

Abstract
Genetically-encoded p-azido-phenylalanine (azF) residues in G protein-coupled receptors (GPCRs) can be targeted with dibenzocyclooctyne (DIBO) modified fluorescent probes using strain-promoted [3+2] azide–alkyne cycloaddition (SpAAC). Here we show that azF residues situated on the transmembrane surface of detergent-solubilized receptors exhibit up to 1000-fold rate enhancement compared with azF residues on water-exposed surfaces. We show that the amphipathic moment of the labeling reagent, consisting of hydrophobic DIBO coupled to hydrophilic Alexa dye, results in strong partitioning of the DIBO group into the hydrocarbon core of the detergent micelle and consequently high local reactant concentrations. The observed rate constant for the micelle-enhanced SpAAC is comparable with the fastest bioorthogonal labeling reactions known. Targeting hydrophobic regions of membrane proteins using the micelle-enhanced SpAAC reaction should expand the utility of bioorthogonal labeling strategies.
Keywords: bioorthogonal labeling, detergent micelle, partition, non-canonical amino acids, rhodopsin
TOC image
Rhodopsin, a G protein-coupled receptor, was tagged with an azido group using non-canonical amino-acid mutagenesis. A fluorophore was site-specifically attached to the fourth transmembrane helix using strain-promoted azide-alkyne cycloaddition, without affecting the ligand binding behavior. We observed greatly enhanced reaction kinetics for the cycloaddition reaction with sites in the transmembrane region exposed to the hydrocarbon core of the micellar environment. Partitioning of amphipathic dye into the micelle results in micelle-enhanced cycloaddition reaction.
INTRODUCTION
The superfamily of seven-transmembrane (TM) G protein-coupled receptors (GPCRs) mediates diverse physiological actions by transmitting extracellular signals across the membrane.[1] In order to understand the conformational states of GPCRs, a number of methods have been developed to attach biophysical probes to receptors.[2] However, methods like fluorescent protein fusion,[3] short peptide/probe pair-based labeling,[4] and SNAP/CLIP labeling[5] generally allow label attachment only to the N-terminus, C-terminus, and the loops, which have the structural flexibility to accommodate a fusion protein or a recognition sequence.[6] Labeling chemistries involving thiols are, in principle, more general and this strategy has yielded important insights into the structure-function relationship of GPCRs. For instance, site-directed spin labeling of the TM helices of the visual photoreceptor rhodopsin (Rho) involving cysteine/methanethiosulfonate chemistry has enabled high-resolution distance mapping of the helix movement of Rho.[7] Nonetheless, site-directed cysteine labeling requires engineering a cysteine-free background in the receptor, which may not be applicable to receptors where cysteines are structurally or functionally critical.
To overcome these limiting factors, we have developed a two-step approach to site-specifically attach a probe to a GPCR. We first incorporate an unnatural amino acid (uaa) carrying either a keto or an azido reactive handle[8] into a chosen site through amber codon suppression,[9] then conjugate a probe to the reactive handle through a suitable bioorthogonal chemistry. After testing different choices of uaas and bioorthogonal chemistries, we concluded that the strain-promoted [3+2] azide–alkyne cycloaddition (SpAAC) of p-azido-L-phenylalanine (azF) and dibenzocyclooctyne (DIBO) reagents is a suitable strategy to label GPCRs (Scheme 1).[10] We showed that Rho can be fluorescently labeled using this approach. We demonstrated that S144azF in the second intracellular (IC) loop and V173azF at the extracellular (EC) end of the fourth TM helix could be stoichiometrically labeled. We also found that Y102azF, a solvent-accessible site based on the crystal structure, could not be efficiently modified using the dibenzocyclooctyne reagent.[10c]
Scheme 1.
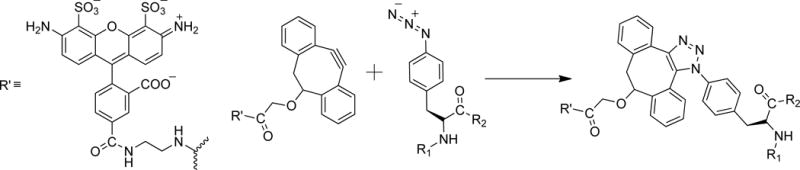
The SpAAC reaction between Alexa488-DIBO and azF.
We observed that the reaction rates of SpAAC between the ~40-kDa biomolecule S144azF-Rho, and the Alexa488-derivatized dibenzocyclooctyne (Alexa488-DIBO) was 2~3 orders of magnitude higher than the reaction rates reported for the SpAAC between two small molecules.[11] Moreover, we found that residues in a hydrophobic pocket of another GPCR (CCR5) are more readily labeled as compared to other water exposed surface residues.[10d] Based on these observations, we postulate that the reactivity of the DIBO reagent is modulated by the local environment on the protein surface, and that the hydrophobicity of the local environment contributes to the observed reactivity enhancement. We further reason that if this hypothesis is true, then the TM region of GPCRs should be amenable to covalent modification by SpAAC.
RESULTS
Fluorescent labeling of the TM region of azF-Rho variants
The structural hallmark of GPCRs is their seven TM helix bundle, which is invariably involved in ligand-dependent activation. Based on biochemical and structural studies of Class A GPCRs, TM4 is positioned distal to the main helix bundle and it is generally not involved in the helix rotation required for GPCR activation.[2a, 12] Compared with all other TMs, the residues of TM4 are least likely to contribute to the ligand binding pocket.[13] Thus, we contend that TM4 is a satisfactory region to test our hypothesis, since modification of residues in TM4 is expected to have minimal effects on the intrinsic functionality of the receptor (Figure 1).
Figure 1.
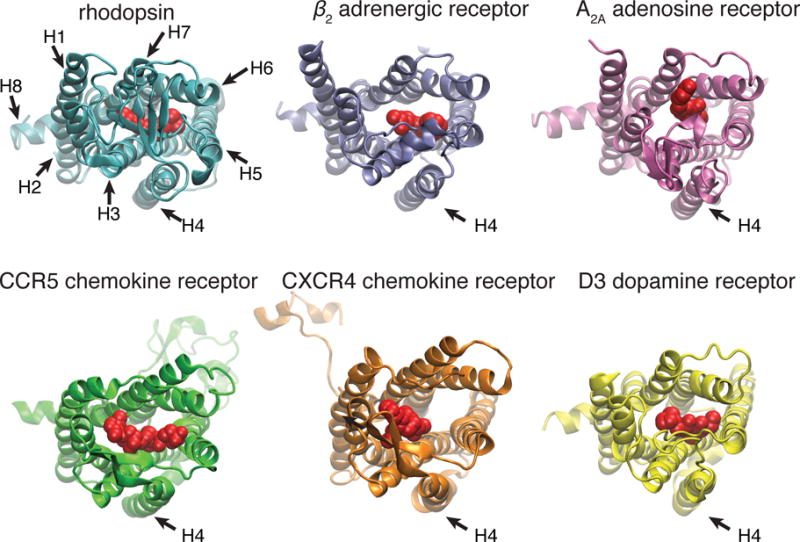
Comparison of the crystal structures of different Class A GPCRs with bound ligands: 1) rhodopsin and 11-cis-retinal (protein data base access code 1GZM);[46] 2) β2 adrenergic receptor and carazolol (2RH1);[47] 3)A2A adenosine receptor and ZM241385 (3EML);[48] 4) CCR5 chemokine receptor and maraviroc (4MBS);[49] 5) CXCR4 chemokine receptor and IT1t (3ODU);[50] 6) D3 dopamine receptor (3PBL),[51] viewed from the EC side, with the ligands shown in red. The TM4 of these receptors (indicated by an arrow) are positioned outside the helix bundle and not in contact with the ligands.
We generated amber stop codon at every turn of TM4 (N151, M155, A158, V162, L165, A169, V173). These residues were chosen because their side chains point outward from the helix bundle (Figure 2). We avoided mutating the residues involved in the conserved non-covalent contact between TM3 and TM4 (in the case of Rho they correspond to W161, A164, A168).[13] Except for L165amb (not shown), all the constructs yielded expression of full-length receptor containing azF (Figure 3A). The azF-Rho variants were solubilized in n-dodecyl-β-D-maltoside (DM) micelles, captured to an immunoaffinity matrix (1D4-sepharose) via its C-terminal epitope, and reacted with Alexa488-DIBO reagent using the labeling protocol described previously.[10a, 10c]
Figure 2.
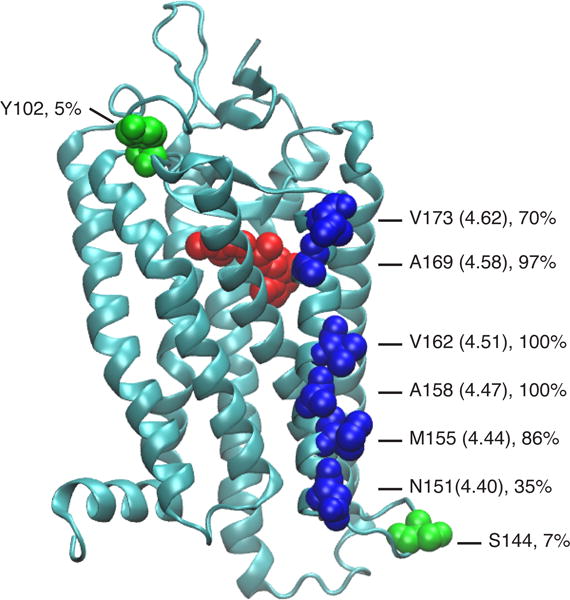
Structure of bovine Rho (PDB 1GZM) with 11-cis-retinal (red) and the tested amino acid residues (blue: TM4 sites; green: sites reported earlier[10c]). For the TM4 sites, the Ballesteros-Weinstein notation is shown in parentheses.[52] The hydrophobicity of the local environment at these sites is estimated by the percent of lipid contacts versus water contacts. The values give the fraction of the surface exposure of these residues to lipids as observed in molecular dynamics simulations of Rho in a phospholipid bilayer membrane.[15]
Figure 3.
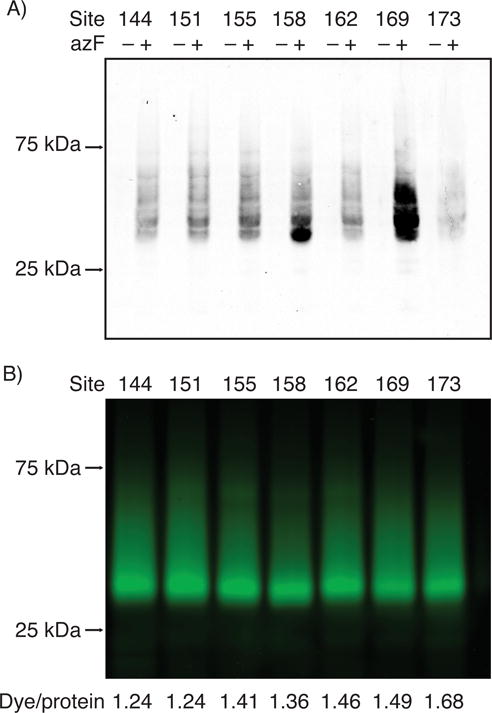
Bioorthogonal labeling of the TM4 sites of Rho. A) The heterologous expression of Rho with single azF substitution at the indicated positions. HEK293F cells were transfected with three plasmids encoding the suppressor tRNA, the amino-acyl tRNA synthetase, and Rho with an amber mutation. The cell lysates were analyzed by Western blot (100 μg total protein per lane). The receptor was probed with 1D4 mAb specific to the C-terminus of Rho, followed by goat-anti-mouse 800 CW (LI-COR). The full-length receptor was detected only when the expression medium was supplemented with azF, indicating specific incorporation of azF into Rho. B) Fluorescent labeling of azF-Rho by Alexa488-DIBO. The cells expressing azF-Rho were regenerated with 11-cis-retinal, solubilized in 1% DM, immunoprecipitated by 1D4 mAb-sepharose resin, and reacted with Alexa488-DIBO (a final concentration of 50 μM in the resin/buffer mixture, 25 °C, for 18 hrs). The purified receptor was analyzed by in-gel fluorescence (50 ng purified receptor per lane) and UV–Vis spectroscopy (Supplementary Figure S1). The labeling stoichiomeries of the azF-Rho variants calculated as dye-to-protein ratios are indicated below.
In support of our hypothesis, we observed robust fluorescent labeling for all the tested azF-Rho variants (Figure 3B and Supplementary Figure S1). This is noteworthy because the hydrophobic alkyl chain of DM might be expected to shield the TM region of Rho in DM micelles. The labeling stoichiometries were determined from the absorbance of the fluorophore Alexa488 and 11-cis-retinal the chromophore utilized by Rho as its native ligand (Supplementary Figure S1). The resulting dye/protein ratios ranged from 1.21 to 1.68.
Accelerated SpAAC in the TM region
We next quantitatively studied how the local environment on the TM surface contributes to the reactivity of SpAAC. We measured the reaction rates of SpAAC at two sites, M155azF and V162azF, both of which are located in the middle of TM4 (Figure 4 and Supplementary Figure S2). The time-courses of the reactions were monitored by in-gel fluorescence of the reaction products at different time points, and the amount of protein loading was quantified by silver staining. The second-order rate constants for the coupling reactions (k2) for M155azF-Rho and V162azF-Rho were (3.2 ± 0.4)×102 M−1 s−1 and (1.9 ± 0.5) ×102 M−1 s−1, respectively (Figure 4A). Previously, we have found that reaction of Y102azF-Rho with 50 μM Alexa488-DIBO for 18 h yielded a labeling stoichiometry of 0.58,[10c] which corresponds to a second order rate constant of 0.27 M−1 s−1 that is similar to the value 0.17 M−1 s−1 observed for the reaction of benzyl azide and DIBO in methanol.[11, 14] The crystal structure of Rho shows that Y102 is a solvent-accessible site with the side chain pointing towards the EC space. Molecular dynamics simulations of Rho in a phospholipid bilayer membrane show that Y102 is water exposed with only minimal (5%) contacts to lipids, whereas M155 and V162 are predominantly lipid exposed (cf. Figure 2).[15] Thus, a 103-fold enhancement of SpAAC kinetics was observed for azF residues located in the TM region as compared with the polar region, demonstrating that the local environment near the TM surface greatly accelerates the cycloaddition reaction.
Figure 4.
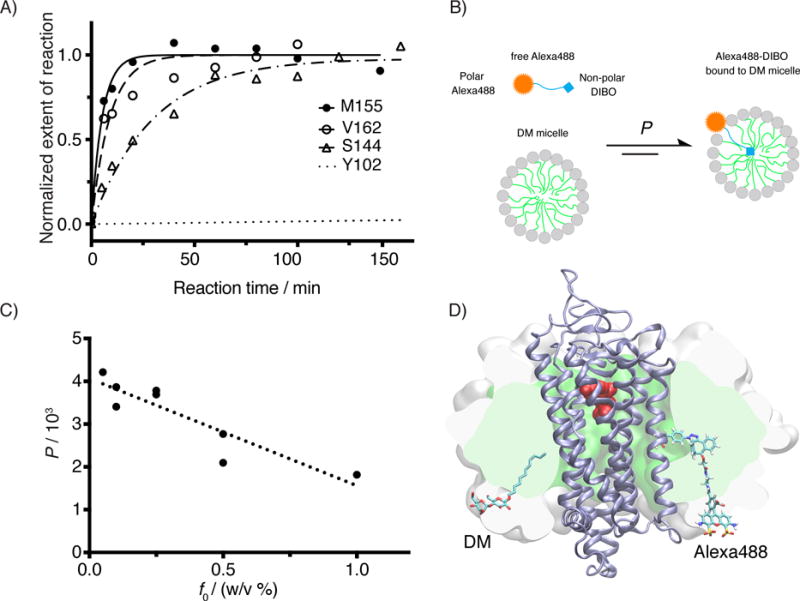
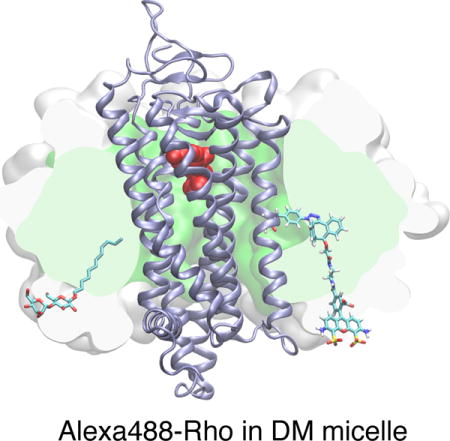
Kinetic study for the SpAAC reaction between azF-Rho and Alexa488-DIBO. A) Reaction time course of four azF-Rho variant with Alexa488-DIBO (10 μM): M155 and V162 (TM4), S144 (IC2) and Y102 (EC2). The data points were fitted using pseudo first-order kinetic model. The calculated k2 is (3.2 ± 0.4)×102 M−1 s−1 for M155azF-Rho, and (1.9 ± 0.5) ×102 M−1 s−1 for V162azF-Rho. The kinetic study for S144azF-Rho (k2 = 62 M−1 s−1) has been described earlier[10c] and included in the plot for comparison. The curve for Y102azF-Rho was simulated based on the k2 (= 0.27 M−1 s−1) estimated from single time point labeling stoichiometry data.[10c] B) Partition equilibrium of Alexa488-DIBO between water and DM micelle. C) Partition coefficients (water to micelle) from ultrafiltration experiment for various concentrations of DM. The observed partition coefficients (P) were plotted as against the mass concentration of DM (f0). We found empirically a linear correlation between P and f0 (R2 = 0.81). D) Atomic model of Rho (PDB:1GZM) embedded in a micelle (150 DM molecules). The SpAAC conjugated V162azF-DIBO-Alexa488 is shown as sticks including hydrogens. For size comparison, one of the 150 DM molecules is shown in sticks but without hydrogens. The protein backbone is shown as a cartoon (ice blue). 11-cis-retinal is shown in spheres (red). The hydrophobic core of the micelle is rendered as a green surface sliced by the image plane just behind the dye. The white surface shows the extension of the head groups of the micelle sliced in the same plane. The model of Alexa488-DIBO was generated with Schrödinger’s small-molecule drug discovery suite, and the model of DM micelle was constructed by charmm-gui micelle builder.[53] A short molecular dynamics simulation (1.3 ns) was performed with namd2.10[54] to relax the DM micelle around the receptor.
The micelles formed by amphiphilic detergents consist of a non-polar core covering the hydrophobic protein surface and a polar envelope surrounding the hydrophobic core. A straightforward explanation for the observed rate enhancement would be that Alexa488-DIBO molecules partition into the DM micelles, resulting in an increased local concentration of the reactive moiety around azF. To test this hypothesis, we designed an ultrafiltration experiment, where Alexa488-DIBO was mixed with various concentrations of DM micelles and centrifuged in spin filter units with regenerated cellulose membrane that has a molecular weight cut-off of 10 kDa. The larger DM micelles, together with the Alexa488-DIBO partitioning into them, are expected to remain in the retentate fraction, while free Alexa488-DIBO will flow through the membrane (Supplementary Scheme S1).[16] The concentration of Alexa488-DIBO in filtrate and retentate were quantified by UV–Vis spectroscopy (Supplementary Figure S3). We showed that the total concentration of Alexa488-DIBO in the retentate increased with the mass concentration of DM (Supplementary Figure S3A and Table S1). We calculated the partition coefficient of Alexa488-DIBO between DM micelles and buffer at different DM concentrations. (Figure 4C and Supplementary Table S2; the detailed calculation is explained in the Supplementary Information). We found that when the concentration of DM is 0.1% (w/v), the effective local concentration of Alexa488-DIBO in DM micelles was increased by a factor of 7.8 ×102, a value in good agreement with the observed rate enhancement (1.2×103-fold for M155azF and 7.0 ×102-fold for V162azF as compared to Y102azF). Therefore, the effect of partition suffices to explain the rate enhancement.
Characterizing the ligand-binding property of Alexa488-Rho by a steady-state fluorescence-quenching assay
We then addressed whether the engineered Rho variants, with the modification at TM4, remained functional. We utilized a fluorescence-quenching assay to assess their photoactivation and ligand binding (Figure 5 and Supplementary Figure S4).[10c] This assay is based on the energy transfer between Alexa488 (the donor), and the bound 11-cis-retinal (the acceptor). The emission spectrum of Alexa488 overlaps with the absorption band of dark-state Rho centered at 500 nm, causing pronounced quenching of Alexa488 fluorescence by 11-cis-retinal. Upon photoactivation, 11-cis-retinal isomerizes to all-trans-retinal to generate the active-state metarhodopsin II (Meta-II), and the retinal absorption peak shifts to 380 nm. This spectral shift results in loss of quenching and an increase in the Alexa488 signal. Then 11-cis-retinal dissociates from the binding pocket, giving opsin with the empty binding pocket.[17] Therefore the Alexa488 signal can serve as a reporter for the recombination reaction between 11-cis-retinal and the apoprotein opsin. Upon addition of exogenous 11-cis-retinal, the Alexa488 signal decrease as retinal enters the binding pocket of opsin and forms the protonated Schiff base linkage. It is worth noting that the excitation light for Alexa488 may also cause photoisomerization of 11-cis-retinal in Rho, which can be further enhanced by the energy transfer between Alexa488 and 11-cis-retinal in Rho. Both effects may result in a lower apparent recombination rate. Therefore it is important to minimize the excitation intensity by using very narrow band-pass filter and a shutter that closes the excitation light path between data points (duty cycle 1/15) to reduce such a systematic error.
Figure 5.
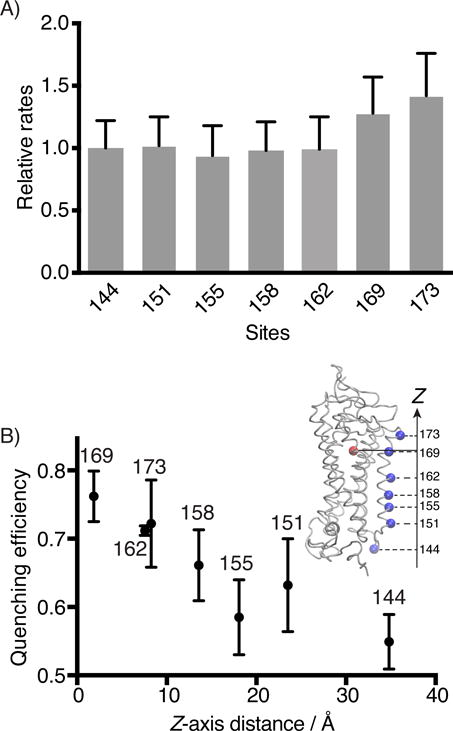
Steady-state fluorescence quenching assay for the binding kinetics of 11-cis-retinal to Alexa488-Rho variants. A) The second-order rate constants (k2) of 11-cis-retinal uptake by different Alexa488-Rho variants. S144-Alexa488 Rho was used as a reference to normalize the reaction rates of other variants. B) The energy transfer efficiencies between 11-cis-retinal (acceptor) and Alexa488 (donor) attached to different sites of Rho plotted versus the Z-axis distance between the alpha carbon of the labeled site (blue dot), and the center of mass of the retinal molecule (red dot), as determined from the crystal structure (inset, PDB 1U19).[55]
We found that the recombination between the TM4-Alexa488 Rho mutants and 11-cis-retinal were essentially complete (> 95%), with the exception of V162-Alexa488 Rho (90%). The regenerated Rho can be repeatedly photoactivated, demonstrating the formation of a functional pigment (Supplementary Figure S4). The rates of recombination reactions between 11-cis-retinal and N151-, M155-, A158-, and V162-Alexa488 Rho are consistent with the value obtained for S144-Alexa488 Rho. The regeneration kinetics of A169- and V173-Alexa488 Rho are slightly faster (30–40%) compared with the rest of Alexa488-labeled Rho variants (Figure 5A).
The energy transfer efficiencies of these Alexa488-labeled variants are dependent on the distance between the site of labeling and the binding pocket (Figure 5B). Among all the Alexa488-labeled azF-Rho variants, S144-Alexa488 Rho showed the lowest quenching efficiency (0.55 ± 0.04), while A169-Alexa488 Rho showed the highest quenching efficiency (0.76 ± 0.04).
DISCUSSION
The specificity of the SpAAC reaction
In our earlier report, we have tested the azF-independent background reaction of Alexa488-DIBO with wt Rho. We utilized UV–Vis spectroscopy, in-gel fluorescence and steady-state fluorescence-quenching experiment to estimate the degree of non-specific labeling. We found the covalent labeling of functional wt Rho to be less than 1% of the specific labeling reaction with azF-Rho.[10c] Therefore, background labeling cannot explain the apparent super-stoichiometry observed for azF-Rho variants. Note that the dye/protein ratios overestimate the actual labeling stoichiometry. The main reason is that the protein is quantified as correctly folded, ligand-bound receptor. However, in addition to dye-labeled, ligand-bound receptor, the dye quantification also includes contaminations from free dye, dye-labeled antibody leaching from the immunoaffinity matrix, and dye-labeled, non-functional receptor, which cannot recombine with 11-cis-retinal. Therefore the dye/protein-labeling ratio overestimates the absolute labeling stoichiometry depending on the relative contribution from these contaminants.
Another concern is the potential reduction of azide to amine in the cellular context, which would decrease the number of reactive handles present in Rho. The in vitro reduction of aryl azide by thiols to aryl amines has been reported,[18] and the reduction product of azF might be utilized by the aminoacyl synthetase.[19] Reduction of genetically encoded azido group to amino group has also been observed by mass spectroscopy.[20] However, in the case of unnatural amino mutagenesis, it is not clear whether the degradation of azF occurred before or after incorporation into protein. Reduction of azF would lead to a decrease of the labeling stoichiometry and might contribute to the apparent sub-stoichiometry for Y102azF Rho. Y102azF is exposed to the oxidizing extracellular environment rather than the reducing environment of the cytosol. Theoretically, compared with Y102azF, S144azF is more likely to be degraded in the cellular environment. Due to the difficulty of mass spectroscopy experiment with GPCRs, we do not have direct experimental evidence to test this possibility. Overall, we believe that the slow reaction kinetics, rather than the reduction of azF, is the major cause for the sub-stoichiometrical labeling for residues like Y102azF.
Accelerated SpAAC in micellar environment
While the reaction kinetics of SpAAC with azF-Rho varies with the specific chemical and structural properties of the labeling reagent and the location of the azF residue, it has proven generally satisfactory for our purpose. Interestingly, we noted in our system a substantial rate enhancement effect (from 4-fold to 400-fold) for the SpAAC reaction (8.6 ± 1.3 M−1 s−1 for DIBO-biotin or 62 ± 12 M−1 s−1 for Alexa488-DIBO and Rho-S144azF) as compared with model reactions of DIBO and benzylazide in methanol (0.17 M−1 s−1) or water/acetonitrile (1:4 v/v) (2.3 M−1 s−1).[11, 14]
The unexpectedly high reaction rates of SpAAC between the ~40-kDa biomolecule S144azF-Rho and DIBO prompted us to postulate that the reactivity of the DIBO reagent is modulated by the local environment on the protein surface, and that the hydrophobicity of the local environment contributes to the observed reactivity enhancement. Therefore, we determined the kinetics for two TM4 azF-Rho variants, M155azF- and V162azF-Rho, and found even greater rate enhancment effect. To explain this observation, we designed an ultrafiltration experiment to determine the partition coefficient of Alexa488-DIBO between micelle and water. We hypothesized that the partitioning of Alexa488-DIBO resulted in a higher local concentration of DIBO around azF and hence accelerated reaction. We found such a simple model suffices to explain the order of magnitude of the rate enhancement effect for SpAAC with azF-Rho in micelles.
A popular method to predict the stickiness of cyclooctynes and fluorescent dyes, and their applicability for fluorescent labeling, is based on in silico calculation of the n-octanol/water partition coefficient (cLogP).[11, 21] We used ChemBioDraw 14 to calculate cLogP = −11.8 for Alexa488-DIBO. Thus the high value of the measured partition coefficient (Log 780 = +2.9) of Alexa488-DIBO between DM micelles and water seems counterintuitive. However, the cLogP of DIBO was calculated as +4.4,[11] suggesting a strong tendency for DIBO to partition into the hydrophobic core of DM micelles. The linker length between the hydrophobic DIBO and the hydrophilic Alexa488 enabled the polar moiety to reside outside the water/detergent interface (Figure 4D). Overall, the amphiphilicity of Alexa488-DIBO makes it thermodynamically favorable to partition into the detergent/water interface of the micelle and adopt an orientation that brings the reactive DIBO moiety into proximity of transmembrane azF sites. Our amphiphilicity interpretation also provides a framework to explain why calculated cLogP values do not robustly predict lipid membrane interactions of fluorescent dyes.[21]
The partition effect helps to interpret differential reactivity of different azF labeling sites. The k2 for S144azF-Rho is 230-fold faster than the reaction rate for the EC site Y102azF, but 3- to 5-fold slower than that of TM4 sites V162azF and M155azF, respectively. Site S144 is located in the IC2 loop that exhibits higher flexibility than the rigid site Y102.[15] Although the residue S144 is largely exposed to water with only 7% lipid contacts (cf. Figure 2), substitution of serine for azF increases the hydrophobicity of this site, and consequently its probability to partition into the lipidic environment.
Recently, we found that a site located in a hydrophobic cavity on the EC side of the CCR5 chemokine receptor was much more efficiently labeled by SpAAC than other EC sites.[10d] Interestingly, another report of labeling experiments with azF-tagged GFP expressed in bacteria showed large variation of SpAAC reaction rates for different labeling positions. The most highly reactive site (k2 = 17.8 M−1 s−1) had the least surface exposure of all tested sites and was in a non-charged but polar local environment with an adjacent aromatic phenylalanine residue.[22]
Taken together, a strong determinant for the SpAAC reactivity of DIBO is the hydrophobicity of the local environment in which azF resides.
Alternate uaas for protein labeling
In the recent years, the pyrrolysyl-tRNA synthetase/tRNA pair[23] has been developed as an alternative to evolved tyrosyl-tRNA synthetase/tRNA pairs for amber suppression, first in E.coli[24], later in yeast[25]and mammalian cells.[25] A major advantage of the pyrrolysyl synthetase is its high substrate side-chain promiscuity.[26] In addition to azido and alkynyl groups for CuAAC or SpAAC,[27] the engineered pyrrolysyl synthetases have enabled a wider range of bioorthogonal reactive group to be genetically encoded into protein, including 2-aminothiols for cyanobenzothiazole condensation,[28] strained alkynes for SpAAC[29]or tetrazine ligation,[30] strained alkenes for Diels-Alder reaction,[31] norbornene that reacts with nitrile-imines[32] or tetrazine,[30a] tetrazines with trans-cyclooctyenes,[33] acrylamide with nitrile imine,[34] etc. The list will continue to grow in the foreseeable future.
An appealing feature about some of these newly developed labeling methods is their ultrafast reaction kinetics, with k2 up to 104 M−1 s−1.[35] However, most of the proof-of concept studies were based on globular proteins that are routine to express and purify, and their applicability to proteins of greater biological significance is yet to be demonstrated.
To prepare fluorescently labeled proteins for research applications, reaction kinetics is certainly one of the important concerns, but not the predominant one. Non-specificity can be an issue as well. The strained alkenes[36], strained alkynes[37] and tetrazines[38] all have been suggested or demonstrated to react with thiols, which raises the possibility that these genetically encoded reactive handle may also suffer from cross-reactivity with neighboring cysteines, or with intracellular cysteines in the course of protein folding and trafficking.
Based on our own experience gleaned from labeling GPCRs, we contend that characterization of reaction specificity, kinetics, and topology-dependent reactivity are essential for successful application of the bioorthogonal chemistries to protein labeling.
The functionality of Alexa488-Rho variants
In the steady-state fluorescence quenching experiment, we found that the regeneration kinetics of A169- and V173-Alexa488 Rho are slightly faster (30–40%) compared with the rest of Alexa488-labeled Rho variants (Figure 5A). While the difference is not significant, we would like to discuss some possible effects. Differences in energy transfer efficiency between Alexa488 and retinal in rhodopsin could change the sensitivity of the experiment bleaching artifacts due to the probe light. Sites 169 and 173 are closer to the binding pocket and Alexa488 attached to these two sites exhibits higher energy transfer efficiency than when the fluorophore was conjugated to site 144. Higher transfer efficiency between Alexa488 and retinal in rhodopsin would lead to higher bleaching rates and consequently reduced observed regeneration kinetics. Thus, we ruled out the possibility that the faster recombination kinetics for A169-Alexa488 and V173-Alexa488 Rho was an artifact embedded in the energy transfer scheme, as greater energy transfer between Alexa488 and retinal should cause the measured kinetics to be even slower. An alternate explanation would be that modification of the sites in proximity to, although not a part of, the binding pocket caused slightly altered ligand binding kinetics.
The measured fluorescence quenching efficiencies are generally consistent with distance measurements based on with the crystal structure of Rho and the site-dependent quenching efficiency thus provides an additional line of evidence for the site-specificity of the labeling strategy. It should be pointed out that here we used the z-axis coordinates of the alpha carbon of the labeled sites and the center of 11-cis-retinal to estimate the distance between Alexa488 and retinal. A rigorous treatment should take into account the dipole orientations of Alexa488 and 11-cis-retinal, the length and conformation of the linker between the alpha carbon and the fluorophore, and the angular distribution of the linker relative to TM4, which is beyond the scope of this study.
The advantage of targeting the TM region of GPCRs
Unlike maleimide/cysteine chemistry, which involves a thiolate anion intermediate and preferably labels sites exposed to aqueous environment,[39] the DIBO/azF combination readily modifies sites in the hydrophobic TM region. This property of SpAAC offers several opportunities for future work. First, the EC surfaces of GPCRs are often critical for receptor-ligand binding, and the IC surface for receptor-G protein interaction. Thus for numerous applications it is desirable to leave the EC and IC surface intact. Second, the sequence of molecular events in GPCR activation has received increasing interest.[7, 40] The capability to anchor a probe to a particular site in the TM region of GPCRs will be a valuable tool for such studies.
Taken together, we present here a systematic evaluation of the site-specific labeling of the TM region of Rho, a prototypical GPCR, using SpAAC. The modification at TM4 did not cause significant alteration in the behavior of Rho with respect to ligand binding and photoactivation. We found that SpAAC exhibited greatly enhanced reactivity with TM4 sites compared with loop sites, which can be explained by the partitioning of the labeling reagent into the micelles. The rate constants are comparable with some of the fastest bioorthogonal reactions known.[35a] The preference of SpAAC with sites embedded in hydrophobic environment provides a unique possibility for studying membrane proteins, which are estimated to have about 5330 members in the human proteome.[41] We anticipate that the micelle-enhanced bioorthogonal labeling strategy reported here will be useful for investigating receptor oligomerization, lipid-receptor interactions, and single-molecule FRET of GPCR-ligand interactions.[42]
EXPERIMENTAL SECTION
Materials
E. coli TOP10 (Invitrogen) was used for plasmid propagation and isolation. Oligonucleotides were obtained from eOligo or Fisher Scientific. Plasmid DNA was purified using standard Maxi Prep Kits from Qiagen. azF (Chem-Impex International) was used without further purification. The plasmid pSVB.Yam is a tRNA expression vector carrying a humanized chimeric gene encoding an amber suppressor tRNA derived from the B. stearothermophilus tRNATyr [9a]. pcDNA.RS-azF is a protein expression vector based pcDNA3.1 (+)/neo (Invitrogen) with a gene encoding E. coli Tyr-RS-Y37L/D182S/F183M/L186A with a C-terminal FLAG tag, the tRNA synthetase for azF[9b] and pMT4 carries the synthetic gene encoding wt bovine Rho[43]. We introduced amber mutations into the Rho gene by QuikChange mutagenesis (Stratagene). All plasmid constructs were confirmed by automated DNA sequencing. The HEK293F cell lines, the transfection reagents (FreeStyle MAX), and expression media (OptiPRO SFM reduced serum medium and serum-free FreeStyle 293 expression medium) were obtained from Invitrogen/Thermo Fisher Scientific. Sepharose 2B was purchased from Sigma. 1D4-Sepharose 2B was prepared from 1D4 mAb and CNBr-activated Sepharose 2B (2 mg IgG per mL packed beads) as described before.[44] Alexa488-DIBO was obtained from Molecular Probes/Thermo Fisher Scientific as dry powder, and dissolved in DMSO at 5 mM and stored at −20°C.
Heterologous expression of azF-tagged Rho in mammalian cell culture
The wt and azF-tagged Rho were expressed in HEK293F suspension cell culture. The HEK293F suspension cells were cultured in serum-free FreeStyle 293 expression medium in a 125-mL disposable, sterile, polycarbonate Erlenmeyer flask (Corning) at 37°C in 5% CO2 atmosphere. The cell culture was shaken on an orbital shaker at 125 rpm. For a transfection experiment in a 30-mL culture, plasmid DNA (38.6 μg in total; for amber codon suppression 18.4 μg of pMT4.Rho containing the amber codon, 18.4 μg of pSVB.Yam, and 1.84 μg pcDNA.RS-azF were mixed together) was added into OptiPRO SFM (a total volume of 0.6 mL) reduced serum medium. In another sterile tube, transfection reagent FreeStyle MAX (38.6 μL) was diluted in into OptiPRO SFM (a total volume of 0.6 mL). The diluted transfection reagent was gently combined with the DNA. The mixture was let stand at room temperature for 10 minutes and then added into the cell culture. Before transfection, the cell culture was diluted to a density of 106 cells/mL in serum-free FreeStyle 293 expression medium supplemented with 1 mM azF. The cells were harvested 96 hours post-transfection. The total cell number upon harvesting normally ranged from 6×107 to 8×107. The harvested cells were resuspended in DPBS (Gibco, supplemented with leupeptin and aprotinin, Sigma) at a density of 107 cells/mL in a 15-mL conical, polypropylene tube (Falcon). In the dark room, 11-cis-retinal (1.4 mM ethanol solution) was added into the cell suspension to a final concentration of 5 μM.[45] The suspension was nutated at 4°C overnight. Excess 11-cis-retinal was removed by spinning down the cells and discarding the supernatant fraction. The regenerated cells can be immediately used, or may be stored at −20°C for several months.
Bioorthogonal labeling of Rho
The 11-cis-retinal regenerated cells expressing azF-rhodopsin variants were lysed with the solubilization buffer (1 mL per 107 cells, 1% (w/v) DM, 50 mM HEPES or Tris-HCl, pH 6.8, 100 mM NaCl, 1 mM CaCl2 with Complete EDTA-free Protease Inhibitor Cocktail, Roche) for at least 1 h at 4°C. The lysate was cleared by centrifugation at 100,000×g for 30 min and incubated overnight at 4°C with 1D4-mAb-sepharose 2B (100 μL). The resin was transferred into a 1.5-mL Eppendorf tube and washed three times for 30 min each with 0.5 mL reaction buffer (0.1% DM in DPBS, pH 7.2). Then the reaction buffer (200 μL) was mixed with the resin (100 μL) to give 300-μL slurry. The Alexa488-DIBO stock solution (5 mM in DMSO) was directly added to into the reaction mixture to give the appropriate final concentration. The reaction was agitated with a thermomixer at 25 °C. For labeling the TM4 sites, we used 50 μM of Alexa488-DIBO and allowed the reaction to proceed for 18 hours, unless otherwise noted.
The reaction was stopped by centrifugation and removal of the supernatant fraction. The resin was then transferred into a microporous centrifugal filtering unit (Microcon-MC pore size 0.48 μm, Millipore). The resin was first washed with the reaction/wash buffer for three times (30 min incubation each time) to deplete the unreacted dyes, and then with a low-salt buffer (0.1% (w/v) DM, 2 mM sodium phosphate buffer, pH 6.0). The receptor was eluted with elution buffer (100 μL, no less than the volume of the resin; 0.33 mg/mL C9 peptide in 0.1% (w/v) DM, 2 mM sodium phosphate buffer, pH 6.0). The resin was incubated with the elution buffer on ice for at least 1 h. The purified receptor was collected in a clean 1.5-mL Eppendorf tube. The elution was repeated a second time. The combined elutions were supplemented with 150 mM NaCl and characterized by UV–Vis spectroscopy and in-gel fluorescence (Supplementary Information). Purified samples were stored at −80°C and thawed on ice before use. The yield from 107 HEK293F cells was typically 0.5–1 μg Alexa488-labeled azF-Rho.
Kinetic study of SpAAC using Alexa488-DIBO
The reaction was performed under the same condition as described above. The initial concentration of the labeling reagents (10 μM) was approximately 20-fold excess molarity. At different time points, an aliquot of the resin/buffer mixture (30 μL) was taken out and added into a clean 1.5-mL Eppendorf tube. The reaction was quenched by adding the wash buffer (0.4 mL; 0.1% DM (w/v) DPBS, pH 7.2) and centrifugation. Washing and elution were done as described in the previous section. The labeled Rho was first separated by SDS-PAGE electrophoresis and then visualized by in-gel fluorescence. Silver staining was performed for the same gel in order to normalize the sample concentration. The experimental details for in-gel fluorescence and silver staining are described in the Supplementary Information.
Determination of the partition coefficient of Alexa488-DIBO between micelle and water
Approximately 5 μM Alexa488-DIBO solution was prepared in 1× DPBS buffer containing various concentrations of DM micelles (up to ~ 1% (w/v)). 400 μL of the solution was placed in an ultrafiltration spin filter (Amicon Ultra 0.5 mL centrifugal filters, 10 kDa MWCO) and centrifuged at 14,000×g for 5~7 min until the volume of the filtrate was approximately 200 μL. The volumes of the filtrate and the retentate were measured with an adjustable pipette. The concentrations of Alexa488-DIBO in the filtrate and retentate were quantified by UV–Vis spectroscopy (ε495nm = 73, 000 M−1 cm−1).
Functional characterization of labeled Rho by steady-state fluorescence
Fluorescence spectroscopy was performed at 28°C on a SPEX Fluorolog spectrofluorometer in photon counting mode. The time course experiments were done by adding Alexa488-labeled receptor elutions (30 μL) at a final concentration of 15–50 nM to the assay buffer (450 μL; 10 mg/mL POPC, 10 mg/mL CHAPS, 125 mM KCl, 25 mM MES, 25 mM HEPES, 12.5 mM KOH, pH 6.0) under constant stirring. During time scan experiments, the fluorescence signal was integrated for 2 s every 30 s with the excitation light path closed in-between data points to minimize photobleaching. The excitation wavelength was 480 nm with 0.2-nm band-pass to further reduce photobleaching, and the emission was measured at a wavelength of 525 nm with 15-nm band-pass. The 11-cis-retinal stock solution in ethanol was diluted in the assay buffer, and 20 μL of this working dilution was added to the cuvette to a final concentration of 1.5 μM (1.48–1.59 μM) retinal. For each measurement, the concentration of freshly diluted retinal (cretinal) was determined from its 378-nm absorbance (extinction coefficient 24,400 M−1 cm−1). The pseudo-first order rate constant (k1) was obtained by fitting the fluorescence signal with monoexponential decay model. The second-order recombination reaction rate constant (k2) was calculated as k1/cretinal.
Supplementary Material
Acknowledgments
We acknowledge the generous support from the Crowley Family Fund, the Danica Foundation, and the NIH R01 EY012049 (T.P.S. and T.H.), as well as the Tri-Institutional Training Program in Chemical Biology for supporting H.T. This work was also supported by an International Research Alliance with Prof. Thue W. Schwartz at The Novo Nordisk Foundation Center for Basic Metabolic Research (http://www.metabol.ku.dk) through an unconditional grant from the Novo Nordisk Foundation to the University of Copenhagen.
ABBREVIATIONS
- azF
p-azido-L-phenylalanine
- DIBO
dibenzocyclooctyne
- DM
n-dodecyl-β-D-maltoside
- EC
extracellular
- GPCR
G protein-coupled receptor
- IC
intracellular
- Rho
rhodopsin
- SpAAC
strain-promoted [3+2] azide–alkyne cycloaddition
- TM
transmembrane
- uaa
unnatural amino acid
- wt
wild-type
References
- 1.Oldham WM, Hamm HE. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 2.a) Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]; b) Cohen BE, Pralle A, Yao X, Swaminath G, Gandhi CS, Jan YN, Kobilka BK, Isacoff EY, Jan LY. Proc Natl Acad Sci USA. 2005;102:965–970. doi: 10.1073/pnas.0409469102. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mathiasen S, Christensen SM, Fung JJ, Rasmussen SGF, Fay JF, Jorgensen SK, Veshaguri S, Farrens DL, Kiskowski M, Kobilka B, Stamou D. Nat Methods. 2014;11:931–934. doi: 10.1038/nmeth.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M. Proc Natl Acad Sci USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Ellisman MH, Lohse MJ. Nat Methods. 2005;2:171–176. doi: 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- 5.Emami-Nemini A, Roux T, Leblay M, Bourrier E, Lamarque L, Trinquet E, Lohse MJ. Nat Protoc. 2013;8:1307–1320. doi: 10.1038/nprot.2013.073. [DOI] [PubMed] [Google Scholar]
- 6.a) Bohme I, Beck-Sickinger AG. Cell Commun Signal. 2009;7 doi: 10.1186/1478-811X-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lohse MJ, Nuber S, Hoffmann C. Pharmacol Rev. 2012;64:299–336. doi: 10.1124/pr.110.004309. [DOI] [PubMed] [Google Scholar]
- 7.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. Proc Natl Acad Sci USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 9.a) Ye S, Kohrer C, Huber T, Kazmi M, Sachdev P, Yan EC, Bhagat A, RajBhandary UL, Sakmar TP. J Biol Chem. 2008;283:1525–1533. doi: 10.1074/jbc.M707355200. [DOI] [PubMed] [Google Scholar]; b) Ye S, Huber T, Vogel R, Sakmar TP. Nat Chem Biol. 2009;5:397–399. doi: 10.1038/nchembio.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Huber T, Naganathan S, Tian H, Ye S, Sakmar TP. Methods Enzymol. 2013;520:281–305. doi: 10.1016/B978-0-12-391861-1.00013-7. [DOI] [PubMed] [Google Scholar]; b) Tian H, Sakmar TP, Huber T. Methods Cell Biol. 2013;117:267–303. doi: 10.1016/B978-0-12-408143-7.00015-3. [DOI] [PubMed] [Google Scholar]; c) Tian H, Naganathan S, Kazmi MA, Schwartz TW, Sakmar TP, Huber T. ChemBioChem. 2014;15:1820–1829. doi: 10.1002/cbic.201402193. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Naganathan S, Ray-Saha S, Park M, Tian H, Sakmar TP, Huber T. Biochemistry. 2015;54:776–786. doi: 10.1021/bi501267x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debets MF, van Berkel SS, Dommerholt J, Dirks AT, Rutjes FP, van Delft FL. Acc Chem Res. 2011;44:805–815. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- 12.a) Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]; b) Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]; c) Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 14.Ning X, Guo J, Wolfert MA, Boons GJ. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber T, Botelho AV, Beyer K, Brown MF. Biophys J. 2004;86:2078–2100. doi: 10.1016/S0006-3495(04)74268-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strop P, Brunger AT. Protein Sci. 2005;14:2207–2211. doi: 10.1110/ps.051543805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon ST, Han M, Sakmar TP. Physiol Rev. 2001;81:1659–1688. doi: 10.1152/physrev.2001.81.4.1659. [DOI] [PubMed] [Google Scholar]
- 18.Staros JV, Bayley H, Standring DN, Knowles JR. Biochem Biophy Res Commun. 1978;80:568–572. doi: 10.1016/0006-291x(78)91606-6. [DOI] [PubMed] [Google Scholar]
- 19.Nehring S, Budisa N, Wiltschi B. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Proc Natl Acad Sci USA. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu WS, Brock A, Chen S, Chen SB, Schultz PG. Nat Methods. 2007;4:239–244. doi: 10.1038/nmeth1016. [DOI] [PubMed] [Google Scholar]
- 21.Hughes LD, Rawle RJ, Boxer SG. PLoS One. 2014;9:e87649. doi: 10.1371/journal.pone.0087649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddington SC, Tippmann EM, Jones DD. Chem Commun. 2012;48:8419–8421. doi: 10.1039/c2cc31887c. [DOI] [PubMed] [Google Scholar]
- 23.a) Srinivasan G, James CM, Krzycki JA. Science. 2002;296:1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]; b) Polycarpo C, Ambrogelly A, Berube A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Soll D. Proc Natl Acad Sci USA. 2004;101:12450–12454. doi: 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blight SK, Larue RC, Mahapatra A, Longstaff DG, Chang E, Zhao G, Kang PT, Green-Church KB, Chan MK, Krzycki JA. Nature. 2004;431:333–335. doi: 10.1038/nature02895. [DOI] [PubMed] [Google Scholar]
- 24.Neumann H, Peak-Chew SY, Chin JW. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- 25.Hancock SM, Uprety R, Deiters A, Chin JW. J Am Chem Soc. 2010;132:14819–14824. doi: 10.1021/ja104609m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan W, Tharp JM, Liu WR. Biochim Biophys Acta. 2014;1844:1059–1070. doi: 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen DP, Lusic H, Neumann H, Kapadnis PB, Deiters A, Chin JW. J Am Chem Soc. 2009;131:8720–+. doi: 10.1021/ja900553w. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen DP, Elliott T, Holt M, Muir TW, Chin JW. J Am Chem Soc. 2011;133:11418–11421. doi: 10.1021/ja203111c. [DOI] [PubMed] [Google Scholar]
- 29.Plass T, Milles S, Koehler C, Schultz C, Lemke EA. Angew Chem Int Ed. 2011;50:3878–3881. doi: 10.1002/anie.201008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW. J Am Chem Soc. 2012;134:10317–10320. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Borrmann A, Milles S, Plass T, Dommerholt J, Verkade JMM, Wiessler M, Schultz C, van Hest JCM, van Delft FL, Lemke EA. ChemBioChem. 2012;13:2094–2099. doi: 10.1002/cbic.201200407. [DOI] [PubMed] [Google Scholar]
- 31.Plass T, Milles S, Koehler C, Szymanski J, Mueller R, Wiessler M, Schultz C, Lemke EA. Angew Chem Int Ed. 2012;51:4166–4170. doi: 10.1002/anie.201108231. [DOI] [PubMed] [Google Scholar]
- 32.Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carell T. Angew Chem Int Ed. 2012;51:4466–4469. doi: 10.1002/anie.201109252. [DOI] [PubMed] [Google Scholar]
- 33.Seitchik JL, Peeler JC, Taylor MT, Blackman ML, Rhoads TW, Cooley RB, Refakis C, Fox JM, Mehl RA. J Am Chem Soc. 2012;134:2898–2901. doi: 10.1021/ja2109745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YJ, Wu B, Raymond JE, Zeng Y, Fang XQ, Wooley KL, Liu WRS. ACS Chem Biol. 2013;8:1664–1670. doi: 10.1021/cb400267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Lang K, Chin JW. ACS Chem Biol. 2014;9:16–20. doi: 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]; b) Lang K, Chin JW. Chem Rev. 2014;114:4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 36.Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 37.van Geel R, Pruijn GJM, van Delft FL, Boelens WC. Bioconjugate Chem. 2012;23:392–398. doi: 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- 38.Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Xu Q, Cortes DM, Perozo E, Laskey A, Karlin A. Proc Natl Acad Sci USA. 2002;99:11605–11610. doi: 10.1073/pnas.192439299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye S, Zaitseva E, Caltabiano G, Schertler GF, Sakmar TP, Deupi X, Vogel R. Nature. 2010;464:1386–1389. doi: 10.1038/nature08948. [DOI] [PubMed] [Google Scholar]
- 41.Almen MS, Nordstrom KJV, Fredriksson R, Schioth HB. BMC Biol. 2009;7 doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huber T, Sakmar TP. Chem Biol. 2014;21:1224–1237. doi: 10.1016/j.chembiol.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Franke RR, Sakmar TP, Oprian DD, Khorana HG. J Biol Chem. 1988;263:2119–2122. [PubMed] [Google Scholar]
- 44.Knepp AM, Grunbeck A, Banerjee S, Sakmar TP, Huber T. Biochemistry. 2011;50:502–511. doi: 10.1021/bi101059w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.a) Starace DM, Knox BE. Exp Eye Res. 1998;67:209–220. doi: 10.1006/exer.1998.0507. [DOI] [PubMed] [Google Scholar]; b) Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Proc Natl Acad Sci USA. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 47.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, Wu B. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 53.Cheng X, Jo S, Lee HS, Klauda JB, Im W. J Chem Inf Model. 2013;53:2171–2180. doi: 10.1021/ci4002684. [DOI] [PubMed] [Google Scholar]
- 54.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.