

Abstract
Fluorescence anisotropy measurements of reagents compartmentalized into individual nanoliter droplets are shown to yield high-resolution binding curves from which precise dissociation constants (Kd) for protein–peptide interactions can be inferred. With the current platform, four titrations can be obtained per minute (based on ∼100 data points each), with stoichiometries spanning more than 2 orders of magnitude and requiring only tens of microliters of reagents. In addition to affinity measurements with purified components, Kd values for unpurified proteins in crude cell lysates can be obtained without prior knowledge of the concentration of the expressed protein, so that protein purification can be avoided. Finally, we show how a competition assay can be set up to perform focused library screens, so that compound labeling is not required anymore. These data demonstrate the utility of droplet compartments for the quantitative characterization of biomolecular interactions and establish fluorescence anisotropy imaging as a quantitative technique in a miniaturized droplet format, which is shown to be as reliable as its macroscopic test tube equivalent.
Protein–ligand interactions interfere with, or promote, essential biological processes such as immune recognition, signal transduction or enzyme inhibition. Their quantitative investigation is the basis for systematic and mechanistic analyses of biological processes and for therapeutic strategies based on selective molecular intervention. A wide range of assays exists to assess the strength of binding interactions, including techniques based on fluorescence probes (e.g., lifetime,1 resonance energy transfer,2 and anisotropy3), on surface immobilization (e.g., surface plasmon resonance, biolayer interferometry), or on calorimetry (isothermal titration calorimetry, ITC). Each of these approaches have shortcomings: surface immobilization may affect the properties of the molecular binding partners (e.g., as a consequence of conformational changes or molecular crowding);4 ITC requires large volumes (typically hundreds of microliters) and highly concentrated reagents, precluding its use for precious and relatively insoluble samples.5 An attractive choice is fluorescence anisotropy (FA) that enables measurements in homogeneous solution (although it still requires one of the binding partners to be fluorescently labeled).
Here we introduce a system for the evaluation of binding interactions by FA in nanoliter water-in-oil droplets, achieving a ∼1000-fold decrease per assay volume (15 nL compared to typically >13 μL used in a 384-well plate), yet circumventing some shortcomings of the methods described above: (i) the assay read-out is independent of signal intensity; (ii) the need to label only one binding partner reduces the potential for interference of the label with protein function (e.g., by blocking binding sites). Furthermore, only a single binding partner has to be chemically conjugated to a fluorescence reporter, greatly simplifying the complexity of sample preparation over readouts where all binding partners have to be labeled (e.g., in assays exploiting fluorescence resonance energy transfer). Flow segmentation and microdroplet technology furthermore decrease assay volumes to the nano- or even femtoliter scale and speed up sample handling operations; they are therefore increasingly used for high-throughput studies.6 Recent attempts to establish fluorescence anisotropy assays in droplets7 involved the averaging the readout of thousands of droplets to obtain signal-to-noise ratios sufficiently precise for deriving quantitative dose–response curves. However, the method still required the total sample volumes to be in the microliter range. An attractive alternative approach to save reagents and increase throughput would be to obtain a quantitative readout from a single droplet.8 Here each droplet would correspond to a specified ligand/target stoichiometry and the evaluation of the binding event in each droplet would give rise to a binding curve. We achieve this by producing a series of single droplets9 with continuously varying reagent stoichiometries in rapid succession and implementing a highly sensitive readout system that provides the fraction of bound vs unbound protein. The resulting dose–response curve is the basis for the quantitative determination of binding constants.
We explore the potential of single droplet anisotropy measurements for the study of structure–activity relationships for protein–protein interactions in nanoliter volumes. Specifically, we address the interactions of the BRC4 peptide, a reductionist model of the recombination mediator BRCA2, with mutants of an archaeal surrogate of the recombinase RAD51.10 The protein interaction pair BRCA2/RAD51 (Figure 1A) is a potential drug target: it is relevant for monitoring cancer progression, as RAD51 levels are often upregulated in cancerous cells, conferring resistance to chemotherapy.11 Inhibition of the interaction of BRCA2 and RAD51 has the potential to sensitize cells to DNA damage, thus potentiating cancer chemotherapies. To facilitate drug discovery, the two binding partners have been converted into analogues that maintain the relevant interactions (i.e., are good functional mimics of RAD51), but for which it is easier to deconvolute the binding interactions and thus elucidate structure–activity relationships: (i) Eight BRC repeats have been identified in BRCA2 as interaction partners for RAD51 and a 30–35 amino acid peptide, corresponding to BRC repeat 4 (BRC4), has been shown to block RAD51 activity, suggesting a role as a cancer suppressor.12 (ii) For a fragment-based drug discovery campaign a monomeric variant, RadA-ct, was derived from an archaeal homologue (RadA) and humanized, to facilitate measurement of small molecule binding constants.13 RadA-ct and its humanized variants (HumRadAs) are much more stable than RAD51, enabling biophysical analysis of interactions and development of small molecule inhibitors of this protein–protein interactions pair. Here, we use a set of previously described HumRadA mutants with varying affinity for BRC4 peptide (from nonbinding to Kd of 6 nM) as representative examples to demonstrate that binding can be quantitatively assessed in nanolitre droplets. In addition to the determination of Kd, our formats for FA measurements in droplets also allow protein expression levels to be determined, so that cell lysates rather than purified components can be used, which simplifies preparation protocols in practical screening efforts significantly. Finally, we demonstrate that the method has the potential to screen libraries of competitive ligands effectively.
Figure 1.

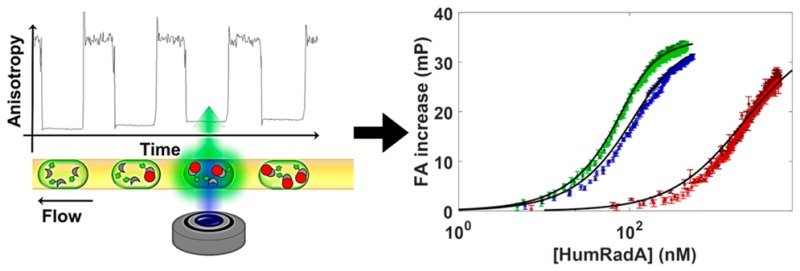
Schematic view of the quantitative binding assay for protein–protein association in nanoliter droplets. (A) Interaction between BRC4 (in gray) and RAD51 (of which “HumRadA” proteins are mimics; in red). A fluorescein tag (not shown) was added to the N-terminus of BRC4. (B) Schematic view of the titration in droplets. The droplets are produced to set up a concentration gradient of the respective HumRadA and are kept in sequence inside PTFE tubing (I.D. 200 μm), so that the ratio of HumRadA to BRC4fl increases. Spatial encoding preserves the concentration gradient: initial droplets contain only labeled peptide (BRC4fl), while final ones have an excess of the RAD51 analogue, i.e., [HumRadA] ≫ [BRC4fl], and thus, contain an increased proportion of HumRadA-BRC4fl complexes. The size difference between free and complexed species results in a change in tumbling rate that can be detected using fluorescence anisotropy. (C) Schematic view of the fluorescence anisotropy detection system developed for affinity determination. The microdroplet production setup includes one or four wells of a 384-well titerplate (1), containing a magnetic stirrer (2), and 40 μL of reagent solution (BRC4fl in CHES buffer, pH 9.5, 1% w/v BSA), a syringe pump injecting a second component (3) into the well (HumRadA premixed with BRC4fl in CHES buffer, pH 9.5, 1% w/v BSA) and a droplet-forming head made with a pipet tip (4) and inserted PTFE tubing (5). This assembly produces 10–20 nL droplets and encapsulates a concentration gradient. The PDMS microchannel device was bonded to a coverslip bottom (7) and droplets in the microchannel were imaged by the microscope with a syringe pump operating in withdrawal mode that pulls that row of droplets across. The optical setup includes a 488 nm diode laser, a linear polarizer, a multiedge dichroic filter (DM), a lens (L1) to focus fluorescence light on a polarization beam splitter, mirrors (M) directing signals with parallel and perpendicular polarizations toward a lens (L2) to generate spatially separate images on a single CCD camera chip. The microfluidic devices featured either one or four parallel channels. The 20× or 10× objectives of the microscope for the single and the four channel device, respectively, and enabled the imaging of the full width of the channels. (c.f. inset, right).
Materials and Methods
Optics
Polarization-resolved fluorescence imaging was performed on a Nikon TE-300 Eclipse microscope with a custom widefield detection system. A detailed description of the applied experimental setup can be found in ref (3). A diode laser (Cobolt, 488 nm emission wavelength) was coupled into the microscope through a fixed polarizer for illumination. Microscopy was performed with a multiedge band-pass dichroic mirror (MEBP, Semrock Di01-R405/488/561/647), a band-pass emission filter (530/43, Semrock), and a 10×, 0.3 NA or 20×, 0.5 NA objective lens. In the detection arm, a rectangular aperture was placed in the conjugate plane of the specimen to serve as a field stop (FS) to restrict the field of view. This was then relayed onto an Andor iXon DV885 EMCCD camera via a polarization beamsplitter as shown in Figure 1. The beamsplitter cube was aligned so that fluorescence polarized along the parallel and perpendicular directions (with respect to the illumination axis) were imaged onto separate areas of the camera. Image data were captured as 16 bit TIF stacks using the Andor Solis software. A frame rate of 84 Hz, with 10 ms exposure, was achieved via 8 × 8 binning of the camera pixels. A Matlab script was written to register the polarization-resolved images and calculate fluorescence anisotropy on a pixelwise basis. The mean anisotropy of image regions within the microdroplet flow cell was then obtained. Anisotropy values for each pixel i, j were obtained by using the following equation:
| 1 |
where I∥ and I⊥ represent the emitted fluorescence intensity measured for the parallel and perpendicular polarizations, respectively. The variable G represents the ratio of the detection sensitivities of the detector and is defined for a specimen with known anisotropy of zero in every pixel (we use a solution of freely tumbling fluorescein dye in water) as3
| 2 |
The G-factor as defined above provides calibration at the detector level so that each pixel would result in the same anisotropy value, when calculated according to eq 1 (see SI, Figure S1). It differs from G-factors used in cuvettes or plate readers for which a sample of known anisotropy is used to obtain absolute anisotropy values via measurement using two laser polarizations oriented at 90° with respect to one another.14
Fluidics
A droplet sampling system (described by Gielen et al.15) was used to set up a concentration gradient: 40 μL of protein in buffer were pipetted into a well in a 384-well plate together with a stir bar (2 × 2 mm, Fisher Scientific). The droplet maker was assembled using polytetrafluoroethylene (PTFE) tubing (I.D. 200 μm, O.D. 400 μm) inserted into a cut gel loading tip (Starlab, 200 μL round, bottom I.D. 360 μm, top I.D. 5 mm). This tip was then inserted into a polydimethylsiloxane (PDMS) slab with a hole of 1 mm in diameter. A total of 10 μL of HFE-7500 + 1% surfactant AZ2C was pipetted into the oil reservoir (see Figure S2).16 A typical flow withdrawal rate of 3 μL/min was used to initiate droplet generation. At this point, a second syringe pump was turned on to continuously increase the concentration of a second component and produce droplets of gradually increasing concentration of this second component. Concentrations for each droplet were derived using eq S1. At the end of the titrations, droplet formation was stopped and the sampling head immersed into an oil-containing well of the plate. The droplet sequence was then transported through the tubing toward the focal point of the microscope by using the withdrawal function of the pump again. PDMS channels were fabricated by soft lithography and bonded to thin coverslips: they allow droplets to transition from tubing to an area where they can be imaged (see Figure S3 for designs). Thus, the chip position did not need to be moved over time, ensuring consistent spatial registration of measurements for all the droplets in the sequence.
Preparation of Cell Lysates for Affinity Screens
HumRadA proteins were produced in 20 mL cultures. After addition of isopropyl-β-d-thiogalactopyranosid (IPTG; 400 μM), the temperature was reduced from previously 37 °C to 35 °C and cell cultures were incubated for 16 h. Next, the cells were centrifuged for 5 min at 10 000 rpm, the pellets resuspended in Luria–Bertani broth (LB) to equal an OD600 nm of 40. 3.7 × 109 cells (assuming an OD600 nm of 1 corresponds to 108 cells/mL) were transferred to a fresh tube, centrifuged (1 min, 14 000 rpm), the supernatant removed, and the pellets stored at −80 °C.
After thawing, the cells were lysed by resuspending the pellet in 200 μL 0.5× BugBuster in 20 mM N-cyclohexyl-2-aminoethanesulfonic acid (CHES), pH 9.5; containing 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) and 1% bovine serum albumin (BSA) and the mixture incubated for 15 min at 25 °C. The addition of BSA prevented significant leakage of BRC4fl into the oil phase (see SI, Figure S4). The cell debris was removed by centrifugation (20 min at 14 000 rpm). The supernatant was diluted with 400 μL of CHES (20 mM, pH 9.5, 100 mM NaCl, 1 mM EDTA, 1% BSA) to reduce destabilization of droplets by detergents in the BugBuster lysis agent. The solution was stored at room temperature until used for the titration. Lysates were always prepared on the day of the experiment.
MBP-BRC4 Fusion Protein Production
The plasmid pRSFDuet2-MBP-BRC4 (KanR), encoding a C-terminal fusion of BRC4 to maltose binding protein (MBP), bearing an N-terminal His6-tag was generously provided by Dr. L. Pellegrini and M. Longo, Cambridge University.17 Rosetta2 (DE3) cells (CmR) were transformed with pRSFDuet2-MBP-BRC4. A 10 mL preculture was prepared and used to inoculate 500 mL of LB with appropriate antibiotics. After incubation at 37 °C (200 rpm) until an OD600 of 0.6 was reached, the cells were induced with 400 μM IPTG. After a further 5 h at 37 °C (200 rpm) the cells were harvested. The pellet was resuspended in 15 mL of phosphate buffered saline (PBS, pH 7.5) and stored at −20 °C.
For protein purification, the cells were thawed, 0.5 μL benzonase added and lysed by sonication. The solution was centrifuged for 40 min at 6,500 rpm. The supernatant was loaded on a Ni-NTA (Qiagen) column previously equilibrated in PBS. The column was washed with 5 bed volumes of 10 mM imidazole, PBS pH 7.5, and 5 bed volumes of 20 mM imidazole in PBS pH 7.5 prior to elution with 2 bed volumes of 250 mM imidazole in PBS pH 7.5. The protein was concentrated with 30 MWCO spin concentrators (Millipore), while the buffer was exchanged to 20 mM CHES pH 9.5, 100 mM NaCl, 1 mM EDTA. The purified protein was stored at 4 °C.
Results and Discussion
Assessment of Binding Affinity by Fluorescence Anisotropy in a Single Droplet
Concentration gradients of one binding partner are necessary for recording binding isotherms. To this end droplets containing varying concentrations of purified HumRadA were generated using a simple microcapillary technique that produces droplets of defined concentrations in a spatially encoded sequence. To enable FA measurements between the binding partners, the smaller component, BRC4, was labeled at its N-terminal cysteine with fluorescein after peptide synthesis to give BRC4fl (see SI, S13.2). While the concentration of HumRadA concentration increased continuously, the concentration of labeled BRC4 was kept constant (100 or 20 nM). This led to a continuously increasing anisotropy value, because the tumbling motion of the complex is slower than that of the individual binding partners. The readout of this series of droplets with a varying ratio of BRC4fl and purified HumRadA represents a “titration” of the steady-state binding event (as schematically represented in Figure 1B).
This droplet sequence representing a concentration gradient of HumRadA was transported through a microchannel mounted onto the stage of an inverted microscope. The microchannel consists of a glass coverslip (thickness 130 μm) and a bonded rectangular PDMS channel of width 150 μm, height 220 μm, to which the droplets were delivered via PTFE tubing inserted into the side of the PDMS block (see Supporting Information, Figures S2B and S3). The side insertion enabled the droplets to flow smoothly from the tubing into the channel without problems caused by flow instabilities or separation of the confined plugs. This is crucial to preserve the spatial encoding of sample stoichiometry. The fluorescence anisotropy signal was read through the glass portion of the channel chip to maintain the polarization state of the emitted fluorescence, which would have been impossible via direct imaging through the PTFE tubing.
Frame-by-frame recordings of parallel/perpendicular fluorescence intensities enabled the extraction of the total intensity and anisotropy maps for a rectangular region of interest (ROI), as defined in Figure 2 (Videos S1 and S2, SI).
Figure 2.

Anisotropy values are extracted frame-by-frame from intensity maps in steps A–C. The subscripts “sc” and “4c” refer to the single channel or four parallel channel configurations, respectively (with the latter quadrupling the throughput). (A) Raw intensity images for parallel and perpendicular fluorescence channels obtained from a rectangular field of view within a single droplet containing 100 nM BRC4fl. (B) Anisotropy maps calculated from the raw data shown in (A). (C) Anisotropy histograms of a single frame used for data quantification. For a setup with four channels, ROIs were defined for each parallel channel. The transformation from intensity maps into anisotropy values was achieved using a Matlab code (https://github.com/quantitativeimaging/icetropy).
Determination of Kd Values for Protein–Protein Interactions
The process of quantifying data from FA measurements from individual droplets and mapping them to HumRadA concentration is shown in Figure 3. HumRadA samples were added to BRC4fl, while keeping the BRC4fl concentration constant. To ensure constant fluorescence intensity, a HumRadA solution containing BRC4fl was slowly titrated to a BRC4fl solution and droplets were generated sequentially (see Figure 3).
Figure 3.

Addition of HumRadA leads to increased anisotropy. (A) Frame-by-frame data extraction of both total intensity (blue) and mean anisotropy (green) for a chosen ROI (as depicted in Figure 2A). The experiment is conducted with increasing [HumRadA] and constant [BRC4fl]. (B) Anisotropy and intensity data corresponding to the dashed rectangle in (A). This trace corresponds to six individual droplets passing through the field of view. Periods of high brightness in the region of interest correspond to the presence of a droplet. The dashed regions in (B) indicate periods when the region of interest is filled by a droplet, during which it is typically possible to obtain 70 frames of image data once the data are trimmed to exclude frames where the region of interest is only partially filled by the droplet. The anisotropy of the droplet and the measurement uncertainty can be estimated as the mean and standard deviation of these 70 values. (C) Anisotropy values plotted as a function of theoretical concentration of protein in each droplet calculated from the time when the droplet picture was taken (frame number). Vertical error bars correspond to the standard deviation of the mean anisotropy for each droplet. The black trace is the best fit obtained using eq 3. (D) Four dose–response curves for HumRadA18 obtained in parallel with the four-channel device.
First, the start of each titration was identified by the first droplet for which fluorescence anisotropy was at least 0.5 mP above that of droplets containing BRC4fl only (Figure 3A). Concentrations for each droplet could be calculated analytically from the known flow parameters (initial volume Vi, flow rates of infusion and withdrawal, qin and qout, respectively), and the time the droplet was generated, using eq S1. For the flow conditions used here, minor mis-assignments of droplets and concentrations lead to negligible errors in calculated Kd. In practice we estimate the assignment to be precise to within 1 s, corresponding to a potential shift in Kd, on the order of 10%. Next, the average droplet-by-droplet anisotropy values were extracted (Figure 3B).
Anisotropy values for the ligand alone were found to be accurately quantified down to 5 nM, below which the signal got too weak (Figure S5). We also found that there was a significant effect of focal position within the droplet volume on the determination of anisotropy values (as seen in Figure S6 of the Supporting Information), which we ascribed to the presence of ligand at the interface oil/water with a high anisotropy contribution. However, the difference in the measured anisotropies between the fully bound and fully unbound scenarios was largely independent of focus and varied by less than 5% for all conditions stated here, as seen in Figure S7. Thus, the measurement of the difference provides a robust readout so that initial anisotropy values with fully unbound ligands can be subtracted from all readings. Experiments yielded comparable results, even when microfluidic devices were removed and replaced or between different flow-chip designs. The standard deviation of the mean anisotropy for each frame was found to be around 12 mP, whereas the standard deviation of the mean anisotropy for the accumulated 70 frames corresponding to a single droplet was around 8 mP (Figure S8). Thus, there is no benefit to be derived from using larger droplets or using multiple droplets containing the same concentration conditions to improve data quality.
Kd values were subsequently extracted using the following equation, in which binding equilibrium is assumed.18
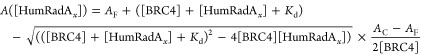 |
3 |
Here AC and AF are the anisotropy values for fully bound (AC), and fully unbound (AF) states, respectively, and [HumRadAx] refers to the concentration of the respective HumRadA protein variants.
The fit for HumRadA18 at 5 nM BRC4fl gave a Kd of 49.3 ± 0.5 nM (Figure 3C). In an analogous HumRadA18 titration in the parallelized setup featuring four channel device the Kd values were extracted as 52.0 ± 2.0, 55.9 ± 2.9, 49.1 ± 1.4, and 56.3 ± 3.0 nM, respectively, for a 100 nM concentration for BRC4fl. All measurements were within the error of the fit, suggesting minimal variation between different series of droplet concentrations and different positions in one device.
Titrations of HumRadA Variants
Next, three representative HumRadA proteins with affinities between 4 and 670 nM were titrated to demonstrate the ability of the method to rank variants. These HumRadA mutants were designed to be increasingly similar to human RAD51 and their affinity for BRC4 varies accordingly. Mutants HumRadA14 and HumRadA16 lack two polar residues, which interact directly with the BRC4 peptide. An A266R mutation in HumRadA16 introduces a salt-bridge with BRC4 residue E1548, while the acidic side chain of residue D198 introduced in HumRadA18 reinstates a polar interaction with BRC4 residue S1528; both of these mutations are found in HumRadA20 and HumRadA22, explaining why the highest BRC4 affinity is expected with these proteins.
To rank the affinity of these proteins, we expressed, purified and titrated them against pure BRC4fl. To this end, the concentration of BRC4fl was kept constant at either 100 or 5 nM for laser powers of 5 and 50 mW, respectively.
Figure 4 summarizes the dose–response curves obtained. As expected, at 100 nM BRC4fl, the dose–responses for HumRadA18 and HumRadA20 were well resolved, whereas HumRadA14 exhibited a significantly higher Kd. On the other hand, reducing BRC4fl concentration and using higher laser power (50 mW) permitted a clear differentiation between HumRadA18 and HumRadA20. The affinity of HumRadA14 was not determined at 5 nM BRC4fl because its Kd is too far from this concentration of ligand and would result in a shallow titration for the same range of protein concentrations with poor prospects for useful data fitting. Table 1 summarizes the Kd values obtained using a standard 96-well plate format in comparison to the data obtained by measurements in droplets.
Figure 4.

Titration of BRC4fl with different HumRadA variants. (A) Duplicate titrations of HumRadA 14 (red), HumRadA18 (blue), and HumRadA20 (green) at constant 100 nM BRC4fl. Kd values extracted from the fits to eq 1 indicate 15.9 ± 0.5, 34.5 ± 1.9, and 2015 ± 102 nM for HumRadA20, HumRadA18, and HumRadA14, respectively. (B) Titrations of HumRadA18 (blue) and HumRadA20 (green) at constant 5 nM BRC4fl. Kd values were found to be 11.0 ± 0.2 and 32.1 ± 0.4 nM for HumRadA20 and HumRadA18, respectively.
Table 1. Comparison of Dissociation Constants Kd Obtained from Fluorescence Anisotropy Measurements in Droplets (Droplets of 10–20 nL) and in a Titerplate (Well Volume: 100μL)a.
| Kd (nM) | dropletc (100 nM BRC4fl) |
||||
|---|---|---|---|---|---|
| HumRadA variants | 96-well plateb (10 nM BRC4fl) | repeat 1 | repeat 2 | avg | dropletc (5 nM BRC4fl) |
| HumRadA14 | 670 ± 12 | 2130 ± 100 | 1901 ± 22 | 2015 ± 102 | not determined |
| HumRadA18 | 10.7 ± 0.4 | 32.9 ± 1.3 | 36.2 ± 1.4 | 34.5 ± 1.9 | 32.1 ± 0.4 |
| HumRadA20 | 3.9 ± 0.2 | 14.6 ± 0.5 | 17.2 ± 0.2 | 15.9 ± 0.5 | 11.0 ± 0.2 |
Conditions in titerplate measurements: 100 μL of 100 nM BRC4fl at varying HumRadA concentrations; [CHES] = 20 mM, pH 9.5, 100 mM NaCl, 1 mM EDTA. Conditions in each droplet: 20 nL at 100 nM BRC4fl at varying HumRadA concentrations; 20 mM, pH 9.5, 100 mM NaCl, 1 mM EDTA, 1% BSA. All measurements were performed at room temperature. Standard deviations come from the fitting algorithm based on a single titration.
See ref (19).
The Kd values derived from droplet experiments are consistently around 3-fold higher than those obtained from titer plate assays, which may indicate residual leakage of BRC4fl as well as affinity of BRC4fl for the oil–water interface. Other oil/surfactant combinations might result in better retention properties for BRC4fl.20Figure S4 shows that fluorescence intensity does not plateau at 1% BSA, suggesting that we have not reached full retention yet and further improvements are possible. However, the ranking is entirely consistent between the droplet and the plate assay. Moreover, it has been reported that variations in Kd values using different techniques can result in up to 1 order of magnitude discrepancy.21
Simultaneous Determination of Protein Expression and Affinity by Screening of Cell Lysates
In order to simplify fluorescence anisotropy assays in droplets further, we designed an experimental setup in which affinity determination of HumRadA variants could be carried out without the need of purifying one of the two protein binding partners. We first verified the assumption that cell lysate components did not interfere with BRC4fl by varying the amount of cell lysate not expressing any HumRadA and measuring the FA increase (Figure S9). The FA signal was found not to change over the course of the titration (up to 45% lysate) within sensitivity limits for 100 nM BRC4fl and increase by 2.5 mP at 30% lysate at 20 nM, indicating little nonspecific interactions between BRC4 and other cellular components (accounting for less than 10% of the dynamic of the assay; ∼34 mP). Instead, lysates of E. coli cells expressing HumRadA were directly assayed. In contrast to the purified protein sample with a known protein concentration, two titrations have to be performed with two distinct concentrations of BRC4fl (Figure 5A). In order to probe the potential of lysate screenings to yield high quality Kd values, all five HumRadA variants, HumRadA14, 16, 18, 20, and 22, were expressed and their cell lysates analyzed. A global fitting algorithm can then be used to infer two unique linear correlations between stock protein concentration and Kd and the intersection of these two correlations gives a unique solution as shown in Figure 5B. Table 2 summarizes the Kd and protein concentrations found for all HumRadA variants (for primary data, see SI, Figure S10).
Figure 5.

Screening of cell lysates to derive dissociation constant Kd and protein concentration. (A) Two HumRadA22 cell lysates were titrated into 100 (blue) and 20 nM (red) BRC4fl. (B) Each curve fit results in a linear relationship between dissociation constant (Kd) and protein concentration ([HumRadA22]lysate). The intersection of the lines gives the only Kd and protein concentration value that solves both line equations derived from the titrations at 100 and 20 nM, respectively. (C) Example fits taken along the two correlation lines of (B). Only the intersect point denoted I gives an accurate fit for both curves ([HumRadA22]lysate = 653 ± 15 nM, Kd = 11 ± 0.5 nM).
Table 2. Dissociation Constants and Protein Concentrations Obtained from FA Measurements of Cell Lysates (SI, Figure S8)a.
| HumRadA | Kdb (nM) | Kdc (nM) | [HumRadA]d (nM) |
|---|---|---|---|
| HumRadA14 | 670 ± 12 | 910 ± 130 | 780 ± 110 |
| HumRadA16 | 294 ± 6 | 250 ± 7 | 273 ± 8 |
| HumRadA18 | 10.3 ± 0.4 | 17 ± 5 | 35 ± 4 |
| HumRadA20 | 3.9 ± 0.2 | 15 ± 2 | 831 ± 30 |
| HumRadA22 | 6.2 ± 0.3 | 11 ± 0.5 | 653 ± 15 |
Conditions: CHES 20 mM, pH 9.5, 100 mM NaCl, 1 mM EDTA, 1% BSA, T = 24 °C. Two titrations were performed at 100 and 20 nM ligand BRC4fl concentration, respectively, so that both Kd and expression levels were derived.
Taken from ref (19). Kd values determined with purified HumRadAs in microtiter plates.
Measured by FA analysis in lysate screens, as described in this work.
Concentration in lysate and determined by the lysate screen described here. Standard deviations for droplets measurements were obtained from the global fit from one single experiment.
An immediate advantage of this setup was that much less HumRadA sample was necessary to perform these measurements. Because only 40 μL cell lysate was needed to obtain a full titration curve, a cell culture of 20 mL provided sufficient material to measure Kd as well as the protein expression yield (c.f. Table 2).
The quality of estimates of the Kd and HumRadA concentration was found to depend on the two chosen BRC4fl concentrations that in turn determine how well the intersection is defined. Thus, linear correlations with very similar slopes lead to larger uncertainty. Therefore, the BRC4fl concentration should differ sufficiently (in our case, 5-fold), and, ideally, one concentration should be above and one below Kd. In addition, expression levels should be ideally much higher than Kd to reach saturation of binding conditions in order to get a good linear relationship between dissociation constant and protein concentration, as was indeed case for [HumRadA22]lysate = 577 nM compared to the measured Kd of 10 nM.
Despite the huge range of expression levels spanning a 30-fold concentration range, we obtained Kd values similar to those measured previously.19 However, as is the case for HumRadA14, HumRadA16, and HumRadA18, low expression or high Kd lead to higher statistical errors (below 25%). While HumRadA20 and 22 reverse their rank order in FA droplet assays compared to plate assays, this is not due to insufficient data quality: a low Kd and high protein expression lead to <6% error in Kd and concentration for these HumRadAs. These results can be rationalized by an error that is larger than the statistical error and which precludes the differentiation of binders as similar as HumRadA20 and 22 (∼2-fold Kd difference). Taken together, these observations suggest that FA measurements in droplets can produce data that match experiments on the microliter scale and also confirm the previous achievements of affinity enhancement of HumRadA variants by humanization of RadA.19 Errors made are larger when measuring affinities with Kd values substantially below or above ligand concentration BRC4fl, because this technique does not yield enough data points to fit the nonlinear data adequately.
Screening for BRC4 Competitors
BRC4 derivatives have potential as modulators of up-regulated RAD51 expression and an efficient method to obtain structure–activity relationships for RAD51 binders would thus be highly interesting. However, producing a large number of fluorescent peptides is expensive and time-consuming. Therefore, we established a competition assay, which evaluates the replacement of preincubated BRC4fl bound to HumRadA protein by unlabeled peptide. Competition by the fusion protein MBP-BRC4 (MBP) was tested, as shown schematically in Figure 6A. Although MBP-BRC4 does not have a higher affinity than BRC4fl for HumRadA it will outcompete BRC4fl at high concentrations. The MBP-BRC4 concentration was gradually increased across a sequence of droplets, while keeping the total concentration of BRC4fl and HumRadA18 constant.
Figure 6.

Competition assay performed in nanoliter droplets. (A) Schematic representation of the competition assay in nanoliter plugs. Both the receptor HumRadA18 and the labeled ligand BRC4fl were kept at constant concentration during the whole titration while the MBP-BRC4 concentration is increasing from 0 to 7 μM. (B) Competition of MBP-BRC4 construct with BRC4fl against HumRadA18. Binding curves were normalized to % of bound HumRadA18-BRC4fl. Two identical repeat titrations are overlaid. The well contained initially 40 μL of 40 nM BRC4fl and 60 nM HumRadA18 in CHES buffer pH 9.5, 1% BSA (w/v). The injection of purified MBP-BRC4 was done at a flow rate of 2.5 μL/min (for 1 min) followed by 17.5 μL/min (for 1 min). Vertical error bars correspond to the standard deviation of the mean anisotropy for each droplet.
The curves were plotted using eq S2 to transform anisotropy readings into percentages of binding (Figure 6B and SI, S11). We used the framework of the complete competitive binding model as described in reference 18. Out-competing BRC4fl at 40 nM for HumRadA18 at 60 nM with MBP-BRC4 gives a Kd of 110 ± 3 nM fitted to eq S3 (SI, S12).
This shows that the assay is able to quantitatively screen HumRadA18 binders with a singly labeled ligand. Even though the starting HumRadA18-BRC4fl bound fraction is below 50% to ensure efficient replacement of BRC4fl by BRC4-MBP, the high sensitivity of the platform is nonetheless capable of a reasonable quantification of interactions.
Summary
We demonstrate that fluorescence anisotropy can be performed with quantitative precision in nanoliter droplets, where each droplet encodes for a different protein/ligand stoichiometry. Each droplet can be analyzed individually and in rapid sequence to establish precise dose–response curves with small sample volumes (30–1000 droplets per titration) on very short time scales (minutes). This is in contrast to continuous droplet flow approaches which rely on massive signal averaging over many monoclonal droplets. Previously, it appeared to be necessary to average signals over very large number of droplets (>10 000)7 to obtain sufficient signal with FA and for the determination of a Kd, which meant that, despite the small volume of one droplet, such experiments consumed microliter total volumes (350 pL × 10 000 = 3.5 μL). Furthermore, to provide a sufficient number of data points for construction of a titration curve with continuous droplet flow approaches requires labor intensive reloading of syringes, frequent adjustment periods to equilibrate flow conditions and to ensure monodisperse droplet formation. Finally, adjusting mixing conditions through actively controlled variations of flow rates permits only a limited dynamic range to be obtained, typically less than 2 orders of magnitude: the droplet-on-demand systems in turn are able span several orders of magnitude.6c,22 Apart from device designs with classical T- or flow focusing junctions,23 the miniaturization of liquid-phase assays using FA below microliter volumes has been demonstrated in nanoliter microwells.24 To obtain binding curves containing 10 data points took 15 min in 48 × 48 nanoliter chamber arrays using a commercial microfluidic device.24 The approach was costly and required complex fluidics connections, while still relying on manual pipetting for each concentration point screened.
By contrast, in our experimental design one set of conditions is represented by a single droplet, so that a 200-fold reduction in reagent volume (3.5 μL/15 nL) is possible to obtain data points of comparable quality in a titration curve. Table 3 contrasts the quantitative descriptors of this design with experiments in microtiter plates and with continuous analysis of flowing droplets. Our approach achieves excellent sensitivity and data quality through careful design of the fluidic device and calibration of the optical anisotropy imaging platform, which incorporates an image-based registration and G-factor calibration.3 The precise determinations of dose response curves require minimal or no requirement for manual operator intervention. More data points can be generated per run (>100) and the overall run time required to perform a full titration experiment is very short (<1 min). The setup is relatively straightforward and can be multiplexed, as we have demonstrated with a simple four-channel device that quadruples the throughput. The feature of sampling droplets from an open well brings flexibility in setting up on-demand concentration gradients,6c,22a for instance, through use of sequential injection patterns.
Table 3. Performance Comparison of Different Formats for Anisotropy Measurementsa.
| format | No. of measurements for each sample composition | volume consumed for one sample composition (nL) | total sample volume (nL) | typical number of sample compositions per curve | total volume per titration (μL) | ref |
|---|---|---|---|---|---|---|
| 96-well plates | 1 | 100 000b | 100 000 | 12e | 1200 | |
| 384-well plates | 1 | 13 000b | 13 000 | 24e | 312 | |
| continuous analysis of multiple droplets | 10 000 | 0.35c | 3500d | 10 | 35 | (7) |
| single droplet on-demand analysis | 1 | 15c | 15 | 100 | 1.5 | this work |
Conventional microtiter plate screen, continuous multidroplet analysis, and the analysis of single nanoliter droplets presented in this work. The term “sample composition” corresponds, for example, to one concentration in a binding curve or a Michaelis-Menten plot.
Microwell volume.
Droplet volume.
Volume of 10000 droplets.
Number of wells in one row of a plate.
Using the current implementation of the droplet sampler, the overall volume consumed for one high-resolution titration was 40 μL of HumRadA variant (at a concentration ideally 10 times above Kd in order to reach signal saturation), spiked with 100 nM BRC4fl and 40 μL of pure BRC4fl (at 100 nM). Each droplet had a volume of 15 nL (compared to 13 μL needed for a standard 384-well titer plate format, ∼1000-fold volume reduction). Such small volumes enabled the screening of cell lysates produced in 20 mL cultures (instead of 1.3 L culture for the equivalent titration in a 96-well screen), further reducing screening cost and effort. We showed that extracting the two key parameters, Kd and level of protein expression, was possible via acquisition of just two high-resolution titrations. In addition, the FA signals from the lysates were found to be highly specific to HumRadA-BRC4fl interactions, potentially paving the way for diagnostic applications from more complex sample matrices (e.g., bodily fluids).25 The ability to determine precise FA signals in single droplets also provides the basis for coupling detection with library selections in formats in which droplets cocompartmentalize genotype and phenotype.26 Anisotropy detection would expand the range of assays that can be used for library selections in protein engineering by directed evolution27 or metagenomic screening,8c to enable not only assays that lead to production of fluorophores (e.g., as leaving groups8b), but also, assays that instead detect size changes by fluorescence anisotropy (e.g., for enzymatic breakdown of macromolecular targets by proteases of glycosidases).
Ultimately the sensitivity of the technique is limited by the optical setup (detector quality, laser power), but we could readily detect low nM binders at high laser powers (50 mW) and 100 ms integration time. The precision of anisotropy measurement (and, hence, the precision with which binding fractions can be estimated) is limited by the fluorescence signal from the reagents, which stray background light levels (e.g., light scattered from the oil droplet interfaces). In practice, this can be achieved down to nM concentration.
Additionally, we showed that day-to-day comparisons of FA binding curves were possible via G-factor calibration and simple subtraction of the “base” anisotropy values of a calibration sample (the freely tumbling labeled ligand), without the need for further normalizations, which have potential to introduce bias.
We established this approach as a mature technology that was able to rank the known affinities of HumRadAs, humanized forms of RadA, and established a method for quickly characterizing further variants using cell lysates. We also showed that an MBP-BRC construct can be used to set up libraries of BRC mutants, potentially able to out-compete the already tight-binding BRC4; a better binder would be highly valuable for benchmarking of drug studies as well as going further toward the understanding of BRCA2-RAD51 interactions.
The affinity determination between a small ligand and proteins is key to drawing structure–activity relationships. The use of fluorescence anisotropy is highly attractive for biological experiments, because only a single labeling step required. As we shown here, it also enables liquid-phase assays in complex mixtures such as cell lysates, quantifying dissociation constants from nanoliter droplets with uncomplicated microfluidics and integration with a standard fluorescence microscope. The technique can readily detect low nM binders. Parallelization of the assays enables the acquisition of 50 titrations per day with hundreds of data points each across over 2 orders of magnitude. The number of potential targets for which FA is routinely used is vast, ranging from the detection of interactions of small binding motifs such as SH2 domains28 or STATs29 to the screening of small molecule drug,30 with particular advantages conferred by the method if the samples are contained in complex solutions.31 More broadly, the rapid advances in high-throughput biology32 and its applications, for example, in directed evolution,27 single cell biology, and diagnostics33 suggest a prominent future role of quantitative single-droplet analysis based on FA.
Acknowledgments
This research was funded by the Engineering and Physical Sciences Research Council (EPSRC), the Wellcome Trust, the Medical Research Council (MRC), and Alzheimer Research U.K. M.B. was supported by a fellowship from the Schweizerischer Nationalfonds. F.H. is an ERC Investigator. The authors thank Stephane Emond, Charlotte Miton, and Liisa van Vliet for helpful discussions and comments and Mike Longo for the plasmid encoding MBP-BRC4.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b02528.
Author Present Address
# AstraZeneca, Innovative Medicines Discovery Science, Mereside, Alderley Park, Macclesfield, Cheshire SK10 4TG, U.K.
Author Contributions
‡ These authors contributed equally (F.G. and M.B.).
The authors declare no competing financial interest.
Supplementary Material
References
- Popleteeva M.; Haas K. T.; Stoppa D.; Pancheri L.; Gasparini L.; Kaminski C. F.; Cassidy L. D.; Venkitaraman A. R.; Esposito A. Opt. Express 2015, 23, 23511–25. 10.1364/OE.23.023511. [DOI] [PubMed] [Google Scholar]
- Chen W. Y.; Avezov E.; Schlachter S. C.; Gielen F.; Laine R. F.; Harding H. P.; Hollfelder F.; Ron D.; Kaminski C. F. Biophys. J. 2015, 108, 999–1002. 10.1016/j.bpj.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdelyi M.; Simon J.; Barnard E. A.; Kaminski C. F. PLoS One 2014, 9, e100526. 10.1371/journal.pone.0100526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sota H.; Hasegawa Y.; Iwakura M. Anal. Chem. 1998, 70, 2019–2024. 10.1021/ac9713666. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Johansson J. S. Biophys. J. 2003, 85, 3279–85. 10.1016/S0006-3495(03)74746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kintses B.; van Vliet L. D.; Devenish S. R.; Hollfelder F. Curr. Opin. Chem. Biol. 2010, 14, 548–55. 10.1016/j.cbpa.2010.08.013. [DOI] [PubMed] [Google Scholar]; b Schneider T.; Kreutz J.; Chiu D. T. Anal. Chem. 2013, 85, 3476–82. 10.1021/ac400257c. [DOI] [PMC free article] [PubMed] [Google Scholar]; c van Vliet L. D.; Colin P. Y.; Hollfelder F. Interface Focus 2015, 5, 20150035. 10.1098/rsfs.2015.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Joensson H. N.; Zhang C.; Uhlen M.; Andersson-Svahn H. Electrophoresis 2012, 33, 436–439. 10.1002/elps.201100350. [DOI] [PubMed] [Google Scholar]; b Choi J. W.; Kang D. K.; Park H.; deMello A. J.; Chang S. I. Anal. Chem. 2012, 84, 3849–3854. 10.1021/ac300414g. [DOI] [PubMed] [Google Scholar]
- a Huebner A.; Srisa-Art M.; Holt D.; Abell C.; Hollfelder F.; deMello A. J.; Edel J. B. Chem. Commun. 2007, 1218–20. 10.1039/b618570c. [DOI] [PubMed] [Google Scholar]; b Kintses B.; Hein C.; Mohamed M. F.; Fischlechner M.; Courtois F.; Laine C.; Hollfelder F. Chem. Biol. 2012, 19, 1001–9. 10.1016/j.chembiol.2012.06.009. [DOI] [PubMed] [Google Scholar]; c Colin P. Y.; Kintses B.; Gielen F.; Miton C. M.; Fischer G.; Mohamed M. F.; Hyvonen M.; Morgavi D. P.; Janssen D. B.; Hollfelder F. Nat. Commun. 2015, 6, 10008. 10.1038/ncomms10008. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Huebner A.; Olguin L. F.; Bratton D.; Whyte G.; Huck W. T.; de Mello A. J.; Edel J. B.; Abell C.; Hollfelder F. Anal. Chem. 2008, 80, 3890–6. 10.1021/ac800338z. [DOI] [PubMed] [Google Scholar]
- Basova E. Y.; Foret F. Analyst 2015, 140, 22–38. 10.1039/C4AN01209G. [DOI] [PubMed] [Google Scholar]
- Pellegrini L.; Yu D. S.; Lo T.; Anand S.; Lee M.; Blundell T. L.; Venkitaraman A. R. Nature 2002, 420, 287–293. 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- a Jeyasekharan A. D.; Liu Y.; Hattori H.; Pisupati V.; Jonsdottir A. B.; Rajendra E.; Lee M.; Sundaramoorthy E.; Schlachter S.; Kaminski C. F.; Rosenfeld Y.; Sato K.; Savill J.; Ayoub N.; Venkitaraman A. R. Nat. Struct. Mol. Biol. 2013, 20, 1191. 10.1038/nsmb.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nomme J.; Renodon-Corniere A.; Asanomi Y.; Sakaguchi K.; Stasiak A. Z.; Stasiak A.; Norden B.; Tran V.; Takahashi M. J. Med. Chem. 2010, 53, 5782–5791. 10.1021/jm1002974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole D. J.; Rajendra E.; Roberts-Thomson M.; Hardwick B.; McKenzie G. J.; Payne M. C.; Venkitaraman A. R.; Skylaris C. K. PLoS Comput. Biol. 2011, 7, e1002096. 10.1371/journal.pcbi.1002096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. E.; Ehebauer M. T.; Pukala T.; Marsh M.; Blundell T. L.; Venkitaraman A. R.; Abell C.; Hyvonen M. ChemBioChem 2013, 14, 332–42. 10.1002/cbic.201200521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan F. T. S.; Kaminski C. F.; Schierle G. S. K. ChemPhysChem 2011, 12, 500–509. 10.1002/cphc.201000833. [DOI] [PubMed] [Google Scholar]
- Gielen F.; Buryska T.; Van Vliet L.; Butz M.; Damborsky J.; Prokop Z.; Hollfelder F. Anal. Chem. 2015, 87, 624–632. 10.1021/ac503336g. [DOI] [PubMed] [Google Scholar]
- Zinchenko A.; Devenish S. R.; Kintses B.; Colin P. Y.; Fischlechner M.; Hollfelder F. Anal. Chem. 2014, 86, 2526–2533. 10.1021/ac403585p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschetti T.; Longo M. A.; Pellegrini L.. 2016, Manuscript in preparation.
- Roehrl M. H. A.; Wang J. Y.; Wagner G. Biochemistry 2004, 43, 16056–16066. 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- Moschetti T.; Sharpe T.; Fischer G.; Marsh M. E.; Ng H.; Morgan M.; Scott D.; Blundell T. L.; Venkitaraman A.; Skidmore J.; Abell C.; Hyvönen M. J. Mol. Biol. 2016, 428 (23), 4589–4607. 10.1016/j.jmb.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner P.; Riechers B.; Semin B.; Lim J.; Johnston A.; Short K.; Baret J. C. Nat. Commun. 2016, 7, 10392. 10.1038/ncomms10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhop D. J.; Latham J. C.; Kussrow A.; Markov D. A.; Jones R. D.; Sorensen H. S. Science 2007, 317, 1732–6. 10.1126/science.1146559. [DOI] [PubMed] [Google Scholar]
- a Gielen F.; van Vliet L.; Koprowski B. T.; Devenish S. R.; Fischlechner M.; Edel J. B.; Niu X.; deMello A. J.; Hollfelder F. Anal. Chem. 2013, 85, 4761–9. 10.1021/ac400480z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Niu X.; Gielen F.; Edel J. B.; deMello A. J. Nat. Chem. 2011, 3, 437–42. 10.1038/nchem.1046. [DOI] [PubMed] [Google Scholar]
- a Theberge A. B.; Courtois F.; Schaerli Y.; Fischlechner M.; Abell C.; Hollfelder F.; Huck W. T. Angew. Chem., Int. Ed. 2010, 49, 5846–68. 10.1002/anie.200906653. [DOI] [PubMed] [Google Scholar]; b Seemann R.; Brinkmann M.; Pfohl T.; Herminghaus S. Rep. Prog. Phys. 2012, 75, 016601. 10.1088/0034-4885/75/1/016601. [DOI] [PubMed] [Google Scholar]
- Cheow L. F.; Viswanathan R.; Chin C. S.; Jennifer N.; Jones R. C.; Guccione E.; Quake S. R.; Burkholder W. F. Anal. Chem. 2014, 86, 9901–9908. 10.1021/ac502605f. [DOI] [PubMed] [Google Scholar]
- Choi K.; Mudrik J. M.; Wheeler A. R. Anal. Bioanal. Chem. 2015, 407, 7467–7475. 10.1007/s00216-015-8913-x. [DOI] [PubMed] [Google Scholar]
- Leemhuis H.; Stein V.; Griffiths A. D.; Hollfelder F. Curr. Opin. Struct. Biol. 2005, 15, 472–8. 10.1016/j.sbi.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Colin P. Y.; Zinchenko A.; Hollfelder F. Curr. Opin. Struct. Biol. 2015, 33, 42–51. 10.1016/j.sbi.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Nieto L.; Tharun I. M.; Balk M.; Wienk H.; Boelens R.; Ottmann C.; Milroy L. G.; Brunsveld L. ACS Chem. Biol. 2015, 10, 2624–32. 10.1021/acschembio.5b00568. [DOI] [PubMed] [Google Scholar]
- Leung K. H.; Liu L. J.; Lin S.; Lu L. H.; Zhong H. J.; Susanti D.; Rao W. D.; Wang M. D.; Che W. I.; Chan D. S. H.; Leung C. H.; Chan P. W. H.; Ma D. L. Methods 2015, 71, 38–43. 10.1016/j.ymeth.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Zhai D. Y.; Godoi P.; Sergienko E.; Dahl R.; Chan X.; Brown B.; Rascon J.; Hurder A.; Su Y.; Chung T. D. Y.; Jin C. F.; Diaz P.; Reed J. C. J. Biomol. Screening 2012, 17, 350–360. 10.1177/1087057111429372. [DOI] [PubMed] [Google Scholar]
- Mathias U.; Jung M. Anal. Bioanal. Chem. 2007, 388, 1147–1156. 10.1007/s00216-007-1351-7. [DOI] [PubMed] [Google Scholar]
- Schaerli Y.; Hollfelder F. Mol. BioSyst. 2009, 5, 1392–404. 10.1039/b907578j. [DOI] [PubMed] [Google Scholar]
- Lach S.; Yoon S. M.; Grzybowski B. A. Chem. Soc. Rev. 2016, 45, 4766–96. 10.1039/C6CS00242K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.