

Abstract
Purpose
Increased inflammatory mediator levels are reported in diabetic retinopathy. We previously reported that β-adrenergic receptor agonists reduced inflammatory mediators in the diabetic retina; however, these agents cannot be given systemically. Here, we investigated whether Epac1 is key to the protective effects of β-adrenergic receptor agonists.
Methods
We cultured primary human retinal endothelial cells (RECs) in normal (5 mM) or high (25 mM) glucose and treated them with an Epac1-specific agonist. Additionally, we generated Epac1 conditional vascular endothelial cell knockout mice by breeding Epac1 floxed mice with Cdh5 Cre mice to investigate the role of Epac1 in the retinal levels of tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), nuclear factor kappa beta (NFκB), and inhibitor of kappa beta (IκB). Confocal microscopy was performed to localize Epac1 in the mouse retina.
Results
Data showed that high glucose increased the TNF-α and IL-1β levels in the RECs, which were reduced cells treated with the Epac1 agonist. The loss of Epac1 in the retinas of the conditional knockout mice resulted in statistically significantly increased levels of TNF-α and IL-1β, as well as NFκB.
Conclusions
These data indicate that Epac1 may be protective to the retina through inhibition of key inflammatory mediators.
Introduction
An ever-increasing number of scientific studies suggest that some form of chronic inflammation is an initiating factor in diabetic retinopathy [1-3]. Researchers have shown that a large number of cytokines are increased in non-proliferative diabetic retinopathy, which can contribute to vascular and neuronal damage in the retina [4-7]. Additionally, researchers have shown that inhibition of the inciting inflammatory mediators is protective for the diabetic retina [8-10]. We have shown that the application of a novel β-adrenergic receptor agonist, Compound 49b, can significantly decrease tumor necrosis factor alpha (TNF-α) in the diabetic rat retina [11]. Compound 49b also significantly reduced toll-like receptor 4 signaling cascades in the diabetic retina [12].
Compound 49b actions in the diabetic retina are likely mediated through increased levels of cAMP, leading to activation of protein kinase A (PKA) and/or exchange protein for cAMP (Epac1). Epac1 can serve as an alternative pathway for β-adrenergic receptor/cAMP activation of downstream pathways [13]. Alternatively, PKA and Epac1 pathways may become activated after β-adrenergic receptor stimulation, leading to the initiation of distinct signaling cascades [14]. Our interest in the potential role of Epac1 in retinal endothelial cells (RECs) and diabetes stems from work showing that Epac1 regulates vascular endothelial cell permeability [15,16]. Further work showed that PKA and Epac1 can regulate macrovascular and microvascular endothelial actions independently [17]. In addition to Epac1 actions in endothelial cell adhesion, other researchers have reported that Epac1 can inhibit suppressor of cytokine signaling 3 (SOCS3), a direct target for TNF-α in human umbilical vein endothelial cells (HUVECs) [18]. As we found that Compound 49b decreased TNF-α and SOCS3 actions in RECs [19,20], it is possible Epac1 may be involved in this protective action of β-adrenergic receptor signaling in RECs. Although Epac1 and Epac2 have been localized in the retina [21], they have only recently been reported in bovine retinal endothelial cells and shown to play a role in leukostasis (Antonetti, ARVO abstract 2015). Additionally, it has been shown that Epac1 can regulate proinflammatory mediators, TNF-α and interleukin-1β (IL-1β), in RAW 264.7 macrophages [22], as well as in rat microglia [23].
Thus, we hypothesized that Epac1 is protective for the retina through reduced TNF-α and IL-1β levels. We investigated this in RECs treated with an Epac1 agonist, as well as in vascular endothelial cell conditional knockout mice for Epac1.
Methods
Retinal endothelial cell culture
Primary human RECs acquired from Cell Systems Corporation (CSC, Kirkland, WA) were grown in Cell Systems medium supplemented with microvascular growth factors (MVGS), 10 μg/ml gentamycin, and 0.25 μg/ml amphotericin B (Invitrogen, Carlsbad, CA). Once the cells reached confluence, some dishes were moved to Cell Systems Medium with supplemented D-glucose to 25 mM. All cells were cultured on attachment factor–coated dishes. Only cells up to passage 6 were used. Cells were quiesced by incubation in high or normal glucose medium without MVGS for 24 h before experimental use.
Cell treatments
The RECs in normal (5 mM) and high glucose (25 mM) were treated with 8-CPT-2’-O-Me-cAMP (an Epac1 agonist) at 10 μM for 2 h to directly stimulate Epac1 following 24 h of starvation without MVGS. Some RECs in normal (5 mM) and high glucose (25 mM) medium were also transfected with Epac1 siRNA (L-007676–00–0005, Dharmacon, Lafayette, CO) or scrambled siRNA at a final concentration of 20 nM using the RNAiMAX transfection reagent according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were processed for enzyme-linked immunosorbent assay (ELISA) or western blot analyses.
Mice
All animal procedures met the Association for Research in Vision and Ophthalmology requirements, were approved by the Institutional Animal Care and Use Committee of Wayne State University, and conformed to National Institutes of Health (NIH) guidelines. The Epac1 floxed mice (B6;129S2-Rapgef3tm1Geno/J mice) and the B6 FVB-Tg (Cdh5-Cre)7Mlia/J Cre mice were purchased from Jackson Laboratories (Bar Harbor, ME). After two generations, the Epac1 floxed mice were bred with the Cdh5-Cre mice to generate conditional knockout mice in which Epac1 is eliminated in vascular endothelial cells. At 3 months of age, the Epac1 floxed and Epac1 Cre-Lox mice were used for these experiments. Euthanasia was performed with carbon dioxide overdose followed by cervical dislocation.
Genotyping
Genomic DNA were extracted from ear punch samples from 2-week-old mice. The ear punches were digested with one-step tail DNA extraction buffer (100 mM Tris, 5 mM EDTA, 200 mM NaCL, 1% Triton) plus proteinase K (10 mg/ml) at 55 °C overnight, followed by enzyme heat inactivation at 85 °C for 45 min. The following sequences of primer pairs were used to screen the Epac1 conditional knockout mice: Epac1: mutant forward: 5′-ATT TGT CAC GTC CTG CAC GAC G-3′, wild-type forward: 5′-CTG GCC TCT CCT GAA TCT TG-3′, common: 5′-CCT CGC TGT TGG TAA GTG GT-3′. Cdh5-Cre forward: 5′-AGG CAG CTC ACA AAG GAA CAA T-3′; reverse: 5′-TCG TTG CAT CGA CCG GTA A-3′; Cdh5-Cre internal positive control forward: 5′-CTA GGC CAC AGA ATT GAA AGA TCT-3′; reverse: 5′-GTA GGT GGA AAT TCT AGC ATC ATC C-3′. The standard PCR reaction was performed using KAPA2G HotStar Genotyping PCR Mix (KK5621, KAPA Biosystems, Wilmington, MA). The PCR reaction was performed with the following temperatures and times: denaturing at 95 °C 3 min, 35 cycles at 95 °C for 15 s, 60 °C for 15 s, and 72 °C sec/kb, with the final extension at 72 °C for 1 min.
Immunohistochemistry for Epac1
Three-month-old male and female Epac1 floxed and Epac1 Cdh5 Cre-Lox mice (three in each group) were euthanized with carbon dioxide followed by cervical dislocation. After confirmation of death by cervical dislocation, the eyes were removed and immediately placed in 4% paraformaldehyde in PBS (1X; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) overnight. The following day, the whole globe was transferred to 0.1 M PBS containing 30% sucrose for cryoprotection. Ten-micron sections were cut using a cryostat and stored at 80 °C until use. The sections were rinsed in PBS and put in 5% normal goat serum for 1 h at room temperature for blocking any nonspecific staining, followed by incubation with Isolectin GS-IB4 (Alexa Fluor 488 conjugate, 1:100, Life Technologies) and rabbit anti-epac1 (1:100, Abcam, San Francisco, CA) at 4 °C overnight. After rising in 0.3% Triton/PBS, the sections were incubated with secondary antibody goat anti-rabbit conjugated to Alexa Fluor 594 (1:1,000, Life Technologies) for 2 h at room temperature. The slides were then rinsed in PBS and counterstained with 4',6-diamidino-2-phenylindole (DAPI). The slides were examined with a Leica (Buffalo Grove, IL) confocal microscope.
Western blotting
Whole retinal lysates were collected in lysis buffer containing protease and phosphatase inhibitors. Equal amounts of protein were separated on a precast Tris-glycine gel (Invitrogen) and blotted on nitrocellulose membrane. After blocking in Tris-buffered saline and Tween 20 (TBST; 10 mM Tris-HCl buffer, pH 8.0, 150 mM NaCl, 0.1% Tween-20) and 5% (w/v) bovine serum albumin, the membranes were treated with Epac1 (ab109415), Epac 2 (ab193665, Abcam), total nuclear factor kappa beta (NFκB; #4764), phosphorylated NFκB (Ser 536, #3303), phosphorylated IκB (Ser32, #2859), total IκB (#4812, Cell Signaling, Danvers, MA), and beta actin (sc-47778, Santa Cruz Biotechnology, Santa Cruz, CA) primary antibodies followed by incubation with secondary antibodies labeled with horseradish peroxidase. Antigen-antibody complexes were detected with a chemiluminescence reagent kit (Thermo Scientific, Pittsburgh, PA), and data were acquired using an Azure C500 (Azure Biosystems, Dublin, CA). Western blot data were assessed using Image Studio Lite software.
ELISA
A TNF-α ELISA (Fisher Scientific, Pittsburgh, PA) was used according to the manufacturer’s instructions with the exception that the sample was exposed to the primary antibody for 24 h. One hundred micrograms of protein were used to ensure equal amounts of protein in all wells. The IL-1β ELISA was completed according to the manufacturer’s instructions with the exception that 120 μg of protein loaded into all wells, with the primary antibody incubated overnight. Both ELISAs were performed on cell or whole retinal lysates.
Statistical analyses
Non-parametric Kruskal–Wallis with Dunn’s post-hoc tests were used for the cell culture data. One-way ANOVA with Student Newman Keul’s post-hoc test was used for the animal work. A p value of less than 0.05 was considered statistically significant.
Results
Epac1 stimulation reduced TNF-α and IL-1β levels
Based on the literature suggesting a role of Epac1 in inflammatory mediators in other cell types [18,22], we first wanted to investigate whether Epac1 could inhibit TNF-α and IL-1β levels in RECs. For these experiments, the specific Epac1 agonist, 8-CPT-2’-O-Me-cAMP, was added to RECs grown in normal glucose or high glucose. Additionally, some cells received either scrambled siRNA or Epac1 siRNA to determine the specificity of the inflammatory mediators’ response to Epac1 changes. ELISA analyses for TNF-α and IL-1β were performed on all groups. An Epac1 western blot was also performed to verify successful knockdown of Epac1 using siRNA. Figure 1A shows that 8-CPT-2’-O-Me-cAMP statistically significantly increased the Epac 1 protein levels, which were blocked when Epac1 siRNA was used. These experiments are used only to show that the agonist is effective in increasing Epac1 signaling in these cells. Figure 1B shows that 8-CPT-2’-O-Me-cAMP does not increase Epac 2 protein levels, thus showing the specificity of the agonist for Epac 1. Figure 1C shows that high glucose statistically significantly increased the TNF-α levels in the RECs, which were reduced by the Epac1 agonist and increased when Epac1 siRNA was used. Figure 1D shows similar results for IL-1β stimulated with the Epac1 agonist or treated with Epac1 siRNA. Figure 1E shows that phosphorylation of NFκB is increased in response to high glucose, which is blocked by the addition of the Epac1 agonist. Figure 1F shows that the Epac 1 agonist decreased phosphorylation of IκB, likely leading to the activation of NFκB.
Figure 1.

Epac1 agonist decreased TNF-α and IL-1β in vitro. Data show results from retinal endothelial cells (RECs) grown in normal glucose (NG) or high glucose (HG) only, grown in NG or HG and treated with CPT-2’-O-Me-cAMP, an exchange protein for cAMP 1 (Epac1)-specific agonist (NG or HG+8CPT-cAMP), and grown in NG and HG and treated with scrambled (NG or HG+Sc) or Epac1 siRNA (NG or HG+Epac1 siRNA). A: Western blot data show successful knockdown of Epac1. B: Western blot data for Epac2 show that CPT-2’-O-Me-cAMP is specific for Epac1. C: Enzyme-linked immunosorbent assay (ELISA) data for tumor necrosis factor alpha (TNF-α). D: Data for interleukin-1β (IL-1β). E: Western blot results for nuclear factor kappa beta (NFκB). F: Western blot results for inhibitor of kappa beta (IκB). n = 4 for each group. Data are mean ± standard error of the mean (SEM). * p<0.05 versus NG, #p<0.05 versus HG, $p<0.05 versus HG+8CPT-cAMP.
Epac1 can be eliminated in vascular endothelial cells
To further investigate whether Epac1 can reduce inflammatory mediators in the retina, we generated conditional knockout mice for Epac1 where the Epac1 floxed mice were crossed with the Cdh5 Cre mice to generate mice with a loss of Epac1 in the vascular endothelial cells. Figure 2A shows the genotyping of the mice. Figure 2B shows that the Epac1 protein levels are statistically significantly reduced in whole retinal lysates. Because these mice were vascular cell–specific knockout mice, it is likely that some Epac1 remained in other retinal cells, but it is clear that the Epac1 Cre-Lox mice have statistically significantly less Epac1 than their floxed littermates. Figure 2C shows Epac1 staining in the whole retina. It is clear that the Epac1 (red) staining is reduced in the Epac1 Cre-Lox mice vasculature (green), while the Epac1 staining remained in other layers of the retina.
Figure 2.

Epac1 can be knocked out in vascular endothelial cells. A: The agarose gel image from the mouse ear punch samples shows the effective knockout of exchange protein for cAMP 1 (Epac1) by Cdh5 Cre. Expected band sizes: Epac mutant 457 bp, wild-type 276 bp, Cdh5 Cre 300 bp. B: A significant reduction in the Epac1 protein levels from the whole retinal lysates. n = 5 mice in each group. C: Imaging data show the cellular localization of Epac1 in the retina from the Epac1 floxed and Epac1 Cre-Lox mice. The top image is from the Epac1 floxed mice, and the bottom image is from the Epac1 Cre-LoxP mice. On the right is a larger image showing the overlap of Epac1 and B4 staining only in the floxed mice. Epac1 staining is in red, vascular staining (isolectin B4) is in green, and cell nuclei is in blue. Scale bar = 50 μm for left images; scale bar = 20 μm for enlarged images.
Loss of Epac1 in endothelial cells increased TNF-α and IL-1β levels in vivo
Using the conditional knockout mice, we showed that Epac1 is key to inhibition of TNF-α (Figure 3A) and IL-1β (Figure 3B). The loss of Epac1 resulted in a statistically significant increase in both proteins.
Figure 3.

Loss of Epac1 increased TNF-α and IL-1β in the mouse retina. Exchange protein for cAMP 1 (Epac1) floxed versus Epac1 Cre-Lox mice retinal samples were processed for enzyme-linked immunosorbent assay (ELISA) analyses for tumor necrosis factor alpha (TNF-α; A) and interleukin-1β (IL-1β; B). n = 6 for each group. Data are mean ± standard error of the mean (SEM). *p<0.05 versus Epac1 floxed.
Loss of Epac1 in vascular cells increased NFκB phosphorylation
Using whole retinal lysates from the Epac1 floxed and conditional knockout mice, we demonstrated that Epac1 regulates NFκB phosphorylation, as loss of Epac1 significantly increased NFκB phosphorylation (Figure 4A). Figure 4B demonstrates that Epac1 decreased phosphorylation of IκB, likely increasing NFκB activation. This suggests that maintenance of Epac1 may be protective for retinal vascular cells through reduction in inflammatory protein signaling.
Figure 4.
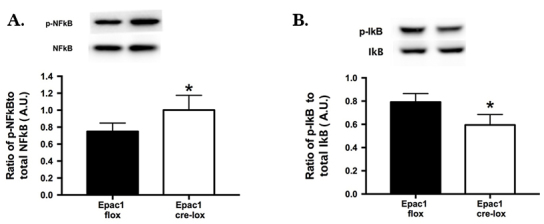
Epac1 regulates NFκB phosphorylation in the mouse retina. Whole retinal lysates from exchange protein for cAMP 1 (Epac1) floxed versus Epac1 Cre-Lox mice were processed for western blot analyses for phosphorylated nuclear factor kappa beta (NFκB (Ser536) versus total NFκB (A) or inhibitor of kappa beta (IκB; B). n = 5 for each group. Data are mean ± standard error of the mean (SEM). *p<0.05 versus Epac1 floxed.
Discussion
There is growing evidence that diabetic retinopathy is associated with increasing levels of key inflammatory mediators [2,7,24-26]. We have previously reported that our novel β-adrenergic receptor agonist, Compound 49b, could significantly reduce TNF-α levels in the retina [11]. Because Compound 49b cannot be given systemically due to cardiovascular side effects, we wanted to investigate downstream pathways that may reduce retinal inflammation.
We have previously reported that Compound 49b can reduce retinal apoptosis through increasing insulin-like growth factor-binding protein 3 (IGFBP-3) levels, which required PKA-induced phosphorylation of DNA-PK [27]. Although we have previously explored the actions of PKA in retinal apoptosis, we wanted to expand our work into other cAMP effectors. Over the past 10 years, novel roles for Epac proteins (Epac1 and Epac2) have come into focus. Epac1 is ubiquitously expressed [28] and has been located in the retina [21]. Figure 1 shows that Epac1 and 2 are expressed in retinal endothelial cells in culture; however, only Epac1 responded to the agonist used in this work. Although little has been done to investigate the physiologic role of Epac1 in the retina, much work has shown that Epac1 has protective anti-inflammatory actions in other targets, including vascular endothelial cells [29]. The early work on Epac1 as an anti-inflammatory agent focused on cardiovascular diseases, such as atherosclerosis [29]. Work on cultured airway smooth muscle cells showed that loss of Epac1 increased the number of inflammatory cells after exposure to cigarette smoke, suggesting that Epac1 is protective against inflammation in the lung [30]. In contrast to the present findings, a differential response to IL-1β in Epac1 knockout mice was not noted in the lungs. The mice exposed to cigarette smoke were complete knockout mice, which are different from the conditional knockout mice used in the present study, providing a potential explanation for the different results. In a renal proximal tubular cell line, Epac1 modulated angiotensin II-mediated inflammatory factors [31], again suggesting that Epac1 is protective against inflammation. Additionally, work in osteoclasts demonstrated that Epac1 regulation of NFκB may be key to a reduction in bone destruction in inflammatory arthritis [32]. Our work extends the previous studies in the lung, heart, bone, and kidney to demonstrate an anti-inflammatory role for Epac1 in the retinal vasculature. We found that the loss of Epac1 via siRNA or in the mouse retina led to decreased phosphorylation of IκB, which would lead to increased stabilization of IκB. This should lead to reduced NFκB activity. However, western blotting for serine 536 on NFκB showed increases in mouse retinal lysates or in RECs treated with Epac1 siRNA. This suggests disparate NFκB signaling in this system. We will dissect other NFκB phosphorylation sites or regulatory pathways in future work.
The histological work showed that Epac1 was expressed throughout the retina and was reduced in the vasculature of the Cre-Lox mice. Because Epac1 and Epac2 were observed in the RECs in culture, one might expect that Epac2 would compensate for the loss of Epac1 in the conditional knockout mice. At least for inflammatory mediators, this does not appear to occur, as there was a statistically significant increase in the TNF-α, IL-1β, and NFκB levels in the conditional knockout retina compared to those of the floxed littermates. We will focus on specific inflammatory cell types and any potential compensatory roles for Epac2 in these mice in future work. We will also expand these studies into diabetic animal models in future work.
Conclusions
These are the first studies to our knowledge to demonstrate that Epac1 can reduce key inflammatory mediators in human retinal endothelial cells and in a conditional knockout mouse model. Using RECs in the cell culture and Epac1 conditional knockout mice, we show Epac1 actions can reduce TNF-α and IL-1β levels in the retinal vasculature, as well as phosphorylation of NFκB in knockout mice.
Acknowledgments
This work was supported by NIH R01EY022045 (JJS), P30EY04068 (Hazlett), Fight for Sight Summer Medical Student Fellowship (EC); and an Unrestricted Grant to the Department of Ophthalmology from Research to Prevent Blindness (Kresge Eye Institute).
References
- 1.Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–58. doi: 10.1016/j.preteyeres.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–2. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 4.Zheng L, Du Y, Miller C, Gubitosi-Klug RA, Kern TS, Ball S, Berkowitz BA. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia. 2007;50:1987–96. doi: 10.1007/s00125-007-0734-9. [DOI] [PubMed] [Google Scholar]
- 5.Tang J, Allen Lee C, Du Y, Sun Y, Pearlman E, Sheibani N, Kern TS. MyD88-dependent pathways in leukocytes affect the retina in diabetes. PLoS One. 2013;8:e68871. doi: 10.1371/journal.pone.0068871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, Poulaki V, Semkova I, Kociok N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–28. [PMC free article] [PubMed] [Google Scholar]
- 7.Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56:224–30. doi: 10.2337/db06-0427. [DOI] [PubMed] [Google Scholar]
- 8.Abcouwer SF, Lin CM, Shanmugam S, Muthusamy A, Barber AJ, Antonetti DA. Minocycline prevents retinal inflammation and vascular permeability following ischemia-reperfusion injury. J Neuroinflammation. 2013;10:149. doi: 10.1186/1742-2094-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS. Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes. 2007;56:337–45. doi: 10.2337/db06-0789. [DOI] [PubMed] [Google Scholar]
- 10.Kern TS, Miller CM, Du Y, Zheng L, Mohr S, Ball SL, Kim M, Jamison JA, Bingaman DP. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes. 2007;56:373–9. doi: 10.2337/db05-1621. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Guy K, Pagadala J, Jiang Y, Walker RJ, Liu L, Soderland C, Kern TS, Ferry R, Jr, He H, Yates CR, Miller DD, Steinle JJ. Compound 49b Prevents Diabetes-Induced Apoptosis through Increased IGFBP-3 Levels. Invest Ophthalmol Vis Sci. 2012;53:3004–13. doi: 10.1167/iovs.11-8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger EA, Carion TW, Jiang Y, Liu L, Chahine A, Walker RJ, Steinle JJ. beta-adrenergic receptor agonist, Compound 49b, inhibits TLR4 signaling pathway in diabetic retina. Immunol Cell Biol. 2016 doi: 10.1038/icb.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kassel KM, Wyatt TA, Panettieri RA, Jr, Toews ML. Inhibition of human airway smooth muscle cell proliferation by beta 2-adrenergic receptors and cAMP is PKA independent: evidence for EPAC involvement. Am J Physiol Lung Cell Mol Physiol. 2008;294:L131–8. doi: 10.1152/ajplung.00381.2007. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Du J, Feng W, Song Y, Lu Z, Xu M, Li Z, Zhang Y. beta-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br J Pharmacol. 2012;166:676–88. doi: 10.1111/j.1476-5381.2011.01785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–72. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 16.Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–5. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- 17.Netherton SJ, Sutton JA, Wilson LS, Carter RL, Maurice DH. Both protein kinase A and exchange protein activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ Res. 2007;101:768–76. doi: 10.1161/CIRCRESAHA.106.146159. [DOI] [PubMed] [Google Scholar]
- 18.Sands WA, Woolson HD, Milne GR, Rutherford C, Palmer TM. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol Cell Biol. 2006;26:6333–46. doi: 10.1128/MCB.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang Y, Pagadala J, Miller D, Steinle JJ. Reduced insulin receptor signaling in retinal Muller cells cultured in high glucose. Mol Vis. 2013;19:804–11. [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Zhang Q, Liu L, Tang J, Kern TS, Steinle JJ. beta2-adrenergic receptor knockout mice exhibit A diabetic retinopathy phenotype. PLoS One. 2013;8:e70555. doi: 10.1371/journal.pone.0070555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitaker CM, Cooper NG. Differential distribution of exchange proteins directly activated by cyclic AMP within the adult rat retina. Neuroscience. 2010;165:955–67. doi: 10.1016/j.neuroscience.2009.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007;19:251–60. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Zhao X, Cao J, Xue Q, Feng X, Liu X, Zhang F, Yu B. Differential roles of PKA and Epac on the production of cytokines in the endotoxin-stimulated primary cultured microglia. J Mol Neurosci. 2011;45:186–93. doi: 10.1007/s12031-010-9426-x. [DOI] [PubMed] [Google Scholar]
- 24.Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–82. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frey T, Antonetti DA. Alterations to the blood-retinal barrier in diabetes: cytokines and reactive oxygen species. Antioxid Redox Signal. 2011;15:1271–84. doi: 10.1089/ars.2011.3906. [DOI] [PubMed] [Google Scholar]
- 26.Koizumi K, Poulaki V, Doehmen S, Welsandt G, Radetzky S, Lappas A, Kociok N, Kirchhof B, Joussen AM. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Invest Ophthalmol Vis Sci. 2003;44:2184–91. doi: 10.1167/iovs.02-0589. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Q, Steinle JJ. DNA-PK phosphorylation of IGFBP-3 is required to Prevent Apoptosis in Retinal Endothelial cells cultured in High Glucose. Invest Ophthalmol Vis Sci. 2013;54:3052–7. doi: 10.1167/iovs.12-11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roscioni SS, Elzinga CR, Schmidt M. Epac: effectors and biological functions. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:345–57. doi: 10.1007/s00210-007-0246-7. [DOI] [PubMed] [Google Scholar]
- 29.Parnell E, Smith BO, Palmer TM, Terrin A, Zaccolo M, Yarwood SJ. Regulation of the inflammatory response of vascular endothelial cells by EPAC1. Br J Pharmacol. 2012;166:434–46. doi: 10.1111/j.1476-5381.2011.01808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oldenburger A, Timens W, Bos S, Smit M, Smrcka AV, Laurent AC, Cao J, Hylkema M, Meurs H, Maarsingh H, Lezoualc’h F, Schmidt M. Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J. 2014;28:4617–28. doi: 10.1096/fj.13-248930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie P, Joladarashi D, Dudeja P, Sun L, Kanwar YS. Modulation of angiotensin II-induced inflammatory cytokines by the Epac1-Rap1A–NHE3 pathway: implications in renal tubular pathobiology. Am J Physiol Renal Physiol. 2014;306:F1260–74. doi: 10.1152/ajprenal.00069.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mediero A, Perez-Aso M, Cronstein BN. Activation of EPAC1/2 is essential for osteoclast formation by modulating NFkappaB nuclear translocation and actin cytoskeleton rearrangements. FASEB J. 2014;28:4901–13. doi: 10.1096/fj.14-255703. [DOI] [PMC free article] [PubMed] [Google Scholar]