

High-performance quasi–solid state Na-CO2 batteries are constructed with polymer electrolyte and a reduced graphene oxide Na anode.
Keywords: Na-CO2 batteries, graphene oxide-Na anodes, composite polymer electrolyte, wide CO2-partial pressure, large energy density
Abstract
Na-CO2 batteries using earth-abundant Na and greenhouse gas CO2 are promising tools for mobile and stationary energy storage, but they still pose safety risks from leakage of liquid electrolyte and instability of the Na metal anode. These issues result in extremely harsh operating conditions of Na-CO2 batteries and increase the difficulty of scaling up this technology. We report the development of quasi–solid state Na-CO2 batteries with high safety using composite polymer electrolyte (CPE) and reduced graphene oxide (rGO) Na anodes. The CPE of PVDF-HFP [poly(vinylidene fluoride-co-hexafluoropropylene)]–4% SiO2/NaClO4–TEGDME (tetraethylene glycol dimethyl ether) has high ion conductivity (1.0 mS cm−1), robust toughness, a nonflammable matrix, and strong electrolyte-locking ability. In addition, the rGO-Na anode presents fast and nondendritic Na+ plating/stripping (5.7 to 16.5 mA cm−2). The improved kinetics and safety enable the constructed rGO-Na/CPE/CO2 batteries to successfully cycle in wide CO2 partial pressure window (5 to 100%, simulated car exhaust) and especially to run for 400 cycles at 500 mA g−1 with a fixed capacity of 1000 mA·hour g−1 in pure CO2. Furthermore, we scaled up the reversible capacity to 1.1 A·hour in pouch-type batteries (20 × 20 cm, 10 g, 232 Wh kg−1). This study makes quasi–solid state Na-CO2 batteries an attractive prospect.
INTRODUCTION
Na-CO2 batteries, which use Na, an element three to five orders of magnitude more abundant on earth than Li, and greenhouse gas CO2, theoretically offer a high energy density of 1.13 kWh kg−1 through the reaction of 4Na + 3CO2↔2Na2CO3 + C (Eθ = 2.35 V) (1–4). In addition, the released CO2 in car exhaust could generate electricity in Na-CO2 batteries to prolong the driven miles of hybrid electric vehicles. Furthermore, 12% of off-gas emissions of thermal power plants (5) and 96% of the atmosphere of Mars (6) is CO2, suggesting the potential usefulness of Na-CO2 batteries for production of clean electric energy, due to the abundance of necessary materials. However, currently available primary and rechargeable Na-CO2 batteries (2, 4, 7) face safety risks, mainly because of liquid electrolyte and the pure Na anode. Na-CO2 batteries with sodium salt/organic solvent liquid electrolyte are plagued by leakage (because of open battery structure) (8–11), volatilization (especially in working conditions at high temperature), and electrochemical instability (caused by large polarization at high current density) (2). Na-CO2 batteries also encounter dendrite formation or surface cracks in the pure Na anodes during cycling, resulting in the possibility of short circuit (12). Therefore, seeking new alternatives for liquid electrolyte and the pure Na anode is vitally important.
To resolve the liquid electrolyte issue, the quasi–solid state polymer electrolyte (QPE) strategy is effective in inorganic and organic Li-ion batteries and Na-ion batteries (13–16). It has been shown that QPE is capable of absorbing liquid electrolyte without leakage (17); it also has a high ion conductivity (10−6 to 10−3 S cm−1 at room temperature) that is comparable to that of liquid electrolyte (10−3 S cm−1) (18, 19). In addition, adding nanosized ceramic fillers into QPE to form composite polymer electrolyte (CPE) would further increase Na+ conductivity and improve mechanical strength of the polymer electrolyte (20). However, so far, to the best of our knowledge, CPE has not been used in Na-air or Na-CO2 batteries. A suitable CPE for Na-CO2 batteries must satisfy three conditions: (i) large amorphous region of polymer matrix in QPE to accelerate the Na+ mobility, (ii) nanosized ceramic fillers with hydrophobic property to increase mechanical strength and avoid H2O contamination of QPE, and (iii) elimination of macropores on QPE to avoid potential contamination from anodic CO2 to cathodic Na.
Recently, graphene oxide (GO) or reduced graphene oxide (rGO) has demonstrated electrochemical and electrocatalytic properties (21, 22). In particular, a stable Li/rGO anode for long-term cycling was synthesized by the reduction of a GO sheet and then the absorption of molten Li metal at 250°C (23). However, there are few reports on methods of Na anode protection. Na has similar physical and chemical properties to Li but has stronger reducibility and a lower melting point (98°C) than Li metal. Because rGO-Na has not been reported in Na-ion, Na-air, or Na-CO2 batteries, making a composite of rGO and Na metal to synthesize an rGO-Na anode is worth investigating. Furthermore, the combination of an rGO-Na anode, CPE, and an activated cathode is also important for rechargeable Na-CO2 batteries.
We report here the integration of an rGO-Na anode, CPE, and an activated multiwall carbon nanotubes (a-MCNTs) cathode to construct rechargeable quasi–solid state Na-CO2 batteries with highly stable performance at room temperature. The CPE is hemicrystalline PVDF-HFP [poly(vinylidene fluoride-co-hexafluoropropylene)]–4% SiO2/NaClO4–TEGDME (tetraethylene glycol dimethyl ether), in which hydrophobic fumed SiO2 (10 to 20 nm) used as ceramic fillers enlarges the amorphous region of the CPE matrix, increases the Na+ conductivity to 1.0 mS cm−1, and improves the mechanical strength to 0.5 to 0.6 GPa. Only scarce nanopores are spread on the CPE, protecting the anode from CO2. The rGO-Na anodes are achieved through reducing three-dimensional GO foam in molten Na metal (98° to 120°C) and adsorbing this molten metal into pore structures of as-prepared rGO. The addition of rGO results in even plating of Na+ on the rGO-Na anode and is also able to improve the mechanical strength and toughness of the Na-based anode. The porous structure and TEGDME-wettable surface of a-MCNT cathodes benefit CO2 transfer and the occurrence of battery reactions. The assembled quasi–solid state Na-CO2 batteries successfully operate in a series of CO2 partial pressure atmospheres [5 to 100%, simulated car exhaust (SCE)]. We scaled up the batteries to a reversible capacity of 1.1 A·hour with an energy density of 232 Wh kg−1. By fixing ~20% depth of discharge and charge (200 mA·hour), Na-CO2 batteries have so far run for 50 cycles with the capacity retention of nearly 100%.
RESULTS
Synthesis and characterization of CPE
A CPE synthesized by integrating polymer matrix of PVDF-HFP, nanosized SiO2, and a plasticizer of NaClO4/TEGDME solution solves the issues of leakage, volatilization, and electrochemical instability of liquid electrolyte in Na-CO2 batteries (Fig. 1A). The large number of F atoms in the PVDF-HFP and TEGDME agent plays a key role in Na+ diffusion. The mass percent of fumed SiO2 with a diameter of 10 to 20 nm (inset of Fig. 1A) as the ceramic filler in CPE is a critical factor for ionic conductivity. The content of fumed SiO2 with 4 weight % (wt %) reaches the highest ion conductivity of 1.0 mS cm−1 (Fig. 1B and figs. S1 and S2). The optimized CPE (namely, PVDF-HFP–4% SiO2/NaClO4–TEGDME) with a thickness of ~160 μm (Fig. 1C) has a high ion conductivity (1.0 mS cm−1). CPE with pores centered at ~30 nm (fig. S3) could suppress CO2 diffusion to the anode and thus protect the Na metal (fig. S4). This CPE has a good toughness with a Young modulus of 0.5 to 0.6 GPa and a roughness of ~80 nm (Fig. 1, D and E, figs. S5 and S6). Suitable toughness and roughness of CPE benefit by tight contact to electrodes. Serious leakage occurs in a traditional Na-CO2 battery containing liquid electrolyte, but the CPE-based Na-CO2 battery successfully suppresses leakage (fig. S7). Moreover, this polymer matrix of CPE does not easily catch fire. Even if it catches fire after long-time ignition (5 s), the fire is soon quenched automatically (1 s; Fig. 1F). We also find that the addition of SiO2 increases both flame resistance and the melting point of the polymer matrix of CPE (fig. S8).
Fig. 1. Optimal characterization of CPE.
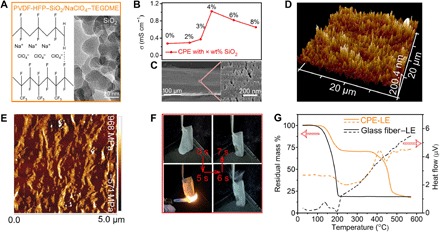
(A) The composition of CPE. Inset: Transmission electron microscopy (TEM) image of fumed SiO2. (B) Ionic conductivity of CPE with different contents of fumed SiO2. (C) Scanning electron microscopy (SEM) images of CPE with a thickness of 160 μm. Inset: SEM image of an enlarged view. (D) Atomic force microscopy (AFM) image of CPE with 3D view. (E) AFM images of Young modulus mapping. (F) Inflammability test of CPE before loading with liquid electrolyte. Fire from a lighter is ~500°C. (G) Thermal gravity analysis and heat flow curves of CPE and glass fiber, both of which are holding liquid electrolyte of TEGDME.
As one component of CPE for Na+ diffusion, TEGDME has a high boiling point (~275°C), but it still volatilizes even at room temperature in the open system of a traditional Na-CO2 battery. Our synthesized CPE can effectively suppress electrolyte volatilization because of robust intermolecular forces between TEGDME and polymer chains of CPE. This intermolecular force was confirmed by Raman spectroscopy, in which the relative peak intensities of both TEGDME and PVDF-HFP were changed (fig. S9). According to thermal gravity analysis and heat flow curves (Fig. 1G), CPE underwent two mass loss in temperature ranges of 126° to 222°C and 370° to 475°C. Within each temperature range, CPE showed endothermic thermal events twice, corresponding to TEGDME volatilization and PVDF-HFP decomposition, respectively. The residual mass of CPE after TEGDME volatilization at 222°C is 70%, indicating that the absorbed liquid electrolyte amount (ALEA) within CPE is 30%. This low ALEA (<50%) demonstrates that the diffusion of Na+ in CPE is mainly attributed to the polymer host of CPE (fig. S10A). In the same way, the ALEA within glass fiber (a traditional separator in an open battery system) is 80%, and the corresponding temperature interval of TEGDME volatilization is 82° to 206°C. The elevated volatilization temperature (126° to 222°C versus 82° to 206°C) of TEGDME proves that CPE can suppress electrolyte volatilization. In addition, the wide electrochemical window of CPE, 1.0 to 4.5 V, should provide the battery system with safe and stable conditions (fig. S10B). The CPE also exhibits long-term stability in terms of morphology and chemical structure (fig. S11). The abovementioned properties indicate the practical values of CPE.
Synthesis and characterization of rGO-Na anode
To achieve a highly stable Na-based anode and economize Na resources, we designed and fabricated an advanced rGO-Na anode for quasi–solid state Na-CO2 batteries. Getting rGO foam with a good Na affinity and infusing molten Na into the abundant nanopores of rGO are vitally important for the fabrication of the rGO-Na anode. When used as a starting material fabricated by the freeze-drying of GO solution, the GO foam displays a porous structure with the mass of GO microsheets (Fig. 2A). When the GO foam was connected in parallel to the molten Na (120°C), an instantaneous reaction occurred across the whole foam (movie S1). In this step, gray GO foam was reduced to dark black rGO, simultaneously resulting in a more porous and stable construction (Fig. 2B). Next, fast and uniform Na intake was achieved simply by placing the edge of the rGO foam into molten Na, finally resulting in a substantial surface with metal luster (Fig. 2C). It takes about 1 min for Na to diffuse across the whole rGO foam (movie S2). This successful synthesis of the rGO-Na anode is correlated to the capillary force produced by the nanopores of rGO foam and the good Na affinity with rGO foam. The extent of the interior filling of the rGO foam by Na is determined by element mapping (fig. S12A), which shows that the host of the rGO foam is totally filled with Na.
Fig. 2. Design and characterization of rGO-Na anode.
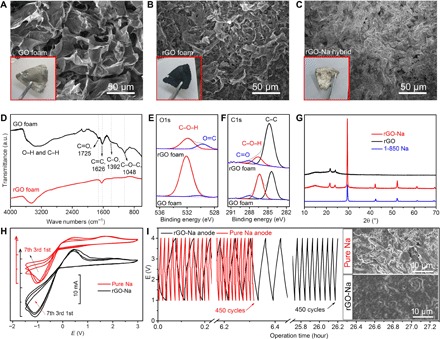
(A to C) SEM images with corresponding inset photographs of GO foam (A), rGO foam reduced by molten Na (B), and rGO-Na anode surface (C). (D) FTIR of GO foam and rGO foam. a.u., arbitrary units. XPS spectra of O1s (E) and C1s (F) of GO foam and rGO foam. (G) XRD of rGO and rGO-Na anode. (H) Cyclic voltammograms of Na+ plating/stripping in a rGO-Na or Na/CPE/stainless steel cell with a sweep speed of 0.5 V s−1. (I) Fast discharge/charge profiles of quasi–solid state Na-CO2 batteries in Ar atmosphere using rGO-Na and pure Na anodes. Rate, 0.3 mA cm−2; voltage range, 1 to 4 V. Inset: SEM images of rGO-Na and pure Na anode surfaces after 450 cycles.
The reduction of GO foam by molten Na is confirmed by Fourier transform infrared spectroscopy (FTIR) and x-ray photoelectron spectroscopy (XPS). Before the reduction reaction, a broad peak beyond 2900 cm−1 corresponding to –OH stretching was detected (Fig. 2D), revealing the presence of adsorbed water or surface –OH. Meanwhile, characteristic peaks of other surface groups, including carbonyl (C=O) (~1725 cm−1) and epoxyl (C–O–C) (~1048 cm−1), were also observed. Obviously, after the reduction reaction, the abovementioned oxygen-containing functional groups became almost undetectable, confirming the high reduction level of rGO. The XPS analysis is consistent with the FTIR result. According to the deconvoluted O1s and C1s spectra (Fig. 2, E and F), the intensity of C–OH dropped from 43.2% (GO foam) to 14.7% (rGO foam), indicating the remarkable removal of –OH groups. The Na (0) in the Na1s spectra of the rGO-Na anode (fig. S12B) arises from Na metal absorbed in rGO. A small quantity of Na (I) is probably correlated with the Na2O or Na2CO3 that is formed during the thermal reduction of GO in molten Na. The oxygen-containing groups on GO may react with molten Na and produce Na (I) species. The successful Na perfusion into rGO foam is verified by x-ray diffraction (XRD), which also suggests the absence of crystalline Na (I) species (Fig. 2G). The tensile strength test and bending test indicate that the rGO foam improves the mechanical strength and toughness of the rGO-Na anode in comparison with the pure Na anode (fig. S13).
In comparison with the pure Na metal anode, the rGO-Na anode has higher cyclic voltammetry current density (5.7 to 16.5 mA cm−2), demonstrating a fast kinetics of Na+ plating/stripping on rGO-Na anodes (Fig. 2H). Fast discharge and charge operation within 1.0 to 4.0 V at 0.3 mA cm−2 under Ar atmosphere (curves in Fig. 2I) smooth the rGO-Na anode surface compared with its initial state (Fig. 2C, inset of Fig. 2I, and fig. S14). The residual voids allow the deposition of Na during the charge process, preventing the formation of Na dendrites even after 450 cycles (inset of Fig. 2I). In sharp contrast, the pure Na anode was seriously cracked after 450 cycles (inset of Fig. 2I). This morphology difference between the pure Na anode and the rGO-Na anode indicates that the addition of rGO foam leads to even plating of Na+ on the rGO-Na anode. In the fast discharge/charge profiles, the capacitance behavior of rGO-Na in Ar is much more obvious than that of pure Na, a difference that can be attributed mainly to the larger specific area of the rGO-Na anode. Symmetric rGO-Na electrodes also exhibit much lower electrochemical polarization (±0.05 V) in the first 90,000 s at 1 mA cm−2 during plating/stripping than that of pure Na electrodes (±0.15 V) (fig. S15), further confirming the superiority of rGO-Na electrodes.
a-MCNT cathodes
To further improve the reaction activity of carbon nanotubes on cathodes, we boiled the MCNTs in TEGDME at 100°C to be a-MCNTs. The a-MCNTs are characterized by Raman and FTIR, but the TEGDME signals only appear in FTIR (Fig. 3, A and B). This suggests that the TEGDME solvent has infiltrated into the internal walls of a-MCNTs. In detail, bands at around 2850 cm−1 indicate the presence of –CH3 and –CH2– in TEGDME, whereas bands at 1100 cm−1 were C–O and C–C stretching signals, which also belong to TEGDME (Fig. 3B). The intensity ratios of D-band and G-band in a-MCNTs and MCNTs are 1.06 and 1.03, respectively (Fig. 3A), indicating a higher disorder degree of carbon atoms in a-MCNTs. In detail, D-band and G-band peaks stand for sp2-type carbon, whereas other peaks at 1317.3 and 1504.8 cm−1 are assigned to the sp3-type carbon (for example, –COOH) in a-MCNTs.
Fig. 3. Synthesis and characterization of a-MCNT cathodes.
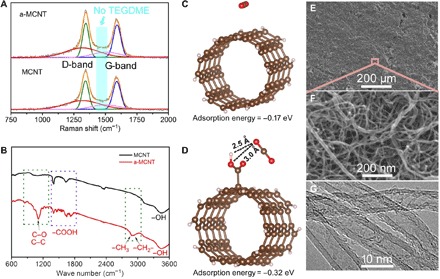
Raman (A) and FTIR (B) comparison of pristine MCNTs and a-MCNTs. The optimized geometries and corresponding adsorption energies of CO2 adsorbed on MCNTs (C) and a-MCNTs (D). SEM (E and F) and TEM (G) images of a-MCNT cathodes. The porous structure of cross-linking a-MCNTs offers open channels for CO2 transfer.
According to the density functional theory (DFT) calculation (Fig. 3, C and D, and fig. S16), the adsorption energy of a-MCNTs (−0.32 eV) is much higher than that of pristine MCNT (−0.17 eV). After activation, the tubular morphology and multiwall structure of carbon nanotubes are not destroyed (Fig. 3, E to G), and the wettability of carbon nanotubes toward TEGDME is enhanced (fig. S17). All the abovementioned merits of a-MCNTs lay a solid foundation for following interface reactions of Na-CO2 batteries. On the basis of the configuration of the Na/NaClO4-TEGDME/cathode, the initial discharge and charge profiles of Na-CO2 batteries with a-MCNT cathodes exhibit smaller overpotentials (1.39 V) than those with MCNT cathodes (2.04 V) (fig. S18), preliminarily confirming the effectiveness of a-MCNT cathodes.
Battery performance
By combining an rGO-Na anode, optimal CPE, and a-MCNT cathodes, quasi–solid state Na-CO2 batteries are first assembled in CR2032 coin-type cells, which exhibit clear discharge-charge voltage plateaus at various rates (Fig. 4A). When the batteries operate at 100, 200, 300, and 500 mA g−1, the charge-discharge voltage gap is 0.74, 0.88, 0.93, and 1.48 V, respectively. However, the initial charge voltage is still low. For example, even at 500 mA g−1, the initial charge voltage is still <3.49 V, which is much lower than the upper limit of the voltage window of CPE. The reaction of 4Na + 3CO2↔2Na2CO3 + C in quasi–solid state Na-CO2 batteries has been demonstrated (figs. S19 and S20), and it is consistent with that in liquid electrolyte–based Na-CO2 batteries (4). In addition, we find that the current densities could control the morphologies of discharge product in Na-CO2 batteries (insets of Fig. 4A). The current densities of 100 and 200 mA g−1 lead to discharge products (nanoparticles) with a round shape (100 to 200 nm in diameter). Meanwhile, larger current densities result in larger sizes of discharge products, namely, angular particles (200 to 300 nm) at 300 mA g−1 and nanobelts (30 × 200 × 500 nm) at 500 mA g−1. This variation in discharge products is different from Li-O2 batteries (24) but similar to Na-O2 batteries (25). The discharge products at various current densities (for example, 100, 200, and 300 mA g−1) pile up rather than just grow on a-MCNT surfaces (fig. S21), suggesting large capacity ability.
Fig. 4. The performance of quasi–solid state Na-CO2 batteries with rGO-Na anodes.
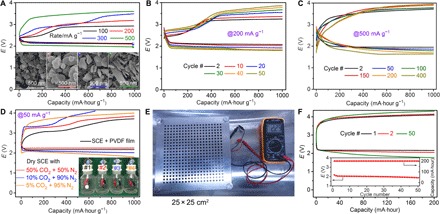
(A) Rate capability. Insets: SEM images of discharged products at different rates. The black, red, blue, and green scale bars correspond to rates of 100, 200, 300, and 500 mA g−1, respectively. (B) Discharge/charge profiles and (C) corresponding variation of the terminal discharge voltage with a cutoff capacity of 1000 mA·hour g−1 at 200 mA g−1. (D) Initial discharge/charge profiles in SCE with different CO2 partial pressure. Rate, 50 mA g−1. Four bottles use different test conditions. #1, SCE atmosphere with PVDF film protection; #2, dry SCE with 50% CO2 and 50% N2; #3, dry SCE with 10% CO2 and 90% N2; #4, dry SCE with 5% CO2 and 95% N2. (E) A photograph of pouch-type battery (20 × 20 cm2, 10 g) packed in a plastic bag with two stainless steel plates (25 × 25 cm2) to fix. (F) Discharge/charge profiles at 10 mA with a reversible capacity of 200 mA·hour. Inset: Corresponding variation of the middle discharge voltage.
Charge-discharge operation at current densities of 200 and 500 mA g−1 demonstrates an impressive stability of quasi–solid state Na-CO2 batteries (Fig. 4, B and C). Especially at 500 mA g−1, the batteries perfectly maintained the specific capacity of 1000 mA·hour g−1 for 400 cycles without obvious fading. Highly reversible capacity of 5000 mA·hour g−1 at 50 mA g−1 is also obtained without controlling capacity, and finally, stable capacity of ~3000 mA·hour g−1 is maintained (fig. S22). This advanced battery performance is mainly due to the sufficient ion conductivity of CPE, fast Na+ plating/stripping kinetics of the rGO-Na anode, and the active surface of a-MCNT cathodes. Excitingly, the performance of quasi–solid state Na-CO2 batteries is comparable to that of other liquid electrolyte–based metal-CO2 batteries (table S1). Moreover, we extend the cathode materials to curly carbon nanosheets that were synthesized by chemical vapor deposition via the reaction of CO2 + 2Mg→C + 2MgO (fig. S23). The MgO was removed from the carbon nanosheets by HCl solution treatment. This carbon material also enables quasi–solid state Na-CO2 batteries to become rechargeable and form nanosized discharge products (fig. S24).
We investigated the battery performance in SCE with different CO2 partial pressure. As is well known, no matter how strict the tailpipe standards [for example, Euro V/VI regulations (26)], all car exhaust must contain a humid mixture of N2 and CO2 (27, 28), which is transformed from initial petrol combustion products by built-in exhaust decontamination equipment. We design a device to fill our coin-type battery vessels with humid (humidity of 100%) or dry SCE (mixture of CO2 and N2; fig. S25), in which the batteries could operate normally even at CO2 partial pressures as low as 0.05 atm (Fig. 4D). The hydrophobic breathable film (PVDF film) provides the battery operation (fig. S26) with relative dry conditions and suppresses side reactions relating to H2O. The batteries in SCE (for example, 50% CO2 and 50% N2) could also run at 50°C for 50 cycles at 200 mA g−1 with overpotentials of 0.79 to 0.81 V (fig. S27). The stable discharge and charge profiles indicate the reliability of the battery system in SCE.
Because batteries with high capacity (mA·hour) and high energy density (Wh kg−1) are highly desirable for practical applications such as electric cars and electrical grid storage, we expanded the battery scale from CR2032 coin-type batteries (2 cm in diameter) to large batteries (20 × 20 cm). One large piece of semitransparent CPE film was integrated with an rGO-Na/Cu foil anode and a carbon cathode in a plastic bag, finally producing a pouch-type battery (Fig. 4E and fig. S28). With controlled voltage of 1.9 to 3.9 V, initial capacity of 1100 mA·hour at 10 mA is reached, corresponding to 232 Wh kg−1 on the basis of the total mass of the pouch-type battery (10.0 g) (fig. S29A). This energy density is higher than that of commercial Li-ion batteries (150 to 180 Wh kg−1) (29), suggesting the possibility of large-scale application of quasi–solid state rechargeable Na-CO2 batteries. At the depth of both 20 and 50% discharge/charge, the capacity retention of quasi–solid state rechargeable Na-CO2 batteries is 100%, without obvious voltage fading (Fig. 4F and fig. S29B).
DISCUSSION
The investigations described here give us an overall picture of quasi–solid state Na-CO2 batteries with the rGO-Na anode, optimized CPE, and a-MCNT cathodes. During discharge, the CO2 absorbs on the a-MCNT cathode surface and reacts with Na+ to generate products of Na2CO3 and carbon at the electrode/electrolyte interface. During charge, the products reversibly decompose and release CO2 gas and Na+. The rGO-Na anode promotes dendrite-free plating/stripping (5.7 to 16.5 mA cm−2) and saves Na metal resources. The CPE suppresses electrolyte volatilization and provides high Na+ conductivity (1.0 mS cm−1 at 25°C) for stable operation of batteries. The a-MCNT cathodes offer the adsorption of CO2 on activated carbon surface and the formation of products. Therefore, all components (anode, electrolyte, and cathode) have irreplacable roles and are vitally important to obtain the long life and stability of quasi–solid state Na-CO2 batteries. Moreover, the uninflammable PVDF-HFP–4% SiO2 as matrix in CPE, abundant Na metal–based anode, and clean utilization of CO2 for energy conversion further improve the practical value of quasi–solid state Na-O2 batteries for future stationary and mobile storage technologies. On the basis of this rGO-Na/CPE/CO2 system, the assembled coin-type batteries can operate at various CO2 partial pressures and, in particular, can run for 400 cycles in pure CO2. The pouch-type battery also exhibits a large capacity of 1.1 A·hour with an energy density of 232 Wh kg−1 and a stable cycling life (50 cycles). This demonstrates the feasibility of safe and stable Na-CO2 batteries, which should accelerate the process of practical application with abundant Na resource and CO2 fuels.
MATERIALS AND METHODS
Polymer electrolyte fabrication
The mixture (2.0 g) of PVDF-HFP copolymer and fumed silica with various mass ratio (0, 2, 3, 4, 6, and 8 wt % SiO2) was dissolved in acetone (15 ml). This solution was heated in oil bath at 50°C for 2 hours with vigorous stirring, followed by resting at the same temperature for 1 hour. The resultant viscous solution was spread as a film on aluminum foil using a graduated blade. Then, the polymer film was dried at 100°C under vacuum. After cooling to room temperature, the polymer film was immersed into a pool of 1 M NaClO4/TEGDME for 48 hours. Before use, the moistened film was wiped with filter paper to remove residual liquid electrolyte. We also synthesized large pieces of CPE by film applicator using a composition of PVDF-HFP–4% SiO2.
Cathode fabrication
MCNTs (42 mg) and TEGDME solvent (20 ml) were added to a round-bottom flask (100 ml), which was then heated in oil bath at 100°C for 2 hours under vigorous stirring. The mixture was then filtered and dried under vacuum at 160°C for 2 hours to obtain TEGDME-activated carbon material (a-MCNTs). The resulting a-MCNTs were mixed with 5 wt % PVDF binder in the solvent of N-methyl pyrrolidone by fully grinding in a mortar. The obtained slurry was pasted onto a piece of Ni mesh (ø10 mm) and dried under vacuum at 100°C for 10 hours. Finally, a-MCNT cathodes were formed with a loading mass of 0.3 mg cm−2. Cathodes with curly carbon nanosheets were prepared by the same method as a-MCNT cathodes. Ag nanowire cathodes were prepared by the following steps. First, Ag nanowires (1 mg) were added to the TEGDME (2 ml) solvent. After continuous sonication for 2 hours, a homogeneous ink was obtained. Second, 10 μl of the ink was added onto a nickel mesh (400 mesh, Emgrid Australia Pty Ltd.) by a pipette. After drying at 25°C under vacuum for 12 hours, Ag nanowire cathodes were obtained.
rGO-Na anode fabrication
The GO solution (5 g liter−1) was prepared by Hummer’s method (30). Briefly, graphite (10 g), concentrated H2SO4 (230 ml), and NaNO3 (5 g) were mixed and cooled down to 0°C. Then, 30 g of KMnO4 powder was added with vigorous stirring for 2 hours. After reaction at 35°C for 3 hours, distilled water (1000 ml, 18.2 megaohm resistance) and H2O2 (400 ml, 30%) were added to remove the unreacted KMnO4 and manganese dioxide. The resulting graphite oxide was separated and washed repeatedly. Afterward, the GO solution was made by the ultrasonication exfoliation of graphite oxides in water (3000 ml) and the centrifugation at a speed of 6000 rpm for 30 min. The centrifuged GO solution was purified by dialysis until its pH value was about 6. Finally, highly stable GO solution was obtained, and the concentration was adjusted to 5 g liter−1. This GO solution was freeze-dried at −47°C under 19 Pa for 24 hours. Then, freestanding GO films were obtained and transferred to an Ar-filled glove box with O2 level <1 parts per million. To realize the sparkle reaction, polished Na bulks (99.9%; Alfa Aesar) were melted in a round stainless steel pan on a hot plate at ~120°C. The GO films were slightly pressed and then brought into contact with the molten Na, and the spark reaction occurred instantaneously. This spark reaction partially transformed the GO into rGO. The molten Na was infused through the edges of the pressed rGO foam. Finally, rGO-Na sheets were obtained. Before being used as Na-based anodes in batteries, rGO-Na sheets were pressed slightly and cut into discs (ø10 mm) for coin-type cells or spliced together into square pieces (20 × 20 cm) for pouch-type batteries.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Programs for NanoKey Project (2016YFA0202500), the National Natural Science Foundation of China (21231005 and 51231003), and the Ministry of Education (B12015 and IRT13R30). Author contributions: X.H. and J.C. conceived and designed this work. X.H. performed the preparation and characterization of CPE. Y.Z., J.S., and Z.L. performed the preparation of the rGO-Na anodes. X.H. and Z.L. characterized the rGO-Na anodes. Z.L. and X.H. constructed and tested the pouch-type batteries. J.W. carried out the DFT calculation. X.H. and J.C. cowrote the paper. All authors contributed to the data analysis. J.C. directed the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1602396/DC1
Chemicals and materials
Fabrication of curly carbon nanosheets
Material characterization
Battery assembly
Electrochemical test
CO2 evolution test
Energy density calculation
Density function theory (DFT) calculation
Explanatory calculation
fig. S1. The raw materials and synthesis procedures of CPE.
fig. S2. The optimization of Na+-CPE.
fig. S3. The pore size distribution of CPE.
fig. S4. The CO2 diffusion test.
fig. S5. SEM images of CPE surface.
fig. S6. Roughness and Young modulus of polymer matrix (PVDF-HFP/4% SiO2) of CPE.
fig. S7. Leakage test of Na-CO2 batteries.
fig. S8. Inflammability test of polymer matrix (PVDF-HFP/SiO2) of CPE.
fig. S9. Raman of CPE, polymer matrix, and liquid electrolyte.
fig. S10. Transporting mechanism of Na+ in polymer chains and work window of CPE.
fig. S11. Long-term stability analysis of CPE.
fig. S12. Element mapping and XPS-Na1s of rGO-Na anode.
fig. S13. Mechanical strength and toughness of pure Na and rGO-Na anodes.
fig. S14. Anode analysis.
fig. S15. Galvanostatic cycling of a symmetric rGO-Na electrodes and pure Na anodes.
fig. S16. The optimized geometries of CO2 adsorbed on MCNT and a-MCNTs.
fig. S17. Soluble inflation of MCNTs and a-MCNTs toward TEGDME solvent.
fig. S18. Initial discharge and charge profiles of Na-CO2 batteries with the configuration of Na/NaClO4-TEGDME/cathode.
fig. S19. Reaction mechanism analysis of Na-CO2 batteries.
fig. S20. Discharge product analysis of quasi–solid state Na-CO2 battery.
fig. S21. SEM image of a-MCNT cathode with discharge capacity of 0.1 mA·hour at 100 mA g−1.
fig. S22. Full discharge and charge of quasi–solid state Na-CO2 batteries.
fig. S23. Characterizations of curly carbon nanosheets.
fig. S24. Discharge/charge curves of quasi–solid state Na-CO2 batteries with new carbon cathodes that contain curly carbon nanosheets.
fig. S25. A device for SCE.
fig. S26. Discharge/charge profiles in SCE with PVDF film protection of quasi–solid state Na-CO2 batteries.
fig. S27. Discharge/charge profiles of quasi–solid state Na-CO2 batteries at 50°C.
fig. S28. The preparation and assembly of large batteries.
fig. S29. Pouch-type battery performance.
table S1. Battery performance comparison.
movie S1. Sudden reaction on GO foam.
movie S2. Molten Na infusion into rGO foam.
REFERENCES AND NOTES
- 1.Armand M., Tarascon J.-M., Building better batteries. Nature 451, 652–657 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Das S. K., Xu S. M., Archer L. A., Carbon dioxide assist for non-aqueous sodium–oxygen batteries. Electrochem. Commun. 27, 59–62 (2013). [Google Scholar]
- 3.Gao S., Lin Y., Jiao X., Sun Y., Luo Q., Zhang W., Li D., Yang J., Xie Y., Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 529, 68–72 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hu X., Sun J., Li Z., Zhao Q., Chen C., Chen J., Rechargeable room-temperature Na–CO2 batteries. Angew. Chem. Int. Ed. 55, 6482–6486 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Ding F., Zhao Y., Mi L., Li H., Li Y., Zhang J., Removal of gas-phase elemental mercury in flue gas by inorganic chemically promoted natural mineral sorbents. Ind. Eng. Chem. Res. 51, 3039−3047 (2012). [Google Scholar]
- 6.D. R. Williams, Mars fact sheet (National Space Science Data Center, NASA, 2016; http://nssdc.gsfc.nasa.gov/planetary/factsheet/marsfact.html.
- 7.Xu S., Wei S., Wang H., Abruña H. D., Archer L. A., The sodium–oxygen/carbon dioxide electrochemical cell. ChemSusChem 9, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Peng Z., Freunberger S. A., Chen Y., Bruce P. G., A reversible and higher-rate Li-O2 battery. Science 337, 563–566 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Cheng F., Chen J., Lithium-air batteries: Something from nothing. Nat. Chem. 4, 962–963 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Li C., Han X., Cheng F., Hu Y., Chen C., Chen J., Phase and composition controllable synthesis of cobalt manganese spinel nanoparticles towards efficient oxygen electrocatalysis. Nat. Commun. 6, 7345–7353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu J., Lee Y. J., Luo X., Lau K. C., Asadi M., Wang H.-H., Brombosz S., Wen J., Zhai D., Chen Z., Miller D. J., Jeong Y. S., Park J.-B., Fang Z. Z., Kumar B., Salehi-Khojin A., Sun Y.-K., Curtiss L. A., Amine K., A lithium–oxygen battery based on lithium superoxide. Nature 529, 377–382 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Zhou W., Gao H., Goodenough J. B., Low-cost hollow mesoporous polymer spheres and all-solid-state lithium, sodium batteries. Adv. Energy Mater. 6, 1501802 (2016). [Google Scholar]
- 13.Huang W., Zhu Z., Wang L., Wang S., Li H., Tao Z., Shi J., Guan L., Chen J., Quasi-solid-state rechargeable lithium-ion batteries with a Calix[4]quinone cathode and gel polymer electrolyte. Angew. Chem. Int. Ed. 52, 9162–9166 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Zhu Z., Hong M., Guo D., Shi J., Tao Z., Chen J., All-solid-state lithium organic battery with composite polymer electrolyte and pillar[5]quinone cathode. J. Am. Chem. Soc. 136, 16461−16464 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Jain H., Thomas J. O., Whittingham M. S., Solid electrolytes: Advances in science and technology. MRS Bull. 25, 11−15 (2000). [Google Scholar]
- 16.Tarascon J. M., Gozdzb A. S., Schmutzb C., Shokoohib F., Warren P. C., Performance of Bellcore’s plastic rechargeable Li-ion batteries. Solid State Ion. 86–88, 49–54 (1996). [Google Scholar]
- 17.Li X., Cheruvally G., Kim J.-K., Choi J.-W., Ahn J.-H., Kim K.-W., Ahn H.-J., Polymer electrolytes based on an electrospun poly(vinylidene fluoride-co-hexafluoropropylene) membrane for lithium batteries. J. Power Sources 167, 491–498 (2007). [Google Scholar]
- 18.Costa C. M., Silva M. M., Lanceros-Méndez S., Battery separators based on vinylidene fluoride (VDF) polymers and copolymers for lithium ion battery applications. RSC Adv. 3, 11404–11417 (2013). [Google Scholar]
- 19.Stephan A. M., Nahm K. S., Review on composite polymer electrolytes for lithium batteries. Polymer 47, 5952–5964 (2006). [Google Scholar]
- 20.Aravindan V., Lakshmi C., Vickraman P., Investigations on Na+ ion conducting polyvinylidenefluoride-co-hexafluoro-propylene/poly ethylmethacrylate blend polymer electrolytes. Curr. Appl. Phys. 9, 1106–1111 (2009). [Google Scholar]
- 21.Li X., Wang X., Zhang L., Lee S., Dai H., Chemically derived, ultrasmooth graphene nanoribbon semiconductors. Science 319, 1229–1232 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Park J.-U., Nam S., Lee M.-S., Lieber C. M., Synthesis of monolithic graphene–graphite integrated electronics. Nat. Mater. 11, 120–125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li D., Liu Y., Liang Z., Lee H.-W., Sun J., Wang H., Yan K., Xie J., Cui Y., Layered reduced graphene oxide with nanoscale interlayer gaps as a stable host for lithium metal anodes. Nat. Nanotechnol. 11, 626–633 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Adams B. D., Radtke C., Black R., Trudeau M. L., Zaghib K., Nazar L. F., Current density dependence of peroxide formation in the Li–O2 battery and its effect on charge. Energy Environ. Sci. 6, 1772–1778 (2013). [Google Scholar]
- 25.Ortiz-Vitoriano N., Batcho T. P., Kwabi D. G., Han B., Pour N., Pierre Claver Yao K., Thompson C. V., Shao-Horn Y., Rate-dependent nucleation and growth of NaO2 in Na−O2 batteries. J. Phys. Chem. Lett. 6, 2636−2643 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Commission Regulation (EU) no. 582/2011 of 25 May 2011 (2016); http://eur-lex.europa.eu/legal-content/en/ALL/?uri=CELEX:32011R0582.
- 27.Matsumoto S., Recent advances in automobile exhaust catalysts. Catal. Today 90, 183–190 (2004). [Google Scholar]
- 28.Twigg M. V., Progress and future challenges in controlling automotive exhaust gas emissions. Appl. Catal. B 70, 2–15 (2007). [Google Scholar]
- 29.Dunn B., Kamath H., Tarascon J.-M., Electrical energy storage for the grid: A battery of choices. Science 334, 928–935 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Hummers W. S. Jr, Offeman R. E., Preparation of graphitic oxide. J. Am. Chem. Soc. 80, 1339–1339 (1958). [Google Scholar]
- 31.Kresse G., Furthmüller J., Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 33.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Ross G. G., Chassé M., Bolduc M., Effect of ageing on wettability of quartz surfaces modified by Ar implantation. J. Phys. D Appl. Phys. 36, 1001–1008 (2003). [Google Scholar]
- 35.Ataollahi N., Ahmad A., Hamzah H., Rahman M. Y. A., Mohamed N. S., Ionic conduction of blend poly (vinylidene fluoride-hexafluoro propylene) and poly (methyl methacrylate)-grafted natural rubber based solid polymer electrolyte. Int. J. Electrochem. Sci. 8, 7875–7884 (2013). [Google Scholar]
- 36.Chiang C.-Y., Reddy M. J., Chu P. P., Nano-tube TiO2 composite PVDF/LiPF6 solid membranes. Solid State Ion. 175, 631–635 (2004). [Google Scholar]
- 37.Xu S., Das S. K., Archer L. A., The Li-CO2 battery: A novel method for CO2 capture and utilization. RSC Adv. 3, 6656–6660 (2013). [Google Scholar]
- 38.Liu Y., Wang R., Lyu Y., Li H., Chen L., Rechargeable Li/CO2–O2 (2:1) battery and Li/CO2 battery. Energy Environ. Sci. 7, 677–681 (2014). [Google Scholar]
- 39.Zhang Z., Zhang Q., Chen Y. N., Bao J., Zhou X., Xie Z., Wei J., Zhou Z., The first introduction of graphene to rechargeable Li–CO2 batteries. Angew. Chem. Int. Ed. 54, 6550–6553 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Zhang X., Zhang Q., Zhang Z., Chen Y., Xie Z., Wei J., Zhou Z., Rechargeable Li–CO2 batteries with carbon nanotubes as air cathodes. Chem. Commun. 51, 14636–14639 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Xu S., Lu Y., Wang H., Abruña H. D., Archer L. A., A rechargeable Na–CO2/O2 battery enabled by stable nanoparticle hybrid electrolytes. J. Mater. Chem. A 2, 17723–17729 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1602396/DC1
Chemicals and materials
Fabrication of curly carbon nanosheets
Material characterization
Battery assembly
Electrochemical test
CO2 evolution test
Energy density calculation
Density function theory (DFT) calculation
Explanatory calculation
fig. S1. The raw materials and synthesis procedures of CPE.
fig. S2. The optimization of Na+-CPE.
fig. S3. The pore size distribution of CPE.
fig. S4. The CO2 diffusion test.
fig. S5. SEM images of CPE surface.
fig. S6. Roughness and Young modulus of polymer matrix (PVDF-HFP/4% SiO2) of CPE.
fig. S7. Leakage test of Na-CO2 batteries.
fig. S8. Inflammability test of polymer matrix (PVDF-HFP/SiO2) of CPE.
fig. S9. Raman of CPE, polymer matrix, and liquid electrolyte.
fig. S10. Transporting mechanism of Na+ in polymer chains and work window of CPE.
fig. S11. Long-term stability analysis of CPE.
fig. S12. Element mapping and XPS-Na1s of rGO-Na anode.
fig. S13. Mechanical strength and toughness of pure Na and rGO-Na anodes.
fig. S14. Anode analysis.
fig. S15. Galvanostatic cycling of a symmetric rGO-Na electrodes and pure Na anodes.
fig. S16. The optimized geometries of CO2 adsorbed on MCNT and a-MCNTs.
fig. S17. Soluble inflation of MCNTs and a-MCNTs toward TEGDME solvent.
fig. S18. Initial discharge and charge profiles of Na-CO2 batteries with the configuration of Na/NaClO4-TEGDME/cathode.
fig. S19. Reaction mechanism analysis of Na-CO2 batteries.
fig. S20. Discharge product analysis of quasi–solid state Na-CO2 battery.
fig. S21. SEM image of a-MCNT cathode with discharge capacity of 0.1 mA·hour at 100 mA g−1.
fig. S22. Full discharge and charge of quasi–solid state Na-CO2 batteries.
fig. S23. Characterizations of curly carbon nanosheets.
fig. S24. Discharge/charge curves of quasi–solid state Na-CO2 batteries with new carbon cathodes that contain curly carbon nanosheets.
fig. S25. A device for SCE.
fig. S26. Discharge/charge profiles in SCE with PVDF film protection of quasi–solid state Na-CO2 batteries.
fig. S27. Discharge/charge profiles of quasi–solid state Na-CO2 batteries at 50°C.
fig. S28. The preparation and assembly of large batteries.
fig. S29. Pouch-type battery performance.
table S1. Battery performance comparison.
movie S1. Sudden reaction on GO foam.
movie S2. Molten Na infusion into rGO foam.