

Abstract
Both turn sequence and interstrand hydrophobic side-chain–side-chain interaction have been suggested to be important determinants of β-hairpin stability. However, their roles in controlling the folding dynamics of β-hairpins have not been clearly determined. Herein, we investigated the structural stability and folding kinetics of a series of tryptophan zippers by static IR and CD spectroscopies and the IR temperature jump method. Our results support a β-hairpin folding mechanism wherein the rate-limiting event corresponds to the formation of the turn. We find that the logarithm of the folding rate depends linearly on the entropic change associated with the turn formation, where faster folding correlates with lower entropic cost. Moreover, a stronger turn-promoting sequence increases the stability of a β-hairpin primarily by increasing its folding rate, whereas a stronger hydrophobic cluster increases the stability of a β-hairpin primarily by decreasing its unfolding rate.
Small size and structural simplicity make short peptides that fold into well defined structures ideal model systems for examining factors that govern protein folding (1). Of particular interest are β-hairpins. With two antiparallel β-strands connected by a turn (or loop), the β-hairpin motif may be regarded as the smallest folding unit that contains tertiary contacts. Although an increasing body of evidence suggests that the β-hairpin can act as a folding nucleus (2–4), the mechanism by which individual β-hairpins fold has remained elusive. This elusiveness is partly due to the fact that so far only the folding kinetics of a few sequence-unrelated β-hairpins have been studied experimentally (5–9). These studies firmly demonstrated that β-hairpins fold on the microsecond time scale; however, the marked difference in the peptide sequence of those systems studied makes it difficult to determine explicitly the key factors that control the rate of β-hairpin folding.
Although experimental measurements of the folding kinetics of β-hairpins are scarce, in the past few years a remarkable number of theoretical and computational studies have been conducted regarding the folding dynamics and energetics of a variety of β-hairpin systems (10–21). Results from these studies generally support the idea that the peptide sequence is an important determinant of the folding rate of β-hairpins. For example, the statistical model of Muñoz et al. (10) predicts that moving the hydrophobic cluster one residue closer to the turn will speed up the folding rate by 4 times, whereas the results of Thirumalai and Klimov (14, 17) suggest that the turn rigidity plays a rather important role in determining the rate as well as the cooperativity of β-hairpin folding. Although it is still under debate whether β-hairpin folding begins with the formation of the turn (5, 10), interstrand hydrogen bond (16), or hydrophobic collapse (11), results from simulations generally support the idea that it involves multiple kinetic events, whereas the rate-limiting step may correspond to the assembly of interstrand native contacts within a collapsed ensemble (14), formation of critical hydrophobic contacts (5, 10), or formation of the turn, depending on the model. Undoubtedly, these theoretical studies provide new insights into our understanding of the folding mechanism of β-hairpins, but many predictions have yet to be demonstrated experimentally.
In light of findings from previous theoretical and simulation studies, it would, therefore, be quite useful to examine the folding kinetics of a series of sequence-related β-hairpins that differ, for example, in the turn composition or the relative position of the hydrophobic cluster. Herein, we studied the thermal stability and folding kinetics of three tryptophan zippers (trpzips): trpzip3, trpzip3-I, and trpzip4 (Table 1). Both trpzip3 and trpzip4 originally were designed by Cochran et al. (22) and have been shown by NMR spectroscopy to adopt β-hairpin conformations in aqueous solution. Thermal unfolding studies of these trpzips by CD spectroscopy indicated that they exhibit thermodynamic properties similar to those of proteins, owing to the strong interactions between cross-strand pairs of tryptophan (Trp) side chains. Following the design of Cochran et al. (22), we also introduced a new trpzip, trpzip3-I (Table 1), which differs from trpzip3 only in the turn sequence. Compared with trpzip4, trpzip3-I has the same turn sequence (DATK) but a shorter chain length and, thus, a shorter distance between the hydrophobic cluster and the turn. Therefore, by studying the folding kinetics of these trpzips, we are aiming to answer the following question: Are the turn (or loop) sequence and the relative position of the hydrophobic cluster strong determinants of the folding rate of β-hairpins?
Table 1. Sequences of trpzips and the GB1 peptide.
| Peptide | Sequence |
|---|---|
| trpzip1 | SWTWEGNKWTWK |
| trpzip2 | SWTWENGKWTWK |
| trpzip3 | SWTWEDPNKWTWK |
| trpzip3-I | SWTWDATKWTWK |
| trpzip4 | GEWTWDDATKTWTWTE |
| GB1 | GEWTYDDATKTFTVTE |
Moreover, trpzip4 is a triple mutant of the most studied β-hairpin, the 16-residue GB1 peptide (23, 24), in which the wild-type residues Y45, F52, and V54 are replaced by Ws (Table 1). Muñoz et al. (5) have shown that the wild-type GB1 hairpin folds in a two-state manner, both thermodynamically and kinetically, with a folding rate constant of ≈(6 μs)–1 at 297 K. Although highly twisted, the β-hairpin conformation formed by trpzip4 is much more stable than that of the GB1 peptide. Using CD spectroscopy, Cochran et al. (22) showed that trpzip4 unfolds in a single cooperative transition with a thermal melting temperature (Tm) of ≈70°C but that the GB1 β-hairpin unfolds with a much lower Tm (≈24°C) as determined by fluorescence spectroscopy (5). Thus, by comparing the folding kinetics of trpzip4 with those of the GB1 peptide, one should be able to determine whether the increased thermal stability of trpzip4 is the result of an increase in its folding rate or a decrease in its unfolding rate.
Consistent with theoretical predications, our laser-induced temperature-jump (T-jump) studies indeed show that the turn sequence has a strong effect on the folding rate of trpzips. One rather surprising result, however, is that the folding rate of trpzip3-I is roughly an order of magnitude slower than that of trpzip4, even though the hydrophobic cluster of the latter is farther away from the turn region. In addition, we found that trpzip4 acquires its higher thermal stability, compared with the GB1 β-hairpin, primarily by decreasing its unfolding rate.
Materials and Methods
The trpzips used in the current study were synthesized based on standard fluorenylmethoxycarbonyl (Fmoc) protocols. All products were purified to homogeneity by reverse-phase chromatography and identified by matrix-assisted laser desorption ionization mass spectroscopy. The residual trifluoroacetic acid (TFA) from peptide synthesis, the absorbance of which overlaps with the amide I′ band of polypeptides, was removed by lyophilization against a 0.1 M DCl solution.
CD thermal melting curves were obtained on a 62A DS spectropolarimeter (Aviv Associates, Lakewood, NJ) with a 1-mm sample holder. The peptide concentration was ≈50 μMin either D2O [uncorrected pH (pH*) 3] (for tripzip3-I) or 20 mM phosphate-D2O buffer solution (pH* 7) (for trpzip3 and trpzip4).
Temperature-dependent Fourier transform infrared (FTIR) spectra were collected on a Magna-IR 860 spectrometer (Nicolet) by using 2-cm–1 resolution. A CaF2 sample cell that was divided into two compartments with a Teflon spacer was used to allow the separate measurements of the sample and the reference under identical conditions. The optical path length of the sample cell was determined by its interference fringes, obtained from the transmittance signal of the empty cell, to be 52 μm. Temperature control with ±0.2°C precision was obtained by a thermostated copper block. To correct slow instrument drifts, both the sample and reference sides of the sample cell were moved in and out of the IR beam alternatively, and each time a spectrum corresponding to an average of eight scans was collected. The final result was usually an average of 32 such spectra, for both the sample and the reference. For both static and time-resolved IR measurements, the sample was prepared by directly dissolving lyophilized solids in either D2O (pH* 3) (for trpzip3-I) or 20 mM phosphate-D2O buffer (pH* 7) (for trpzip3 and trpzip4). The final concentration was ≈1–2 mM, as estimated by Trp absorbance.
The T-jump IR setup is described in detail in refs. 25 and 26. Briefly, a heating pulse at 1.9 μm (3 ns and 10 mJ) was used to generate a T-jump of ≈10°C. Transient absorbance change induced by the T-jump pulse was probed by a continuous-wave IR diode laser and a 50-MHz HgCdTe detector. As in the static FTIR measurement, a sample cell with dual compartments was used to allow the separate measurements of the absorbance change of the sample and reference under identical conditions. Measurements with the reference provided the information needed for both T-jump amplitude calibration and background subtraction.
All curve fittings were carried out in excel (Microsoft) by using its Solver capability. Simulations of the amide I′ band of the folded and extended structures of tripzip4 were performed with matlab (Mathworks, Natick, MA). The method used to generate the amide I′ band of proteins is based on the transition dipole coupling mechanism (27, 28), which attributes the bandwidth and splitting of the amide I′ manifold to the dipolar interactions or couplings among amide groups. Following Torii and Tasumi (29), we denoted the transition dipole moment of the jth (1 ≤ j ≤ n) peptide group to δμj [in D·Å–1·atomic mass units (amu)–1/2], the magnitude of which was set to be 3.70 D·Å–1·amu–1/2, and n was the number of peptide bonds. The direction of the transition dipole also was determined according to the method of Torii and Tasumi (29). Finally, the force-constant matrix (the F matrix) was constructed as follows:
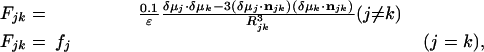 |
where each diagonal force constant, fj, is determined by the intrinsic vibrational frequency of an isolated oscillator [in mdyne·Å–1·amu–1 (1 dyne = 10 μN)], and Rjk is the length (in Å) of the line connecting the jth and kth transition dipoles; njk is the unit vector along this line, and ε is the dielectric constant, which was assumed here to be unity. The eigenvalues of the F matrix correspond to the eigenmodes of amide I′ vibrational motions. Therefore, the IR intensity for each eigenmode was obtained by taking the magnitude square of the product of the transition dipole and eigenvectors of the F matrix. For trpzip4, two fundamental vibrational frequencies, 1.61 mdyne· Å–1·amu–1 (1,653 cm–1) and 1.59 mdyne·Å–1·amu–1 (1,641 cm–1), were used for the buried and solvated amide groups, respectively. For the extended conformation, 1.59 mdyne·Å–1·amu–1 was used as the fundamental vibrational frequency for all amide groups.
Results
First, the thermal unfolding transitions of trpzip3, trpzip3-I, and trpzip4 were studied by CD spectroscopy according to the method of Cochran et al. (22). Although most β-hairpins do not show CD signatures that can be used reliably to monitor their thermal folding/unfolding transitions, trpzips exhibit a rather unique exciton-coupled CD peak around 229 nm because of the strong interaction between the paired Trp side chains (30). For trpzip3 and trpzip4, our CD data (data not shown), as well as the resulting thermodynamic parameters from a two-state fit, are very similar to those obtained by Cochran et al. (22) (Table 2). Interestingly, however, the thermal unfolding transition of trpzip3-I, as suggested by CD, is less cooperative and has a much lower Tm, indicating that replacing the native turn residues of trpzip3, i.e., EDPNK (in which DP represents d-Pro), with DATK severely compromises its thermal stability.
Table 2. Thermodynamic unfolding parameters obtained from equilibrium CD and IR measurements.
| CD
|
IR
|
|||||
|---|---|---|---|---|---|---|
| Parameter | trpzip3 | trpzip3-I | trpzip4 | trpzip3 | trpzip3-I | trpzip4 |
| ΔHm, kcal·mol-1 | 13.0 ± 1.3 | 8.7 ± 0.9 | 20.2 ± 1.6 | — | 8.0 ± 0.7 | 22.0 ± 2.1 |
| ΔSm, cal·K-1·mol-1 | 36.8 ± 2.6 | 30.0 ± 3.1 | 58.8 ± 4.1 | — | 27.6 ± 1.4 | 65.1 ± 3.8 |
| ΔCp, cal·K-1·mol-1 | 194 ± 19 | 90 ± 14 | 374 ± 35 | — | 201 ± 26 | 402 ± 37 |
| Tm, °C | 80.1 ± 2.0 | 16.8 ± 1.6 | 70.4 ± 1.8 | — | 16.7 ± 2.1 | 64.8 ± 1.5 |
—, Not measured.
Second, the thermal unfolding transitions of these trpzips were further investigated by FTIR spectroscopy, wherein the temperature-dependent amide I′ bands of these peptides were analyzed. The amide I′ band of polypeptides arises mainly from the stretching vibration of backbone carbonyls and is an established indicator of protein secondary structures (31) because of its sensitivity to structural parameters. As shown in Fig. 1a, the resolution-enhanced (32) FTIR spectrum of trpzip4 at 14.4°C exhibits three resolvable spectral features, centered at ≈1,633, 1,655, and 1,676 cm–1, respectively. In particular, the pair of low- and high-frequency bands (the 1,633- and 1,676-cm–1 components, in this case), which originates from vibrational couplings among interstrand amide C Os, is considered characteristic of antiparallel β-sheets (27, 28). Consistent with these experimental results, the calculated amide I′ band of trpzip4, which corresponds to the average of 20 calculated spectra based on the transition dipole coupling model and using the 20 NMR structures of trpzip4 in the Protein Data Bank (ID code 1LE3), also shows three major spectral components (Fig. 1b), centered at 1,634, 1,654, and 1,674 cm–1, respectively. For comparison, we have also calculated the amide I′ absorption profile of extended trpzip4, which was generated by changing the native φ and ϕ angles randomly to values obtained from a Gaussian distribution centered at (180, 180) with a width of 20°. As expected, the simulated spectrum (Fig. 1b), which corresponds to the average of the calculated amide I′ bands of 10 extended conformations of trpzip4, contains mostly a broad band, centered at around 1,651 cm–1.
Os, is considered characteristic of antiparallel β-sheets (27, 28). Consistent with these experimental results, the calculated amide I′ band of trpzip4, which corresponds to the average of 20 calculated spectra based on the transition dipole coupling model and using the 20 NMR structures of trpzip4 in the Protein Data Bank (ID code 1LE3), also shows three major spectral components (Fig. 1b), centered at 1,634, 1,654, and 1,674 cm–1, respectively. For comparison, we have also calculated the amide I′ absorption profile of extended trpzip4, which was generated by changing the native φ and ϕ angles randomly to values obtained from a Gaussian distribution centered at (180, 180) with a width of 20°. As expected, the simulated spectrum (Fig. 1b), which corresponds to the average of the calculated amide I′ bands of 10 extended conformations of trpzip4, contains mostly a broad band, centered at around 1,651 cm–1.
Fig. 1.

Properties of the amide I′ band of trpzip4. (a) FTIR spectrum of trpzip4 (in the amide I′ region) collected at 14.5°C and resolution-enhanced spectrum [i.e., Fourier self-deconvolution (FSD) spectrum], as indicated in the plot. The resolution enhancement was done by the FSD method (32) with k = 2 and a full width at half maximum (FWHM) of 18 cm–1.(b) Calculated amide I′ bands of the native (solid) and extended (dashed) conformations of trpzip4. In these calculations, the Gaussian broadening width was set at 5 cm–1. a.u., arbitrary units.
The above results suggest that the integrated intensity of either the 1,633- or 1,677-cm–1 band may be used to monitor the population change of the β-hairpin conformation in thermal unfolding studies of trpzips. Although quantitatively resolving all overlapping features in a measured amide I′ profile is not always feasible, previously we have shown that the integrated area of the high-frequency component of an antiparallel β-sheet can be obtained by globally analyzing a set of difference FTIR spectra in the vicinity of 1,680 cm–1 (7, 33). For example, the difference FTIR spectra of trpzip4 (Fig. 2), which were generated by subtracting the FTIR spectrum collected at 14.4°C from the spectra collected at higher temperatures, exhibit a negative-going feature around 1,680 cm–1, because of the decreasing β-hairpin population when temperature is increased. Globally fitting these difference spectra with a Gaussian function whose position is allowed to shift linearly with temperature, plus a monotonic, nonlinear background, allowed us to obtain for each temperature the integrated area of the high-frequency component (33). It can be shown quite easily that the latter is proportional to the population of the folded β-hairpin conformation, and its dependence on temperature can further be used to characterize quantitatively the thermal unfolding transition of trpzip4 (Fig. 3). Similar analysis also was carried out for trpzip3 and trpzip3-I. As shown in Table 2, the unfolding thermodynamic parameters obtained by this method for both trpzip4 and trpzip3-I are similar to those obtained by CD spectroscopy, although the Tm of trpzip4 reported by IR is somewhat lower than that reported by CD, a trend that also has been observed for trpzip2 (33, 34).
Fig. 2.

Difference FTIR spectra (○) of trpzip4. These spectra were generated by subtracting the FTIR spectrum collected at 14.4°C from those collected at higher temperatures (T = 14.4 + n * 6.0°C, n = 1–11) and adding an offset. Solid lines are fits to a Gaussian function plus a monotonic, nonlinear baseline. As an example, the baseline corresponding to the difference spectrum at 80.4°C is shown (+). As discussed in the text, the area of the Gaussian function at each temperature T is proportional to the β-hairpin population difference between T and 14.4°C.
Fig. 3.

Integrated areas of the high-frequency component of trpzip4, trpzip3, and trpzip3-I (open symbols). Fitting the data of trpzip4 and trpzip3-I to a two-state model (solid lines) yields those thermodynamic parameters summarized in Table 2. The solid line for trpzip3 is present to guide the eyes. a.u., arbitrary units.
Interestingly, the IR data of trpzip3 (Fig. 3) do not show a simple symmetric sigmoid transition, as suggested by CD. Instead, these data reveal two unfolding transitions, at ≈45°C and ≈80°C, respectively. The latter result is similar to that obtained with CD spectroscopy. A plausible interpretation of these results is that the low-temperature transition arises from the melting of a small amount of soluble aggregates present in the solution, whereas the high-temperature transition corresponds to the thermal unfolding of the monomeric trpzip3 β-hairpin. Consistent with this explanation is that the Fourier self-deconvolution spectrum of trpzip3 exhibits an additional band at ≈1,615 cm–1 (data not shown), a feature that is typical for β-aggregates (35). Furthermore, Decatur and coworkers (36) recently have shown that β-aggregates formed by short peptides can be dissociated reversibly by heat. Nonetheless, we cannot rule out the possibility that this complex behavior results from the intrinsic complexity of the folding energy landscape of trpzip3 (34).
The T-jump-induced relaxation kinetics of these trpzips were monitored by time-resolved IR spectroscopy (25, 26). As shown in Fig. 4, these relaxations exhibit two distinct phases. The fast phase is instrumentation-limited and is likely due to temperature-induced spectral changes, such as shift and broadening, and/or imperfect background subtraction, whereas the slow phase can be modeled by first-order kinetics. When the final temperature in T-jump experiments increases, the amplitude of the slow phase first increases and then decreases, indicating that this component probes the cooperative thermal folding–unfolding transition reported by the static IR measurements. Therefore, the observed relaxation rate constants (kobs) were further separated into folding (kf) and unfolding (ku) rate constants (Fig. 5), based on the following relationships: kobs = kf + ku and Keq = kf/ku. Keq is the equilibrium constant, calculated by using the unfolding thermodynamic parameters obtained from either IR (for trpzip4 and trpzip3-I) or CD (for trpzip3) thermal unfolding studies. It is evident that the folding rates of these trpzips exhibit non-Arrhenius behavior and also weak temperature dependence (Fig. 5). Fitting these data to the Eyring equation indeed yields a relatively small ΔH≠ for folding (note that the ΔS≠ cannot be determined because the preexponential factor is not known), suggesting that the energetic barrier for the formation of β-hairpins is small, in agreement with other studies (5, 7).
Fig. 4.

A representative relaxation trace of trpzips, measured on trpzip4 at 1,630 cm–1 in response to a T-jump of ≈10°C, from 45.0°C to 55.3°C. The smooth line is the fit to the following function: ΔOD(t) = A * [1–B * exp(–t/τ)], with A = –0.0031, B = 0.64, and τ = 2.9 μs.
Fig. 5.

Arrhenius plot of the observed relaxation (•), folding (○), and unfolding (▵) rate constants of trpzip4 (a), trpzip3-I (b), and trpzip3 (c). Lines are fits to the Eyring equation, i.e., ln(k) = ln(D) – ΔG≠/RT, where D was set to 1.0 × 1010 s–1 and ΔG≠ is the temperature-dependent free energy of activation.
Discussion
It has long been recognized that several factors can influence the stability of the folded state of β-hairpins. These include hydrogen bonds, electrostatic interaction, turn preference, and hydrophobic packing of side chains (37–41). The wide range of thermal melting temperatures observed for trpzips (including the GB1 β-hairpin) is consistent with the idea that a stable β-hairpin results from an intricate interplay among several factors. For instance, the low thermal stability of trpzip3-I may be attributed mainly to the lack of a crucial stabilizing interaction provided by key side chains in the loop. Munekata and coworkers (39) recently have shown that a characteristic hydrogen bond network in the loop region (i.e., DDATKT) of the GB1 β-hairpin, which involves mostly D46, T49, and K50, is important for its stability because most single mutations in this region destabilize the β-hairpin conformation. A similar hydrogen bond network can also be found in the GB1 turn region of its parent protein, protein G (42). The study of Munekata and coworkers (39) further suggests that it seems that D46 is particularly crucial for maintaining the rigidity and consequently the stability of the loop region of the GB1 β-hairpin by restricting rotational freedoms of the main chain and/or the side chains. A similar conclusion also was reached recently by Tsai and Levitt (40) in a molecular dynamics simulation. Therefore, it is not surprising that trpzip3-I is less stable than trpzip4, because the former lacks a pair of critical residues, i.e., D46 and T51. Nevertheless, because of the stronger hydrophobic interactions provided by the pairs of cross-strand Trp side chains, the stability of Trpzip3-I is still comparable to that of the GB1 β-hairpin.
Despite its small size, trpzip4 exhibits exceptional stability and folding cooperativity. In fact, its folding thermodynamic properties are similar to those of proteins (22). Because trpzip4 and the GB1 β-hairpin differ from each other only in the composition of the hydrophobic cluster, we can compare their folding kinetics meaningfully. Using the method of T-jump fluorescence, Eaton and coworkers (5) have shown that the GB1 β-hairpin folds in ≈6 μs at 297 K. Although trpzip4 is more stable, it folds with a similar rate. At 297 K, its folding time constant is extrapolated to be ≈15 μs (Table 3). On the contrary, the unfolding rates of the GB1 β-hairpin and trpzip4 differ significantly. At 297 K, the GB1 β-hairpin unfolds in ≈6 μs (5), whereas the unfolding time constant of trpzip4 is ≈234 μs.
Table 3. Folding/unfolding rate constants and equilibrium entropic changes of the 12-residue trpzips at 23°C and those of trpzip4 and the GB1 peptide at 24°C.
These results have strong implications regarding the mechanism of β-hairpin folding. Taken together, they allow us to determine the kinetic role of the hydrophobic clusters found in these β-hairpins. The similar folding rates exhibited by the GB1 β-hairpin and trpzip4 suggest that their folding free-energy barriers are also quite similar. However, the slower unfolding rate of trpzip4 indicates that it has a larger unfolding free-energy barrier than does the GB1 β-hairpin. Furthermore, a quantitative analysis indicates that the φ value (43) of trpzip4 for folding, because of the triple Trp mutations in the GB1 β-hairpin, is approximately –0.11‡, suggesting that mutations to the hydrophobic side chains in the GB1 peptide do not alter significantly the free energy of the folding transition state. In other words, the native hydrophobic side-chain–side-chain contacts have not been formed in the transition state structural ensemble. Therefore, the role of a strong cross-strand hydrophobic cluster, such as the one found in trpzips, primarily is to prevent the unfolding of the folded β-hairpin conformation. Conceptually, this conclusion is similar to the zipper model of Muñoz et al. (5). Results obtained on other trpzips also corroborate this idea.
It is well established that the turn sequence is an important determinant of the turn conformation and, thereby, other features of β-hairpin conformation, such as the pattern of interstrand hydrogen bonding and residue pairing (44). Moreover, a relatively low entropic cost associated with the turn (or loop) formation is another characteristic feature of strong turn-promoting sequences, such as those containing a DP residue (45, 46). For example, Gellman and coworkers (46) have recently shown that the high turn-forming propensity of the DPG segment decreases the equilibrium entropic cost of β-hairpin formation relative to the more flexible NG segment. Although many studies have contributed greatly to our understanding of the thermodynamic role of many different turns, the kinetic role of the turn (or loop) in β-hairpin folding has not been explicitly analyzed. Therefore, systematically analyzing the folding kinetics of a series of 12-residue trpzips, such as trpzip1, trpzip2, trpzip3, and trpzip3-I, which differ only in the turn sequence, should provide a unique opportunity for us to elucidate the kinetic role of the turn in β-hairpin formation.
The folding kinetics of the four 12-residue trpzips mentioned above have been studied by T-jump IR, T-jump fluorescence, or both. For trpzip2 and trpzip3, both T-jump IR and T-jump fluorescence yielded nearly identical results. For example, at 296 K, the obtained folding rate constant for trpzip3 from T-jump IR was ≈(1.7 μs)–1, whereas a T-jump fluorescence study reported a value of ≈(0.83 μs)–1 at 294 K (21). As shown in Table 3, although the unfolding rate constants of these 12-residue trpzips are different only by a factor of 3 or less, their folding rate constants differ by as much as 30 times (e.g., comparing trpzip3 with trpzip3-I). Taken together, these results suggest that the turn sequence is a strong determinant of the β-hairpin folding rate but has a rather small effect on the unfolding rate, consistent with the idea proposed above that the rate-limiting event in β-hairpin folding is the formation of the turn.
Interestingly, trpzip3, which contains a strong turn-promoting sequence, DPN, exhibits the fastest folding rate among the 12-residue trpzips studied. According to Gellman and coworkers (46), the excellent turn preference of DPN may be attributed to the conformational rigidity of the DP residue, which effectively reduces the entropic penalty, ΔSturn, upon turn formation. As indicated by results from this and other studies (5–7), the free-energy barrier of β-hairpin folding is dominated by the unfavorable entropic cost for the formation of the transition state. Therefore, within the framework of the folding transition state theory, it is reasonable to assume that kf ∝ exp(α·ΔSturn/R), where α is constant. Although it is difficult to explicitly determine ΔSturn for a given β-hairpin, the following relationship is valid: ΔSf = ΔSturn + ΔSstrand, where ΔSf is the total folding entropic change obtained from thermal melting experiments and ΔSstrand is the entropic change associated with the folding of the strands. Thus, kf ∝ exp[α·(ΔSf – ΔSstrand)/R]. It is easy to show that the latter equation leads to the following relationship: ln(kf) = ρ + α·(ΔSf – ΔSstrand)/R, where ρ is constant. For the four 12-residue trpzips discussed above, an approximate but quite reasonable assumption is that they have the same ΔSstrand. Such an assumption would lead directly to a relationship wherein ln(kf) is linearly proportional to ΔSf. Indeed, such a linear correlation is observed for the four 12-residue trpzips (Fig. 6), for which faster folding correlates with smaller entropic change. These results further strengthen our argument that the turn (or the turn region) is a key determinant of the folding rate of β-hairpins and clearly demonstrate that stronger turn-promoting sequences, such as those that contain a DP residue, increase the stability of β-hairpins by increasing the folding rate. Finally, it is worth pointing out that one should not simply interpret the term ΔSturn that is invoked here as the differences in disordered loop entropy; it only corresponds to a fraction of the entropic change determined from the thermal unfolding studies.
Fig. 6.

ln(kf) vs. ΔSf at 296 K for the 12-residue trpzips (see Table 3).
The slower folding rate observed for trpzip3-I, when compared with that of trpzip4, is also consistent with the picture that the formation of the turn is rate-limiting in β-hairpin folding. Frank et al. (47) have shown that even in 7.4 M urea, the protein GB1 domain still shows residual structures that involve D46, T49, and T51, indicating that the loop region of the GB1 β-hairpin is quite stable and rigid. It is reasonable to assume that the unfolded state of trpzip4 also contains similar residual structures. Thus, the faster folding rate of trpzip4 can be attributed to the existence of such residual structures because they effectively reduce the entropic cost corresponding to the turn formation. On the other hand, such residual structures are not expected to exist in the unfolded state of trpzip3-I, because it lacks a pair of critical residues, D46 and T51.
Conclusion
In summary, we have studied the thermal stability and folding kinetics of a series of sequence-related β-hairpins by using both static and time-resolved IR spectroscopy. Our results provide insight into the understanding of the kinetics of β-hairpin folding. In particular, our results suggest a folding mechanism in which the turn plays a key role in determining the rate of β-hairpin folding. In addition, a good turn-promoting sequence or a strong interstrand hydrophobic cluster can help to stabilize the folded conformation of β-hairpins. However, the former increases the stability of a β-hairpin primarily by increasing its folding rate, whereas the latter increases the stability of a β-hairpin primarily by decreasing its unfolding rate.
Acknowledgments
We thank the National Science Foundation for Grant CHE-0094077 and the National Institutes of Health for Grants GM065978 and RR01348.
Author contributions: F.G. designed research; D.D. and C.-Y.H. performed research; Y.Z. analyzed data; and F.G. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: amu, atomic mass units; DP, d-Pro; FTIR, Fourier transform infrared; T-jump, temperature-jump; Trp, tryptophan; trpzip, Trp zipper.
Footnotes
A recent NMR study by Andersen and coworkers (41) suggested that at 298 K the percentage of the folded state of the GB1 β-hairpin is ≈30%, which corresponds to a folding equilibrium constant (Keq) of ≈0.43. Such a Keq and a 3-μs relaxation time (5) would result in a kf of (10 μs)–1 for the GB1 peptide at ≈298 K. Using this folding rate constant for the GB1 peptide and the folding rate constant of trpzip4, we calculated the φ value based on the following equation: 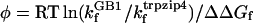 , where
, where 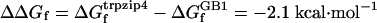 .
.
References
- 1.Ferguson, N. & Fersht, A. R. (2003) Curr. Opin. Struct. Biol. 13, 75–81. [DOI] [PubMed] [Google Scholar]
- 2.Freund, S. M., Wong, K. B. & Fersht, A. R. (1996) Proc. Natl. Acad. Sci. USA 93, 10600–10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCallister, E. L., Alm, E. & Baker, D. (2000) Nat. Struct. Biol. 7, 669–673. [DOI] [PubMed] [Google Scholar]
- 4.Walkenhorst, W. F., Edwards, J. A., Markley, J. L. & Roder, H. (2002) Protein Sci. 11, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muñoz, V., Thompson, P. A., Hofrichter, J. & Eaton, W. A. (1997) Nature 390, 196–199. [DOI] [PubMed] [Google Scholar]
- 6.Maness, S. J., Franzen, S., Gibbs, A. C., Causgrove, T. P. & Dyer, R. B. (2003) Biophys. J. 84, 3874–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu, Y., Oyola, R. & Gai, F. (2003) J. Am. Chem. Soc. 125, 15388–15394. [DOI] [PubMed] [Google Scholar]
- 8.Chen, R. P., Huang, J. J., Chen, H.-L., Jan, H., Velusamy, M., Lee, C.-T., Fann, W., Larsen, R. W. & Chan, S. I. (2004) Proc. Natl. Acad. Sci. USA 101, 7305–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyer, R. B., Maness, S. J., Peterson, E. S., Franzen, S., Fesinmeyer, R. M. & Anderson, N. H. (2004) Biochemistry 43, 11560–11566. [DOI] [PubMed] [Google Scholar]
- 10.Muñoz, V., Henry, E. R., Hofrichter, J. & Eaton, W. A. (1998) Proc. Natl. Acad. Sci. USA 95, 5872–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pande, V. S. & Rokhsar, D. S. (1999) Proc. Natl. Acad. Sci. USA 96, 9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinner, A. R., Lazaridis, T. & Karplus, M. (1999) Proc. Natl. Acad. Sci. USA 96, 9068–9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roccatano, D., Amadei, A., Di Nola, A. & Berendsen, H. J. (1999) Protein Sci. 10, 2130–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klimov, D. K. & Thirumalai, D. (2000) Proc. Natl. Acad. Sci. USA 97, 2544–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia, A. E. & Sanbonmatsu, K. Y. (2001) Proteins 42, 345–354. [DOI] [PubMed] [Google Scholar]
- 16.Zhou, R., Berne, B. J. & Germain, R. (2001) Proc. Natl. Acad. Sci. USA 98, 14931–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klimov, D. K. & Thirumalai, D. (2002) J. Mol. Biol. 315, 721–737. [DOI] [PubMed] [Google Scholar]
- 18.Zhou, Y. & Linhananta, A. (2002) Proteins 47, 154–162. [DOI] [PubMed] [Google Scholar]
- 19.Ma, B. & Nussinov, R. (2003) Protein Sci. 12, 1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolhuis, P. G. (2003) Proc. Natl. Acad. Sci. USA 100, 12129–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snow, C. D., Qiu, L., Du, D., Gai, F., Hagen, S. J. & Pande, V. S. (2004) Proc. Natl. Acad. Sci. USA 101, 4077–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cochran, A. G., Skelton, N. J. & Starovasnik, M. A. (2001) Proc. Natl. Acad. Sci. USA 98, 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanco, F. J., Rivas, G. & Serrano, L. (1994) Nat. Struct. Biol. 1, 584–590. [DOI] [PubMed] [Google Scholar]
- 24.Honda, S., Kobayashi, N. & Munekata, E. (2000) J. Mol. Biol. 295, 269–278. [DOI] [PubMed] [Google Scholar]
- 25.Huang, C.-Y., Getahun, Z., Zhu, Y., Klemke, J. W., DeGrado, W. F. & Gai, F. (2002) Proc. Natl. Acad. Sci. USA 99, 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang, C.-Y., Klemke, J. W., Getahun, Z., DeGrado, W. F. & Gai, F. (2001) J. Am. Chem. Soc. 123, 9235–9238. [DOI] [PubMed] [Google Scholar]
- 27.Krimm, S. & Abe, Y. (1972) Proc. Natl. Acad. Sci. USA 69, 2788–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore, W. H. & Krimm, S. (1975) Proc. Natl. Acad. Sci. USA 72, 4933–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torii, H. & Tasumi, M. (1992) J. Chem. Phys. 96, 3379–3387. [Google Scholar]
- 30.Grishina, I. B. & Woody, R. W. (1994) Faraday Discuss. 99, 245–262. [DOI] [PubMed] [Google Scholar]
- 31.Krimm, S. & Bandekar, J. (1986) Adv. Protein Chem. 38, 181–364. [DOI] [PubMed] [Google Scholar]
- 32.Kauppinen, J. K., Moffatt, D. J., Mantsch, H. H. & Cameron, D. G. (1981) Appl. Spectrosc. 35, 271–276. [Google Scholar]
- 33.Wang, T., Xu, Y., Du, D. & Gai, F. (2004) Biopolymers 75, 163–172. [DOI] [PubMed] [Google Scholar]
- 34.Yang, W. Y., Pitera, J. W., Swope, W. C. & Gruebele, M. (2004) J. Mol. Biol. 336, 241–251. [DOI] [PubMed] [Google Scholar]
- 35.Timasheff, S. N., Susi, H. & Stevens, L. (1967) J. Biol. Chem. 242, 5467–5473. [PubMed] [Google Scholar]
- 36.Silva, R. A., Barber-Armstrong, W. & Decatur, S. M. (2003) J. Am. Chem. Soc. 125, 13674–13675. [DOI] [PubMed] [Google Scholar]
- 37.Blanco, F., Ramirez-Alvarado, M. & Serrano, L. (1998) Curr. Opin. Struct. Biol. 8, 107–111. [DOI] [PubMed] [Google Scholar]
- 38.Gellman, S. H. (1998) Curr. Opin. Chem. Biol. 2, 717–725. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi, N., Honda, S., Yoshii, H. & Munekata, E. (2000) Biochemistry 39, 6564–6571. [DOI] [PubMed] [Google Scholar]
- 40.Tsai, J. & Levitt, M. (2002) Biophys. Chem. 101–102, 187–201. [DOI] [PubMed] [Google Scholar]
- 41.Fesinmeyer, R. M., Hudson, F. M. & Andersen, N. H. (2004) J. Am. Chem. Soc. 126, 7238–7243. [DOI] [PubMed] [Google Scholar]
- 42.Gronenborn, A. M., Filpula, D. R., Essig, N. Z., Achari, A., Whitlow, M., Wingfield, P. T. & Clore, G. M. (1991) Science 253, 657–661. [DOI] [PubMed] [Google Scholar]
- 43.Matouschek, A., Kellis, J. T., Serrano, L. & Fersht, A. R. (1989) Nature 340, 122–126. [DOI] [PubMed] [Google Scholar]
- 44.de Alba, E., Jiménez, M. A. & Rico, M. (1997) J. Am. Chem. Soc. 119, 175–183. [Google Scholar]
- 45.Rose, G. D., Gierasch, L. M. & Smith, J. A. (1985) Adv. Protein Chem. 37, 1–109. [DOI] [PubMed] [Google Scholar]
- 46.Espinosa, J. F., Syud, F. A. & Gellman, S. H. (2002) Protein Sci. 11, 1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank, M. K., Clore, G. M. & Gronenborn, A. M. (1995) Protein Sci. 4, 2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]