptotic capacity in PBLs is associated with an increased risk of SCCHN. If our results are validated in large, prospective cohort studies, this apoptotic phenotype could be used as a biomarker for identifying individuals at risk of SCCHN, who might benefit from targeted cancer prevention programs.
Supplementary Material
Acknowledgments
We thank Margaret Lung and Jessica Fiske for their assistance in recruiting the subjects and gathering the questionnaire information, and Qiming Wang and Jianzhong He for their laboratory assistance.
Funding
National Institutes of Health grants R01 ES011740 and R01 CA131274 (to Q. W.), P50 CA097007 (Scott Lippman), and CA 16672 (to M.D. Anderson Cancer Center), and the start-up funds (to Q. Wei) from Duke Cancer Institute. QW was also supported from the Duke University Medical Center and the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (NIH CA014236). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AC
apoptotic capacity
- CPT
camptothecin
- PBLs
peripheral blood lymphocytes
- SCCHN
squamous cell carcinoma of the head and neck
- OR
odds ratio
- CI
confidence interval
Footnotes
Conflict of interest statement
None declared.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.De Petrini M, Ritta M, Schena M, Chiusa L, Campisi P, Giordano C, et al. Head and neck squamous cell carcinoma: role of the human papillomavirus in tumour progression. New Microbiol. 2006;29(1):25–33. [PubMed] [Google Scholar]
- 4.Hashibe M, Brennan P, Chuang SC, Boccia S, Castellsague X, Chen C, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2009;18(2):541–50. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee YC, Boffetta P, Sturgis EM, Wei Q, Zhang ZF, Muscat J, et al. Involuntary smoking and head and neck cancer risk: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2008;17(8):1974–81. doi: 10.1158/1055-9965.EPI-08-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21(48):7435–51. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 7.Lachenmeier DW, Przybylski MC, Rehm J. Comparative risk assessment of carcinogens in alcoholic beverages using the margin of exposure approach. Int J Cancer. 2012;131(6):E995–1003. doi: 10.1002/ijc.27553. [DOI] [PubMed] [Google Scholar]
- 8.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 9.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 10.Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13(24):7254–63. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- 11.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22(53):8543–67. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 12.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5(11):876–85. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 13.Zeiss CJ. The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol. 2003;40(5):481–95. doi: 10.1354/vp.40-5-481. [DOI] [PubMed] [Google Scholar]
- 14.Andersen MH, Becker JC, Straten P. Regulators of apoptosis: suitable targets for immune therapy of cancer. Nat Rev Drug Discov. 2005;4(5):399–409. doi: 10.1038/nrd1717. [DOI] [PubMed] [Google Scholar]
- 15.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284(33):21777–81. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta. 2011;1813(1):238–59. doi: 10.1016/j.bbamcr.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1):92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang LE, Cheng L, Spitz MR, Wei Q. Fas A670G polymorphism, apoptotic capacity in lymphocyte cultures, and risk of lung cancer. Lung Cancer. 2003;42(1):1–8. doi: 10.1016/s0169-5002(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 20.Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845. doi: 10.1155/2014/150845. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Barber JB, West CM, Kiltie AE, Roberts SA, Scott D. Detection of individual differences in radiation-induced apoptosis of peripheral blood lymphocytes in normal individuals, ataxia telangiectasia homozygotes and heterozygotes, and breast cancer patients after radiotherapy. Radiat Res. 2000;153(5 Pt 1):570–8. doi: 10.1667/0033-7587(2000)153[0570:doidir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 22.Han Z, Wei W, Dunaway S, Darnowski JW, Calabresi P, Sedivy J, et al. Role of p21 in apoptosis and senescence of human colon cancer cells treated with camptothecin. J Biol Chem. 2002;277(19):17154–60. doi: 10.1074/jbc.M112401200. [DOI] [PubMed] [Google Scholar]
- 23.Silva MF, Khokhar AR, Qureshi MZ, Farooqi AA. Ionizing radiations induce apoptosis in TRAIL resistant cancer cells: in vivo and in vitro analysis. Asian Pac J Cancer Prev. 2014;15(5):1905–7. doi: 10.7314/apjcp.2014.15.5.1905. [DOI] [PubMed] [Google Scholar]
- 24.Hsieh WC, Hsu PC, Liao YF, Young ST, Wang ZW, Lin CL, et al. Overexpression of ornithine decarboxylase suppresses thapsigargin-induced apoptosis. Mol Cells. 2010;30(4):311–8. doi: 10.1007/s10059-010-0120-1. [DOI] [PubMed] [Google Scholar]
- 25.Tan C, Cai LQ, Wu W, Qiao Y, Imperato-McGinley J, Chen GQ, et al. NSC606985, a novel camptothecin analog, induces apoptosis and growth arrest in prostate tumor cells. Cancer Chemother Pharmacol. 2009;63(2):303–12. doi: 10.1007/s00280-008-0740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Z, Li C, Chen K, Wang LE, Sturgis EM, Spitz MR, et al. Single Nucleotide Polymorphisms in Selected Apoptotic Genes and BPDE-Induced Apoptotic Capacity in Apparently Normal Primary Lymphocytes: A Genotype-Phenotype Correlation Analysis. J Cancer Epidemiol. 2008;2008:147905. doi: 10.1155/2008/147905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng CW, Zhang XJ, Lin KY, Ye H, Feng SY, Zhang H, et al. Camptothecin induces apoptosis in cancer cells via microRNA-125b-mediated mitochondrial pathways. Mol Pharmacol. 2012;81(4):578–86. doi: 10.1124/mol.111.076794. [DOI] [PubMed] [Google Scholar]
- 28.Paquet C, Schmitt E, Beauchemin M, Bertrand R. Activation of multidomain and BH3-only pro-apoptotic Bcl-2 family members in p53-defective cells. Apoptosis. 2004;9(6):815–31. doi: 10.1023/B:APPT.0000045791.55282.91. [DOI] [PubMed] [Google Scholar]
- 29.Furuta T, Hayward RL, Meng LH, Takemura H, Aune GJ, Bonner WM, et al. p21CDKN1A allows the repair of replication-mediated DNA double-strand breaks induced by topoisomerase I and is inactivated by the checkpoint kinase inhibitor 7-hydroxystaurosporine. Oncogene. 2006;25(20):2839–49. doi: 10.1038/sj.onc.1209313. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert DC, Chalmers AJ, El-Khamisy SF. Topoisomerase I inhibition in colorectal cancer: biomarkers and therapeutic targets. Br J Cancer. 2012;106(1):18–24. doi: 10.1038/bjc.2011.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 2001;61(15):5926–32. [PubMed] [Google Scholar]
- 32.Jayasooriya RG, Choi YH, Hyun JW, Kim GY. Camptothecin sensitizes human hepatoma Hep3B cells to TRAIL-mediated apoptosis via ROS-dependent death receptor 5 upregulation with the involvement of MAPKs. Environ Toxicol Pharmacol. 2014;38(3):959–67. doi: 10.1016/j.etap.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Li C, Lu J, Liu Z, Wang LE, Zhao H, El-Naggar AK, et al. The six-nucleotide deletion/insertion variant in the CASP8 promoter region is inversely associated with risk of squamous cell carcinoma of the head and neck. Cancer Prev Res (Phila) 2010;3(2):246–53. doi: 10.1158/1940-6207.CAPR-08-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fanzo JC, Hu CM, Jang SY, Pernis AB. Regulation of lymphocyte apoptosis by interferon regulatory factor 4 (IRF-4) J Exp Med. 2003;197(3):303–14. doi: 10.1084/jem.20020717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Zhao H, Hu Z, Liu Z, Wang LE, Gershenwald JE, et al. Genetic variants and haplotypes of the caspase-8 and caspase-10 genes contribute to susceptibility to cutaneous melanoma. Hum Mutat. 2008;29(12):1443–51. doi: 10.1002/humu.20803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang LE, Hu Z, Sturgis EM, Spitz MR, Strom SS, Amos CI, et al. Reduced DNA repair capacity for removing tobacco carcinogen-induced DNA adducts contributes to risk of head and neck cancer but not tumor characteristics. Clin Cancer Res. 2010;16(2):764–74. doi: 10.1158/1078-0432.CCR-09-2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–45. [PubMed] [Google Scholar]
- 38.Purdue MP, Hashibe M, Berthiller J, La Vecchia C, Dal Maso L, Herrero R, et al. Type of alcoholic beverage and risk of head and neck cancer–a pooled analysis within the INHANCE Consortium. Am J Epidemiol. 2009;169(2):132–42. doi: 10.1093/aje/kwn306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9(7):501–7. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- 40.Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;18(9):1441–9. doi: 10.1038/cdd.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1(1):19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 42.Jia LT, Chen SY, Yang AG. Cancer gene therapy targeting cellular apoptosis machinery. Cancer Treat Rev. 2012;38(7):868–76. doi: 10.1016/j.ctrv.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 43.Zhao H, Spitz MR, Tomlinson GE, Zhang H, Minna JD, Wu X. Gamma-radiation-induced G2 delay, apoptosis, and p53 response as potential susceptibility markers for lung cancer. Cancer Res. 2001;61(21):7819–24. [PubMed] [Google Scholar]
- 44.Camplejohn RS, Gilchrist R, Easton D, McKenzie-Edwards E, Barnes DM, Eccles DM, et al. Apoptosis, ageing and cancer susceptibility. Br J Cancer. 2003;88(4):487–90. doi: 10.1038/sj.bjc.6600767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imyanitov EN. Gene polymorphisms, apoptotic capacity and cancer risk. Hum Genet. 2009;125(3):239–46. doi: 10.1007/s00439-009-0636-7. [DOI] [PubMed] [Google Scholar]
- 46.Zheng R, Dahlstrom KR, Wei Q, Sturgis EM. Gamma radiation-induced apoptosis, G2 delay, and the risk of salivary and thyroid carcinomas–a preliminary report. Head Neck. 2004;26(7):612–8. doi: 10.1002/hed.20053. [DOI] [PubMed] [Google Scholar]
- 47.Docherty Z, Georgiou A, Langman C, Kesterton I, Rose S, Camplejohn R, et al. Is chromosome radiosensitivity and apoptotic response to irradiation correlated with cancer susceptibility? Int J Radiat Biol. 2007;83(1):1–12. doi: 10.1080/09553000600932968. [DOI] [PubMed] [Google Scholar]
- 48.Klatka J, Rolinski J, Kupisz K, Klonowski S, Skomra D. Expression of bcl-2 protein in lymphocytes of patients with laryngeal carcinoma. Eur Arch Otorhinolaryngol. 1999;256(6):299–302. doi: 10.1007/s004050050250. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Zhang W, Wang YF, Liu B, Zhang WF, Zhao YF, et al. Dual induction of apoptotic and autophagic cell death by targeting survivin in head neck squamous cell carcinoma. Cell Death Dis. 2015;6:e1771. doi: 10.1038/cddis.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58(23):5315–20. [PubMed] [Google Scholar]
- 51.Saito T, Kuss I, Dworacki G, Gooding W, Johnson JT, Whiteside TL. Spontaneous ex vivo apoptosis of peripheral blood mononuclear cells in patients with head and neck cancer. Clin Cancer Res. 1999;5(6):1263–73. [PubMed] [Google Scholar]
- 52.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89(2):213–28. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 54.Chung CH, Gillison ML. Human papillomavirus in head and neck cancer: its role in pathogenesis and clinical implications. Clin Cancer Res. 2009;15(22):6758–62. doi: 10.1158/1078-0432.CCR-09-0784. [DOI] [PubMed] [Google Scholar]
- 55.Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20(54):7874–87. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- 56.Steller MA, Zou Z, Schiller JT, Baserga R. Transformation by human papillomavirus 16 E6 and E7: role of the insulin-like growth factor 1 receptor. Cancer Res. 1996;56(21):5087–91. [PubMed] [Google Scholar]
- 57.Pan H, Griep AE. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995;9(17):2157–69. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 58.Lacko M, Braakhuis BJ, Sturgis EM, Boedeker CC, Suarez C, Rinaldo A, et al. Genetic susceptibility to head and neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2014;89(1):38–48. doi: 10.1016/j.ijrobp.2013.09.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.