

Abstract
Although interactions between superoxide (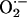 ) and nitric oxide underlie many physiologic and pathophysiologic processes, regulation of this crosstalk at the enzymatic level is poorly understood. Here, we demonstrate that xanthine oxidoreductase (XOR), a prototypic superoxide
) and nitric oxide underlie many physiologic and pathophysiologic processes, regulation of this crosstalk at the enzymatic level is poorly understood. Here, we demonstrate that xanthine oxidoreductase (XOR), a prototypic superoxide 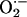 -producing enzyme, and neuronal nitric oxide synthase (NOS1) coimmunoprecipitate and colocalize in the sarcoplasmic reticulum of cardiac myocytes. Deficiency of NOS1 (but not endothelial NOS, NOS3) leads to profound increases in XOR-mediated
-producing enzyme, and neuronal nitric oxide synthase (NOS1) coimmunoprecipitate and colocalize in the sarcoplasmic reticulum of cardiac myocytes. Deficiency of NOS1 (but not endothelial NOS, NOS3) leads to profound increases in XOR-mediated 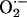 production, which in turn depresses myocardial excitation–contraction coupling in a manner reversible by XOR inhibition with allopurinol. These data demonstrate a unique interaction between a nitric oxide and an
production, which in turn depresses myocardial excitation–contraction coupling in a manner reversible by XOR inhibition with allopurinol. These data demonstrate a unique interaction between a nitric oxide and an 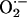 -generating enzyme that accounts for crosstalk between these signaling pathways; these findings demonstrate a direct antioxidant mechanism for NOS1 and have pathophysiologic implications for the growing number of disease states in which increased XOR activity plays a role.
-generating enzyme that accounts for crosstalk between these signaling pathways; these findings demonstrate a direct antioxidant mechanism for NOS1 and have pathophysiologic implications for the growing number of disease states in which increased XOR activity plays a role.
There is accumulating evidence that xanthine oxidoreductase (XOR), an enzyme involved in the final steps of purine metabolism and an important source of superoxide (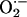 ), plays essential signaling roles within the cardiovascular system. XOR activity is up-regulated within the cardiovascular system of both experimental animals (1–3) and humans (4) with congestive heart failure leading to increased oxidative stress (OS) (5), vascular dysfunction (6), depressed myofilament contractility (7), whole heart contractile depression (8), and mechanoenergetic uncoupling (ref. 1 and see ref. 9 for review). As such, inhibition of XOR has emerged as a potential therapeutic target in the treatment of heart failure (4). Despite these key roles and pathophysiologic implications, relatively little is known about the cellular/molecular regulation of cardiac XOR and its dysregulation in disease.
), plays essential signaling roles within the cardiovascular system. XOR activity is up-regulated within the cardiovascular system of both experimental animals (1–3) and humans (4) with congestive heart failure leading to increased oxidative stress (OS) (5), vascular dysfunction (6), depressed myofilament contractility (7), whole heart contractile depression (8), and mechanoenergetic uncoupling (ref. 1 and see ref. 9 for review). As such, inhibition of XOR has emerged as a potential therapeutic target in the treatment of heart failure (4). Despite these key roles and pathophysiologic implications, relatively little is known about the cellular/molecular regulation of cardiac XOR and its dysregulation in disease.
Recently, we demonstrated important crosstalk between XOR and cardiac nitric oxide (NO) signaling activity (1). In intact animals, nonspecific inhibition of NO synthase (NOS) stimulated XOR activity causing cardiac dysfunction. Indeed, earlier studies had suggested that NO inhibits XOR activity (10, 11). Accordingly, the aims of the present study were to test the hypothesis that endogenous cardiac NOSs exert tonic inhibition over cardiac XOR. Here, we show selective biochemical and functional regulation by neuronal NOS (NOS1) of cardiac XOR and demonstrate spatially confined interactions between NOS1 and XOR. Importantly, endothelial NOS (NOS3) exerted no apparent regulation of XOR. These findings offer important insights into mechanisms by which the physiologic balance between 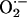 and NO becomes disrupted in the cardiovascular system.
and NO becomes disrupted in the cardiovascular system.
Methods
Preparation of Purifed Cardiac Sarcoplasmic Reticulum (SR). We prepared SR fractions by using modifications of the methods of Xu et al. (12). Whole hearts from C57BL6 mice (WT, n = 6) were pooled and homogenized three times (15 s each time) in a polytron in three volumes of ice-cold 1× lysis buffer (Cell Signaling Technology, Beverly, MA; catalog no. 9803) with a protease inhibitor mixture (Roche Applied Science; catalog no. 1836170). Homogenates were centrifuged at 1,000 × g for 20 min, and the supernatant was filtered through three layers of gauze, centrifuged at 10,000 × g for 20 min, and then centrifuged again at 200,000 × g. The final pellet containing the crude microsomal fraction was resuspended in the lysis buffer and further fractionated on a five-step gradient consisting of 44%, 40%, 36%, 32%, and 28% sucrose. The gradients were centrifuged for 6 h at 103,700 × g, and fractions were removed, diluted in two volumes of 0.4 M KCl, and recentrifuged for 90 min at 100,000 × g. Pellets were resuspended in lysis buffer and stored at –70°C. Purified SR fractions were resolved electrophoretically and probed with anti-XOR (NeoMarkers, Fremont, CA), anti-SR Ca2+ ATPase (anti-SERCA2a, Affinity Bioreagents, Golden, CO), and anti-NOS1 (BD Transduction Laboratories, Lexington, KY) antibodies.
Immunoprecipitation and Western Blots. To analyze whether XOR is associated with either NOS1 or NOS3, we first immunoprecipitated total heart proteins (lysates) with antibodies directed against these proteins (Transduction Laboratories) by using protein G, resolved these proteins by electrophoresis, and then probed with a monoclonal anti-XOR raised against the C-terminal 358-aa fragment of human xanthine oxidase (NeoMarkers) antibody.
To compare levels of XOR protein expression, we performed Western blot analysis on total heart proteins from WT, NOS3–/–, and NOS1–/– mice (2–3 months old) as described in detail by our group (13), and probed with the monoclonal anti-XOR antibody (1:1,000 dilution). A polyclonal anti-p38 mitogen-activated protein kinase antibody (1:1,000 dilution, Cell Signaling Technology) was used separately to normalize potential differences across samples.
mRNA Expression. To compare the levels of XOR mRNA, we performed quantitative PCR on hearts from WT (n = 3), NOS3–/– (n = 3), and NOS1–/– (n = 3) mice (2–3 months old). Total RNA was isolated, cDNA was synthesized, and each sample was run in duplicate on a GeneAmp 7900 Sequence Detection System (PE Applied Biosystems, Foster City, CA) and was analyzed by using sds 2.0 software (Applied Biosystems) as described (14, 15). The primer sequences for XOR and GAPDH (a housekeeping gene was used to standardize the input cDNA and provide a reference for our gene of interest) are as follows: XOR (GenBank accession no. NM_011723), forward (F) (1693–1712) TCCAGCTAACGTCCAGCTTT and reverse (R) (1910–1889) TGGCTTCTGAAGTGTCGAT; GAPDH (GenBank accession no. XM_146433), F (639–660) GGTTGTCTCCTGCGACTTCAAC and R (741–717) ATACCAGGAAATGAGCTTGACAAAG.
Isolated Myocyte Preparation. Myocytes were isolated and prepared from 2-month-old mouse hearts as described in detail by Khan et al. (13). Sarcomere length (SL) and Ca2+ transient ([Ca2+]i) were measured in myocytes stimulated at 2–8 Hz. Caffeine (10 mM) was rapidly infused after a 2- to 3-s pause after steady-state stimulation at increasing rates. Experiments were then repeated with the addition of allopurinol (10–4 M, DSM Pharmaceuticals, Greenville, NC) at a concentration where selective XOR inhibition occurs. All experiments were conducted at 37°C and in room air (PO2 ≈ 20%); increased O2 concentrations are known to disrupt NO signaling particularly its influence over the ryanodine receptor (RYR) (16).
Measurement of 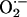 Production. We used lucigenin-enhanced chemiluminescence to measure
Production. We used lucigenin-enhanced chemiluminescence to measure 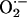 levels in cardiac tissue from WT (n = 9), NOS3–/– (n = 7), and NOS1–/– (n = 7) mice. Cardiac tissue (1 × 3 mm slices of the left ventricle) was transferred into scintillation vials containing Krebs-Hepes buffer (pH 7.4 after 60 min aeration with 95% O2/5%CO2) and 5 μmol/liter lucigenin (bis-N-methylacridinium nitrate), which illuminates in the presences of superoxide. Chemiluminescence was recorded by a luminometer (Monolight 2010) reporting relative light units (RLU) emitted over 2-min intervals. Basal levels of
levels in cardiac tissue from WT (n = 9), NOS3–/– (n = 7), and NOS1–/– (n = 7) mice. Cardiac tissue (1 × 3 mm slices of the left ventricle) was transferred into scintillation vials containing Krebs-Hepes buffer (pH 7.4 after 60 min aeration with 95% O2/5%CO2) and 5 μmol/liter lucigenin (bis-N-methylacridinium nitrate), which illuminates in the presences of superoxide. Chemiluminescence was recorded by a luminometer (Monolight 2010) reporting relative light units (RLU) emitted over 2-min intervals. Basal levels of 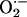 , the chemiluminescence of lucigenin-containing buffer with tissue minus background, as well as responses to xanthine (10–4 M; Sigma) and allopurinol (10–4 M; 1-min incubation) were reported.
, the chemiluminescence of lucigenin-containing buffer with tissue minus background, as well as responses to xanthine (10–4 M; Sigma) and allopurinol (10–4 M; 1-min incubation) were reported. 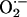 production is expressed as RLU per mg of dry tissue.
production is expressed as RLU per mg of dry tissue.
Oxidative Fluorescent Microtopography Using the Fluorescent Probes Dihydroethidium (DHE) and Diaminofluorescein (DAF). Fresh, unfixed heart segments from 2- to 3-month-old NOS1–/–, NOS3–/–, and WT mice were frozen. Transverse sections (14 μm thick) were cut in a cryostat and placed on glass slides. Samples were incubated at room temperature for 5 min with DHE (0.1 mmol/liter) and protected from light. After washing with PBS, images were obtained by using an Olympus fluorescent microscope. The fluorescence excitation/emission spectrum for ethidium bromide was used during the imaging process (488 and 610 nm, respectively). Fluorescence was detected with a 585-nm long-pass filter. Allopurinol (0.2 mg/ml) was applied in the buffer together with DHE and incubated for 5 min, and then images were obtained. Additionally, images dual labeled with DHE (0.1 mmol/liter) and DAF (a NO-specific probe; 10 μmol/liter) were obtained after a 10-min incubation.
Statistical Analysis. Data are reported as mean ± SEM. Statistical significance was determined by ANOVA and Student–Newman–Keuls post hoc tests (GraphPad instat and sas statistical software). P values < 0.05 were considered significant.
Results
Subcellular Localization of Xanthine Oxidase. Although XOR is present in the cardiac myocyte, its subcellular location and protein associations are not known. We first tested whether XOR and NOSs coimmunoprecipitate in the cardiac myocyte. Indeed, XOR was detectable in NOS1 but not NOS3 immunoprecipitate (Fig. 1a). To further address subcellular location, we probed cardiac SR fractions, the organelle in which NOS1 is physiologically located, for XOR. Indeed, SR fractions prepared by means of sucrose gradient centrifugation contained the 150-kDa XOR monomer, NOS1, and SR Ca2+ ATPase (SERCA2a) (Fig. 1b). Thus, XOR and NOS1 are both physiologically present in the SR and coimmunoprecipitate, suggesting a protein–protein interaction.
Fig. 1.

Subcellular localization of XOR. (a) Coimmunoprecipitation demonstrates association between NOS1 and XOR proteins. XOR coimmunoprecipitates with NOS1 (bands 1, 4, and 7) but not with NOS3 (bands 3, 6, or 9). The lysate bands (bands 2, 5, and 8) represent total heart protein extract revealing little XOR protein. (b) Purified SR fractions were probed with anti-XOR, anti-SERCA2a, and anti-NOS1 antibodies. As depicted, the 150-kDa monomer of XOR appears in the SR, with highest intensity in lane 4. Both NOS1 and SERCA2a are identified in the same fractions. TP, total heart protein; M, microsomal fraction; RC, rat cerebellum.
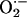 Production. We next examined XOR-directed
Production. We next examined XOR-directed 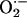 production with lucigenin-enhanced chemiluminescence. Basal chemiluminescence recordings in whole heart extracts were ≈60% higher in NOS1–/– (124 ± 16 arbitrary units) compared to WT (75 ± 12, P < 0.05 vs. NOS1–/–) hearts (Fig. 2a). Incubation with the XOR substrate xanthine produced dramatic increases (≈100-fold) in
production with lucigenin-enhanced chemiluminescence. Basal chemiluminescence recordings in whole heart extracts were ≈60% higher in NOS1–/– (124 ± 16 arbitrary units) compared to WT (75 ± 12, P < 0.05 vs. NOS1–/–) hearts (Fig. 2a). Incubation with the XOR substrate xanthine produced dramatic increases (≈100-fold) in 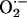 production, which was prevented by the XOR inhibitor allopurinol. Importantly, the increase due to xanthine was 4-fold greater in NOS1–/– compared with WT hearts, indicating that NOS1 deficiency augments cardiac XOR
production, which was prevented by the XOR inhibitor allopurinol. Importantly, the increase due to xanthine was 4-fold greater in NOS1–/– compared with WT hearts, indicating that NOS1 deficiency augments cardiac XOR 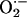 production. (Fig. 2b; P < 0.05). This increase was selective for NOS1 deficiency, because NOS3–/– mice had basal and xanthine-stimulated values similar to WT. Increased
production. (Fig. 2b; P < 0.05). This increase was selective for NOS1 deficiency, because NOS3–/– mice had basal and xanthine-stimulated values similar to WT. Increased 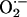 production in NOS1–/– cardiac myocytes was also demonstrated by oxidative fluorescent microtopography using the fluorescent probe DHE (orange staining) (Fig. 2c). Panels combining DHE with an additional NO-specific probe diaminofluorescein (DAF, green staining) demonstrate that NOS1–/– hearts have decreased NO activity accompanying increased OS. Increased XOR-stimulated
production in NOS1–/– cardiac myocytes was also demonstrated by oxidative fluorescent microtopography using the fluorescent probe DHE (orange staining) (Fig. 2c). Panels combining DHE with an additional NO-specific probe diaminofluorescein (DAF, green staining) demonstrate that NOS1–/– hearts have decreased NO activity accompanying increased OS. Increased XOR-stimulated 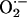 production in NOS1–/– was not caused by increased expression or abundance of XOR (Fig. 3 a and b), suggesting direct modulation of XOR activity by NOS1.
production in NOS1–/– was not caused by increased expression or abundance of XOR (Fig. 3 a and b), suggesting direct modulation of XOR activity by NOS1.
Fig. 2.

Superoxide production in cardiac tissue from NOS knockout mice. (a) Basal lucigenin-enhanced chemiluminescence recordings were elevated in NOS1–/– (n = 7) but not NOS3–/– (n = 7) compared to WT (n = 9) hearts (*, P < 0.05 vs. WT). (b) Incubation with xanthine (filled bars) produced dramatic increases in lucigenin-detected 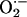 . Importantly, this increase was 4-fold greater in NOS1–/– compared with WT and NOS3–/– hearts (†, P < 0.05; n = 4 mice of each strain), indicating that NOS1 deficiency leads to augmented cardiac XOR
. Importantly, this increase was 4-fold greater in NOS1–/– compared with WT and NOS3–/– hearts (†, P < 0.05; n = 4 mice of each strain), indicating that NOS1 deficiency leads to augmented cardiac XOR 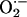 production. Inhibition by allopurinol (striped bars) demonstrates that this increase is caused by XO production of
production. Inhibition by allopurinol (striped bars) demonstrates that this increase is caused by XO production of 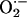 . (c) Oxidative fluorescent microtopography using the fluorescent probe DHE demonstrates increased staining in NOS1–/– (orange-staining nuclei) relative to WT. (Lower) A combination of DHE with the NO probe (DAF, green staining) indicating decreased NO activity accompanying increased oxidative stress in NOS1–/–hearts.
. (c) Oxidative fluorescent microtopography using the fluorescent probe DHE demonstrates increased staining in NOS1–/– (orange-staining nuclei) relative to WT. (Lower) A combination of DHE with the NO probe (DAF, green staining) indicating decreased NO activity accompanying increased oxidative stress in NOS1–/–hearts.
Fig. 3.

XOR expression and abundance. Increased XOR stimulated 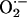 production in NOS1–/– was not caused by increased production or abundance of XOR. (a) XOR mRNA expression (by quantitative PCR) is similar among hearts from WT, NOS3–/–, and NOS1–/– mice (n = 3 mice for each strain). (b) Cardiac XOR protein abundance, measured by Western blot analysis, is not different between the three mouse stains. (c) Optical density bar chart depicting quantification of bands corresponding to both XOR products, XDH (150 kDa) and XO (130 kDa and 85 kDa).
production in NOS1–/– was not caused by increased production or abundance of XOR. (a) XOR mRNA expression (by quantitative PCR) is similar among hearts from WT, NOS3–/–, and NOS1–/– mice (n = 3 mice for each strain). (b) Cardiac XOR protein abundance, measured by Western blot analysis, is not different between the three mouse stains. (c) Optical density bar chart depicting quantification of bands corresponding to both XOR products, XDH (150 kDa) and XO (130 kDa and 85 kDa).
Cardiac Myocyte Contractile Responses. OS depresses myocardial contractility by inhibiting contractile myofilament responsiveness to activator Ca2+ (17). Thus, to examine the functional consequences of increased OS in NOS1–/–, we measured myocyte contraction and Ca2+ transients over a wide range of stimulation rates. Baseline sarcomere shortening and calcium transients were similar among myocytes from NOS1–/–, NOS3–/–, and WT mice at 2 Hz (Table 1; sample transients are shown in Fig. 4a). As reported (13), increasing stimulation rate augments myocyte contraction and [Ca2+]i (Fig. 4 b and c), producing a positive force-frequency response (FFR). Both SL shortening and [Ca2+]i increased in parallel with higher pacing frequencies in WT and NOS3–/– myocytes (P < 0.01 for increase from 2 to 6 Hz); however, as reported (13), the FFR was attenuated in NOS1–/– (P < 0.05, NOS1–/– vs. WT and NOS3–/–). Incubation with allopurinol (10–4 M; concentration where selective XOR inhibition occurs) restored SL shortening in the NOS1–/– myocytes to the level of WT, but did not alter SL shortening or [Ca2+]i in the WT or NOS3–/– myocytes. The increase in SL shortening in NOS1–/– caused by allopurinol was not accompanied by increases in either [Ca2+]i or SR Ca2+ stores, consistent with an acute calcium myofilament-sensitizing effect in NOS1–/–.
Table 1. Baseline myocyte characteristics at 2 Hz.
| WT | NOS3-/- | NOS1-/- | |
|---|---|---|---|
| Mice, n | 5 | 5 | 6 |
| Diastolic SL, μm | 1.73 ± 0.01 | 1.70 ± 0.01 | 1.71 ± 0.01 |
| SL shortening, % | 1.88 ± 0.13 | 2.10 ± 0.15 | 2.23 ± 0.22 |
| [Ca2+]i, % | 20.4 ± 1.06 | 18.6 ± 1.05 | 21.6 ± 1.11 |
Four to five cells were studied per heart; SL shortening, (diastolic SL — systolic SL)/diastolic SL; [Ca2+]i, change in the ratio of the photon live count detected by excitation at 365 nm compared to 380 nm during contraction.
Fig. 4.

Calcium sensitizing effect of allopurinol in NOS1–/– myocytes. (a) Sample transients are shown at 2-Hz baseline, 6 Hz (solid line), and 6 Hz with allopurinol treatment (dashed line). NOS1–/– myocytes have attenuated sarcomere shortening (b) and calcium transient (c) compared to WT and NOS3–/– as pacing frequency is raised from 2 to 6 Hz (solid bar; *, P < 0.05, NOS1–/– vs. WT and NOS3–/–). Allopurinol infusion (10–4 M; striped bar) augments contractility in NOS1–/– to the level of WT (†, P < 0.05 vs. NOS1–/– without allopurinol), without increasing systolic Ca2+ transients, representing a Ca2+ sensitizing effect. In contrast, neither sarcomere shortening nor calcium transient increase with allopurinol treatment (striped bar) in WT and NOS3–/– myocytes.
SR Ca2+ Stores. Because SR stores are a primary determinant of frequency-dependent responses, we measured SR Ca2+ stores by rapidly infusing caffeine (10 mM) at rising pacing frequencies (Fig. 5). As described (13), calcium stores at low (1 Hz) pacing frequency were similar in WT [arbitrary units [Ca2+]i, 38 ± 2%, n = 3], NOS3–/– (43 ± 3%, n = 3), and NOS1–/– (35 ± 3%, n = 3). Increasing stimulation frequency to 6 Hz augmented SR Ca2+ stores in WT (69 ± 4%) and NOS3–/– (60 ± 6%), but not NOS1–/– myocytes (41 ± 5%, P < 0.05 vs. WT and NOS3–/–). Importantly, allopurinol (10–4 M) treatment did not increase SR Ca2+ stores in WT, NOS3–/–, or NOS1–/– myocytes.
Fig. 5.

SR Ca2+ stores. SR Ca2+ stores did not increase with rising pacing frequencies (1–6 Hz) in NOS1–/– myocytes, as it did in WT and NOS3–/– myocytes (*, P < 0.05 vs. 1-Hz baseline; †, P < 0.05, NOS1–/– vs. WT and NOS3–/–). Importantly, allopurinol (10–4 M, open symbols) had no effect on SR Ca2+ stores in NOS1–/–, NOS3–/–, or WT myocytes.
Discussion
The major findings of this study are that (i) XOR and NOS1 colocalize to the SR and coimmunoprecipitate, suggesting a possible protein–protein interaction, (ii) deficiency of NOS1 but not NOS3 stimulates XOR-mediated 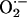 production without affecting XOR mRNA or protein abundance, and (iii) enhanced XOR activity inhibits myocyte contractility in NOS1-deficient myocytes by affecting the myofilament contractile apparatus, and this can be restored toward normal by acute XOR inhibition. Taken together, these findings demonstrate a NOS1-selective, spatially confined mechanism for NOS-XOR crosstalk within the cardiac myocyte that regulates cardiac excitation–contraction coupling. These findings demonstrate a mechanism for contractile depression, and suggest that NOS1 deficiency or inactivation may participate in cardiac disease by unleashing XOR-mediated oxidative stress.
production without affecting XOR mRNA or protein abundance, and (iii) enhanced XOR activity inhibits myocyte contractility in NOS1-deficient myocytes by affecting the myofilament contractile apparatus, and this can be restored toward normal by acute XOR inhibition. Taken together, these findings demonstrate a NOS1-selective, spatially confined mechanism for NOS-XOR crosstalk within the cardiac myocyte that regulates cardiac excitation–contraction coupling. These findings demonstrate a mechanism for contractile depression, and suggest that NOS1 deficiency or inactivation may participate in cardiac disease by unleashing XOR-mediated oxidative stress.
These findings are notable as a demonstration of a protein–protein association between a NOS isoform and a reactive oxygen species-generating enzyme. Spatially confined NO signaling mediated by protein–protein interactions between NOS and either effector or regulatory molecules is emerging as a major theme in NO biology (13, 18–22). The current findings now suggest a similar theme to the reciprocal manner in which NOS may regulate 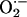 production within biological systems.
production within biological systems.
We have previously shown that NOS1 and NOS3 are located in unique subcellular compartments within the cardiac myocyte and exert independent, and, in some cases, opposite effects on cardiac function (18). Our findings in this study demonstrate that interactions between NOSs and sources of reactive oxygen species are also regulated in a spatially confined manner, likely by protein–protein interactions. These two enzymes may thus undergo direct protein–protein interactions, or may complex via another scaffolding protein, such as CAPON (23). Together, these results demonstrate that NOS1 exerts tonic inhibition of XOR-mediated 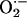 production that protects the cardiac myocyte from contractile depression.
production that protects the cardiac myocyte from contractile depression.
Mechanisms for NO inhibition of XOR have been described (10). XOR contains multiple redox centers essential for enzyme activity that are plausible targets of NO attack (10). In this regard, NO directly binds with an essential sulfur of the reduced molybdenum center of XOR to produce desulfo-type inactive enzymes (9, 10). Because protein nitrosylation is emerging as a major theme for posttranslational modification (24, 25), it is attractive to speculate that this may also play a role in NOS–XOR crosstalk. Consistent with prior studies, our observed increase in XOR activity in NOS1–/– occurs in the absence of any changes in XOR mRNA expression (11).
In addition to inhibiting XOR activity, there are several other chemical mechanisms by which NO exerts its antioxidant activity, but their role in physiologic situations remain unknown (see ref. 26 for review). NO can abate Fenton-type reactions by directly scavenging oxidants, preventing peroxidase reactions, or scavenging reducing equivalents supplied by 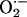 (26, 27). Furthermore, NO produced in a controlled manner can convert oxidative reactive nitrogen oxide species to nitrosating species, thereby providing an antioxidant environment (28).
(26, 27). Furthermore, NO produced in a controlled manner can convert oxidative reactive nitrogen oxide species to nitrosating species, thereby providing an antioxidant environment (28).
Interactions between NO and 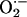 allow for precise physiological regulation of proteins involved in excitation–contraction (E-C) coupling and mitochondrial respiration (29). We predict that localization of NOS1 and XOR in the SR allows for beat-to-beat regulation of the RyR and SERCA2a. Whether these proteins are modulated reversibly as to preserve physiological signaling or irreversibly so as to cause toxicity is determined by the relative balance of NO/
allow for precise physiological regulation of proteins involved in excitation–contraction (E-C) coupling and mitochondrial respiration (29). We predict that localization of NOS1 and XOR in the SR allows for beat-to-beat regulation of the RyR and SERCA2a. Whether these proteins are modulated reversibly as to preserve physiological signaling or irreversibly so as to cause toxicity is determined by the relative balance of NO/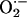 (25, 28, 30, 31). For example, NO augments the RyR activity by protein nitrosylation in a reversible manner capable of optimizing SR activity throughout the cardiac cycle (32). NO may additionally influence SERCA activity, although this remains controversial with studies demonstrating opposite results (16, 33). Excessive NO, however, (even under low
(25, 28, 30, 31). For example, NO augments the RyR activity by protein nitrosylation in a reversible manner capable of optimizing SR activity throughout the cardiac cycle (32). NO may additionally influence SERCA activity, although this remains controversial with studies demonstrating opposite results (16, 33). Excessive NO, however, (even under low 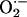 levels) can be detrimental, causing cellular dysfunction and even death (31, 34). Oxidative stress also activates the RyR but promotes maximal channel activity in an unregulated manner, reducing the ability of NO to exert feedback regulation of SR calcium release (32, 35, 36). As we have shown in the NOS1–/– mice, higher
levels) can be detrimental, causing cellular dysfunction and even death (31, 34). Oxidative stress also activates the RyR but promotes maximal channel activity in an unregulated manner, reducing the ability of NO to exert feedback regulation of SR calcium release (32, 35, 36). As we have shown in the NOS1–/– mice, higher 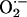 (with or without higher NO production) may disrupt physiologic signaling (26, 31, 37). On the other hand,
(with or without higher NO production) may disrupt physiologic signaling (26, 31, 37). On the other hand, 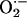 (under physiological levels) is an important intermediate for nitrosylating species (38). We have demonstrated the physiological relevance of NO/
(under physiological levels) is an important intermediate for nitrosylating species (38). We have demonstrated the physiological relevance of NO/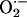 balance both in vitro and in vivo as the positive inotropic effect of the NO donor 3-morpholinosydnonimine (SIN-1) was abrogated by indiscriminate
balance both in vitro and in vivo as the positive inotropic effect of the NO donor 3-morpholinosydnonimine (SIN-1) was abrogated by indiscriminate 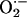 scavenging (39).
scavenging (39).
Our functional observations are consistent with allopurinol improving myofilament calcium sensitivity in NOS1–/– myocytes, as contraction increases without concomitant rises in systolic Ca2+ influx. This finding is in agreement with previous studies demonstrating that XOR inhibition improves myofilament sensitivity to activator calcium, an effect magnified in tissues with increased XOR activity, such as stunned or failing myocardium (2, 40, 41). It is conceivable that oxidant signaling may directly regulate cross-bridge cycling kinetics, possibly by degrading troponin I (42), thereby modulating the efficiency of contraction. XOR inhibition with allopurinol then likely decreases oxidative stress to augment myofilament sensitivity. Direct effects of allopurinol on the myofilaments cannot be excluded; however, actions unrelated to XOR have previously not been described.
Notably, we did not detect increases in [Ca2+]i or SR Ca2+ stores with XOR inhibition in NOS1–/– myocytes. To the extent that Ca2+ cycling proteins of the SR are oxidized in the NOS1–/– myoyctes, acute incubation of these cells with allopurinol fails to increase Ca2+ cycling. There are at least two potential explanations for this finding. First, despite the reduction of oxidative stress by allopurinol, SR ion channel function may continue to be impaired as long as there is disrupted protein nitrosylation in NOS1 deficiency. An alternative explanation may be that chronic SR protein changes in NOS1–/– (such as altered SERCA/phospholamban ratios; refs. 13 and 20) are not acutely reversed with allopurinol. It is possible that chronic XOR inhibition may influence the transcription and translation of SR proteins by altering redox influences over transcription factors; this is an avenue of investigation for future studies (43).
There are several technical issues that warrant mention. Although the frequencies used for isolated myocyte studies were below physiological rodent heart rates, they are comparable to, or above, most published murine myocyte studies (20), and the results from these studies qualitatively resembled those obtained in intact animals (13). In addition, we have previously shown that hearts from both NOS1–/– and NOS3–/– mice develop cardiac hypertrophy (18). To address this potential confounding factor, we studied isolated myocytes from 2-month-old mice, an age that preceded any structural changes (18).
In conclusion, we have uncovered a mechanism by which cardiac oxidative stress is regulated. Our findings demonstrate that NOS1, as opposed to NOS3, directly interacts with XOR to regulate cardiac excitation-contraction coupling. This finding is consistent with the evolving concept that intracrine signaling pathways are spatially compartmentalized with unique effector molecules in the regulation of cardiac contractility. Thus, NOS1 not only regulates the SR Ca2+ cycle, but also represents an important antioxidant system, inhibiting XOR activity. NOS1 and XOR likely interact in the physiologic regulation of the SR, but the ill effects of NOS1 loss from the SR are intensified by a concomitant and resultant increase in XOR generated formation of oxygen free radicals.
Acknowledgments
This work was supported by National Institutes of Health Grants RO1 HL-65455 (to J.M.H.) and RO1 AG021523 (to D.E.B.) and a Paul Beeson Physician Faculty Scholars in Aging Research Award (to J.M.H.).
Author contributions: S.A.K., K.L., K.M.M., D.E.B., and J.M.H. designed research; S.A.K., K.L., K.M.M., D.R.G., S.V.Y.R., A.D.T., and D.L. performed research; D.L. and J.M.H. contributed new reagents/analytic tools; S.A.K., K.L., K.M.M., D.R.G., S.V.Y.R., and D.L. analyzed data; and S.A.K., D.E.B., and J.M.H. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: XOR, xanthine oxidoreductase; NOS, nitric oxide synthase; NOS1, neuronal NOS; NOS3, endothelial NOS; SR, sarcoplasmic reticulum; SERCA, SR Ca2+ ATPase; SL, sarcomere length; DHE, dihydroethidium; DAF, diaminofluorescein; OS, oxidative stress; [Ca2+]i, Ca2+ transient.
References
- 1.Saavedra, W. F., Paolocci, N., St. John, M. E., Skaf, M. W., Stewart, G. C., Xie, J. S., Harrison, R. W., Zeichner, J., Mudrick, D., Marbán, E., et al. (2002) Circ. Res. 90, 297–304. [DOI] [PubMed] [Google Scholar]
- 2.Ekelund, U. E. G., Harrison, R. W., Shokek, O., Thakkar, R. N., Tunin, R. S., Senzaki, H., Kass, D. A., Marbán, E. & Hare, J. M. (1999) Circ. Res. 85, 437–445. [DOI] [PubMed] [Google Scholar]
- 3.de Jong, J. W., Schoemaker, R. G., de Jonge, R., Bernocchi, P., Keijzer, E., Harrison, R., Sharma, H. S. & Ceconi, C. (2000) J. Mol. Cell Cardiol. 32, 2083–2089. [DOI] [PubMed] [Google Scholar]
- 4.Cappola, T. P., Kass, D. A., Nelson, G. S., Berger, R. D., Rosas, G. O., Kobeissi, Z. A., Marbán, E. & Hare, J. M. (2001) Circulation 104, 2407–2411. [DOI] [PubMed] [Google Scholar]
- 5.McCord, J. M. (1985) N. Eng. J. Med. 312, 159–163. [DOI] [PubMed] [Google Scholar]
- 6.Landmesser, U., Spiekermann, S., Dikalov, S., Tatge, H., Wilke, R., Kohler, C., Harrison, D. G., Hornig, B. & Drexler, H. (2002) Circulation 106, 3073–3078. [DOI] [PubMed] [Google Scholar]
- 7.Pérez, N. G., Gao, W. D. & Marbán, E. (1998) Circ. Res. 83, 423–430. [DOI] [PubMed] [Google Scholar]
- 8.Charlat, M. I., O'Neill, P. G., Egan, J. M., Abernethy, D. R., Michael, L. H., Myers, M. L., Roberts, R. & Bolli, R. (1987) Am. J. Physiol. 252, H566–H577. [DOI] [PubMed] [Google Scholar]
- 9.Berry, C. E. & Hare, J. M. (2004) J. Physiol. (London) 555, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ichimori, K., Fukahori, M., Nakazawa, H., Okamoto, K. & Nishino, T. (1999) J. Biol. Chem. 274, 7763–7768. [DOI] [PubMed] [Google Scholar]
- 11.Hassoun, P. M., Yu, F. S., Zulueta, J. J., White, A. C. & Lanzillo, J. J. (1995) Am. J. Physiol. 268, L809–L817. [DOI] [PubMed] [Google Scholar]
- 12.Xu, K. Y., Zweier, J. L. & Becker, L. C. (1995) Circ. Res. 77, 88–97. [DOI] [PubMed] [Google Scholar]
- 13.Khan, S. A., Skaf, M. W., Harrison, R. W., Lee, K., Minhas, K. M., Kumar, A., Fradley, M., Shoukas, A. A., Berkowitz, D. E. & Hare, J. M. (2003) Circ. Res. 92, 1322–1329. [DOI] [PubMed] [Google Scholar]
- 14.Cappola, T. P., Cope, L., Cernetich, A., Barouch, L. A., Minhas, K., Irizarry, R. A., Parmigiani, G., Durrani, S., Lavoie, T., Hoffman, E. P., et al. (2003) Physiol. Genomics 14, 25–34. [DOI] [PubMed] [Google Scholar]
- 15.Berkowitz, D. E., White, R., Li, D., Minhas, K. M., Cernetich, A., Kim, S., Burke, S., Shoukas, A. A., Nyhan, D., Champion, H. C., et al. (2003) Circulation 108, 2000–2006. [DOI] [PubMed] [Google Scholar]
- 16.Eu, J. P., Sun, J., Xu, L., Stamler, J. S. & Meissner, G. (2000) Cell 102, 499–509. [DOI] [PubMed] [Google Scholar]
- 17.Gao, W. D., Liu, Y. & Marbán, E. (1996) Circulation 94, 2597–2604. [DOI] [PubMed] [Google Scholar]
- 18.Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., Hobai, I. A., Lemmon, C. A., Burnett, A. L., O'Rourke, B., et al. (2002) Nature 416, 337–339. [DOI] [PubMed] [Google Scholar]
- 19.Ashley, E., Sears, C., Bryant, S., Watkins, H. & Casadei, B. (2002) Circulation 105, 3011–3016. [DOI] [PubMed] [Google Scholar]
- 20.Sears, C., Bryant, S. M., Ashley, E., Lygate, C. A., Rakovic, S., Wallis, H. L., Neubauer, S., Terrar, D. A. & Casadei, B. (2003) Circ. Res. 92, e52–e59. [DOI] [PubMed] [Google Scholar]
- 21.Ziolo, M. T. & Bers, D. M. (2003) Circ. Res. 92, 1279–1281. [DOI] [PubMed] [Google Scholar]
- 22.Hare, J. M. (2004) Lancet 363, 1338–1339. [DOI] [PubMed] [Google Scholar]
- 23.Jaffrey, S. R., Benfenati, F., Snowman, A. M., Czernik, A. J. & Snyder, S. H. (2002) Proc. Natl. Acad. Sci. USA 99, 3199–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumoto, A., Comatas, K. E., Liu, L. M. & Stamler, J. S. (2003) Science 301, 657–661. [DOI] [PubMed] [Google Scholar]
- 25.Stamler, J. S., Lamas, S. & Fang, F. C. (2001) Cell 106, 675–683. [DOI] [PubMed] [Google Scholar]
- 26.Wink, D. A., Miranda, K. M., Espey, M. G., Pluta, R. M., Hewett, S. J., Colton, C., Vitek, M., Feelisch, M. & Grisham, M. B. (2001) Antioxid. Redox Signal. 3, 203–213. [DOI] [PubMed] [Google Scholar]
- 27.Rubbo, H., Radi, R., Trujillo, M., Telleri, R., Kalyanaraman, B., Barnes, S., Kirk, M. & Freeman, B. A. (1994) J. Biol. Chem. 269, 26066–26075. [PubMed] [Google Scholar]
- 28.Wink, D. A., Cook, J. A., Kim, S. Y., Vodovotz, Y., Pacelli, R., Krishna, M. C., Russo, A., Mitchell, J. B., Jourd'heuil, D., Miles, A. M., et al. (1997) J. Biol. Chem. 272, 11147–11151. [DOI] [PubMed] [Google Scholar]
- 29.Hare, J. M. (2003) J. Mol. Cell Cardiol. 35, 719–729. [DOI] [PubMed] [Google Scholar]
- 30.Stamler, J. S. & Hausladen, A. (1998) Nat. Struct. Biol. 5, 247–249. [DOI] [PubMed] [Google Scholar]
- 31.Joshi, M. S., Ponthier, J. L. & Lancaster, J. R., Jr. (1999) Free Radical Biol. Med. 27, 1357–1366. [DOI] [PubMed] [Google Scholar]
- 32.Xu, L., Eu, J. P., Meissner, G. & Stamler, J. S. (1998) Science 279, 234–237. [DOI] [PubMed] [Google Scholar]
- 33.Xu, K. Y., Huso, D. L., Dawson, T., Bredt, D. S. & Becker, L. C. (1999) Proc. Natl. Acad. Sci. USA 96, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wink, D. A. & Mitchell, J. B. (1998) Free Radical Biol. Med. 25, 434–456. [DOI] [PubMed] [Google Scholar]
- 35.Kawakami, M. & Okabe, E. (1998) Mol. Pharmacol. 53, 497–503. [DOI] [PubMed] [Google Scholar]
- 36.Eu, J. P., Xu, L., Stamler, J. S. & Meissner, G. (1999) Biochem. Pharmacol. 57, 1079–1084. [DOI] [PubMed] [Google Scholar]
- 37.Bloodsworth, A., O'Donnell, V. B. & Freeman, B. A. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 1707–1715. [DOI] [PubMed] [Google Scholar]
- 38.Espey, M. G., Thomas, D. D., Miranda, K. M. & Wink, D. A. (2002) Proc. Natl. Acad. Sci. USA 99, 11127–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paolocci, N., Ekelund, U. E. G., Isoda, T., Ozaki, M., Vandegaer, K., Georgakopoulos, D., Harrison, R., Kass, D. A. & Hare, J. M. (2000) Am. J. Physiol. 279, H1982–H1988. [DOI] [PubMed] [Google Scholar]
- 40.Ukai, T., Cheng, C. P., Tachibana, H., Igawa, A., Zhang, Z. S., Cheng, H. J. & Little, W. C. (2001) Circulation 103, 750–755. [DOI] [PubMed] [Google Scholar]
- 41.Kogler, H., Fraser, H., McCune, S., Altschuld, R. & Marbán, E. (2003) Cardiovasc. Res. 59, 582–592. [DOI] [PubMed] [Google Scholar]
- 42.Gao, W. D., Atar, D., Liu, Y., Perez, N. G., Murphy, A. M. & Marbán, E. (1997) Circ. Res. 80, 393–399. [PubMed] [Google Scholar]
- 43.Kim, S. O., Merchant, K., Nudelman, R., Beyer, W. F., Keng, T., DeAngelo, J., Hausladen, A. & Stamler, J. S. (2002) Cell 109, 383–396. [DOI] [PubMed] [Google Scholar]