

Abstract
Imprinted genes are expressed in a parent of origin manner. Dysregulation of imprinted genes expression causes various disorders associated with abnormalities of growth, neurodevelopment, and metabolism. Molecular mechanisms leading to imprinting disorders and strategies for their diagnosis are discussed in this review article.
Keywords: genomic imprinting, epigenetics, DNA methylation
Mechanisms of Genomic Imprinting
Genomic imprinting is an epigenetic mechanism that results in monoallelic expression of a gene in a parent of origin-specific manner. It provides the classic example of lifelong epigenetic memory of parental origin. To date, there have been ∼100 imprinted genes identified in humans and many more are predicted to be imprinted ( http://www.geneimprint.com/ ). Many imprinted genes are expressed in placenta and are important for fetal growth. They play an important role in postnatal growth and development, behavior, sleep, feeding, maintenance of body temperature, metabolic regulation, and stem cell maintenance and renewal. 1 The vast majority of imprinted genes are clustered in domains that vary in size from less than 100 kb to several megabases. 1 An imprinted domain generally includes both paternally and maternally expressed genes, as well as protein-coding genes and noncoding RNAs. Parent of origin-specific expression of multiple imprinted genes within a domain is under overall control of cis -acting imprinting control regions (ICRs), which are also often referred to as primary/gametic differentially methylated regions (DMRs). 1 The ICRs exhibit parent of origin-specific epigenetic modifications including DNA methylation and histone modifications, which determine whether or not the genes within the imprinted domain are expressed. In this review, we will focus on DNA methylation as it is the epigenetic modification used for molecular diagnosis due to its chemical stability and easy accessibility. DNA methylation occurs at cytosines followed by guanines (CpG dinucleotides). It is usually associated with gene silencing if located in the promoter. The allele-specific DNA methylation at ICRs is established in the germ line by the DNA methyltransferase DNMT3A and its cofactor DNMT3L. 2 3 After fertilization, the DNA methylation patterns at ICRs are stably inherited through mitotic divisions with the help of maintenance DNA methyltransferase DNMT1; also involved are other proteins such as ZFP57, 4 TRIM28, 5 and DPPA3. 6 ICRs are resistant to genome-wide demethylation and remethylation processes that occur in preimplantation embryos. 1 7 In primordial germ cells, the methylation at ICRs is erased by both passive loss through cell division in female gametes and active demethylation in male gametes. 8 The majority of known ICRs are methylated on the maternal allele and only two of clinically relevant ICRs, ICR1 at 11p15.5 and IG-DMR at 14q32, are methylated on the paternal allele ( Table 1 ). There are structural differences between maternal and paternal ICRs in terms of their genomic position within imprinted domains and CpG density. Usually, maternal ICRs are markedly CpG–rich, overlapping CpG islands. They are located at the promoters of long noncoding RNAs (lncRNAs). When a maternal ICR is unmethylated, lncRNA may be expressed, silencing protein-coding imprinted genes in cis through various mechanisms such as recruitment of histone modifying enzymes and transcriptional interference. On the other hand, when a maternal ICR is methylated the lncRNA is silenced permitting expression of protein-coding imprinted genes within the domain. 1 In contrast, paternal ICRs are typically less CpG dense and located in intergenic regions. 1 9 An insulator model was proposed for the paternal ICR1 of the well-studied IGF2-H19 domain 10 ( Fig. 1A ).
Table 1. Human imprinting disorders, their underlying molecular causes and strategies for genetic testing.
| Syndrome/OMIM#/population frequency | Imprinted domain | Clinical features | Known molecular causes | Underlying genetic/epigenetic change | Molecular genetic testing | Multiple imprinting defect |
|---|---|---|---|---|---|---|
| Beckwith-Wiedemann syndrome (BWS) OMIM#130650 1:13,700 |
11p15.5 | Macroglossia, macrosomia, abdominal wall defects, visceromegaly, neonatal hypoglycemia, hemihyperplasia. Increased risk (7.5%) for childhood tumors. The same molecular alterations as in BWS are found in approximately one-third of patients with isolated hemihyperplasia (OMIM: #235000). Incidence of BWS is increased in children born to subfertile couples and couples using ART, due to increased frequency of LOM at ICR2. 13 |
Overexpression of paternally expressed growth promoting gene
IGF2.
Loss of expression/function of maternally expressed growth suppressor gene CDKN1C. |
• LOM at ICR2–50–60% (∼2% due to micro CNVs) 54 • GOM at ICR1 −5–10% (∼35% due to micro CNVs, rarely point mutations in ICR1 OCT-binding sites) 10 54 • UPD (11) pat (usually segmental, can be mosaic)-20% • Loss of function mutations on maternal CDKN1C allele −5% (40% in familial cases) 10 • 11p15.5 CNVs up to 8.4% 54 • Cytogenetically visible alterations including paternal duplications and maternal balanced inversions and translocations involving 11p15.5 <1% 13 • Unknown ∼20% 13 |
Sporadic cases: • MS-MLPA of 11p15.5 locus • If MS-MLPA is positive without CNV, UPD analysis to delineate molecular mechanism • If MS-MPLA is negative, CDKN1C sequencing Familial cases: • CDKN1C sequencing • ICRs microdeletion/ microduplication testing If all tests are negative, consider testing in another cell type: skin fibroblasts, buccal cells, tumor biopsy, due to possibility of mosaic UPD or methylation defect. 13 Karyotype analysis can be initiated at the same time as molecular testing. 13 |
MID found in ∼26% of BWS cases with isolated ICR2 LOM.
15
Maternal NLRP2 and NLRP5 mutations are found in several families with BWS and MID. 22 26 Possible association with ART. 10 |
| Russell-Silver (RSS) OMIM#180860 1:10,000–30,000 |
11p15.5 7p11.2-p13 7q31 |
IUGR, postnatal growth retardation with normal head circumference (relative macrocephaly); failure to thrive, low BMI, typical facial features with triangular facies characterized by broad forehead and narrow chin, limb and body asymmetry present in > 50% of cases. Risk for developmental delay and learning difficulties. |
Loss of expression of paternally expressed growth promoting gene
IGF2
and/or overexpression of maternally expressed growth suppressor gene
CDKN1C
11p15.5.
Altered expression of imprinted genes on chromosome 7p11.2-p13 and 7q31. Rare genomic and eigenomic alterations at other loci. 35 64 65 66 67 |
• LOM at ICR1 at 11p15.5 −35–45% • 11p15.5 maternal duplications −1–2% • UPD (11) mat –rare • Gain of function mutation of maternal allele of CDKN1C – one RSS family 68 • UPD(7) mat- including segmental UPD of 7q-10% • Maternal duplications of 7p11.2-p13- rare • Other (sub)-microscopic imbalances −1% • Unknown ∼48% 69 • Other rare causes: LOM at IG-DMR and the MEG3- DMR at the 14q32.2, 64 66 HMGA2 mutation, 65 gain of methylation at IGF2R at 6q25.3. 35 |
• MS-MLPA of 11p15.5 locus • If MS-MLPA is positive without CNV, UPD analysis to delineate molecular mechanism at 11p15.5 • If MS-MLPA is negative, UPD 7 testing • If both 11p15.5 and UPD 7 tests are negative - array CGH 69 If all tests are negative, consider methylation testing in another cell type: skin fibroblasts, buccal cells, due to possibility of mosaic LOM at ICR1 69 |
MID is found in ∼10% of RSS patient with ICR1 hypomethylation. 15 Maternal NLRP5 mutation is found in several families with SRS and MID. 26 |
| IMAGe syndrome OMIM #614732 Unknown (rare) |
11p15.5 | IUGR, metaphyseal dysplasia, adrenal hypoplasia congenita, and genitourinary abnormalities (in males) | Gain of function of maternally expressed growth suppressor gene CDKN1C . | • Gain of function missense mutations on maternally inherited allele of CDKN1C . All pathogenic mutations described to date are located within a five amino acid region of the PCNA -binding domain. 70 71 | Targeted mutation testing of the PCNA-binding domain or sequencing of the entire CDKN1C gene. | Not applicable |
| Prader-Willi syndrome (PWS) OMIM #176270 1:17,500, Prader-Willi like syndrome (PWLS) OMIM#615547 caused by paternal MAGEL2 mutations |
15q11-q13 | Hypotonia and feeding difficulties in early infancy, global developmental delay, overeating, obesity, mild intellectual disability, hypogonadism, and specific behavioral problems including temper tantrums, stubbornness, manipulative behavior, obsessive-compulsive characteristics. | Loss of paternally expressed genes at 15q11-q13: protein-coding MKRN3, MAGEL2, NDN, NPAP1, SNURF-SNRPN and a cluster of six snoRNA genes. No single gene responsible for PWS was identified, however paternal deletions of SNORD116 snoRNAs 72 73 and paternal truncating mutations in MAGEL2 74 were reported in small proportion of PWS and PWS-like cases. | • Paternal deletion of 15q11-q13 -75–80%: a) Class I (BP1–BP3)∼30% b)Class II (BP2-BP3) ∼60% c) Atypical deletions ∼8% 75 d) Detectable chromosomal rearrangement resulting in 15q11-q13 deletion −1% • UPD(15) mat −20–25% • Imprinting defect-1%: a) Idiopathic GOM −85–90% b) Paternal ICR microdeletions deletions −10–15% • Balanced chromosomal rearrangement at 15q11–13 < 1% 76 • Mutations on paternal allele of MAGEL2 < 1% 74 |
• MS-MLPA of 15q11-q13 will detect all mechanisms except some atypical small deletions and
MAGEL2
mutations.
• If MS-MLPA test is positive without CNV, UPD analysis to delineate molecular mechanism. • If MS-MLPA test is negative, PWS diagnosis is very unlikely, however SNORD116 copy number testing and MAGEL2 sequencing can be considered. |
Single patient with clinical features of PWS and BWS, and LOM consistent with BWS and AS, and several other imprinted loci. 77 |
| Angelman (AS) OMIM#105830 1:16,000 |
15q11-q13 | Severe developmental delay/intellectual disability, severe speech impairment, gait ataxia/tremulousness of the limbs, unique behavior with an inappropriate happy demeanor that includes frequent laughing, smiling, and excitability. Seizures and microcephaly are common. | Loss of expression or function of maternally expressed gene UBE3A in the brain ( UBE3 A expression is restricted to maternal allele in brain and is biallelic in other tissues). | • Maternal deletions of 15q11–13 −70–75%: a) Class I (BP1-BP3) ∼ 40% b) Class II (BP2-BP3 ∼50% c) Longer deletions ∼10% 78 • UPD (15) pat −3–7% • Imprinting defect −2–3%: a)Maternal ICR microdeletions-10–15% b) Idiopathic LOM at ICR −85–90% (approximately one-third exhibit somatic mosaicism) • Mutations on maternal allele of UBE3A ∼10% Deletions of maternal allele of UBE3A < 1% • Balanced chromosome rearrangements <1% • Unknown-∼10% 79 80 |
• MS-MLPA of 15q11-q13 will detect all mechanisms except
UBE3A
mutations.
• If MS-MLPA is positive without CNV, UPD analysis to delineate molecular mechanism. • If MS-MLPA is negative, UBE3A sequencing. • Somatic mosaicism for LOM at ICR was reported; 81 thus, testing of another cell type can be considered if methylation test is negative. |
Not reported |
| Duplication 15q11-q13 syndrome OMIM: #608636 1:12,000 |
15q11-q13 | Autism spectrum disorder, hypotonia, developmental delay, intellectual disability, seizures | Unknown, but likely overexpression of UBE3A and nonimprinted genes located within duplicated region, such as GABA receptors. | • Maternal tandem duplication/triplication of PWS/AS critical region or supernumerary marker chromosome 15. The pathogenecity of paternal gains is uncertain. 57 | • CMA, karyotype, methylation-based test to determine parent of origin. | Not applicable |
| Transient diabetes mellitus type 1 (TNDM1) OMIM #601410 1:400,000 |
6q24 | IUGR and neonatal hyperglycemia resolving by 18 months. ∼50% of cases have relapse of diabetes in adolescence or early adulthood. Macroglossia and umbilical hernia can be present. Patients with TNDM1-MID and ZFP57 mutation can have structural brain anomalies, cardiac malformations, and developmental delay. |
Overexpression of paternally expressed genes
PLAGL1
and
HYMA1
.
PLAGL1 is transcription factor and tumor suppressor gene. HYMA1 is noncoding RNA. |
• UPD (6) pat −40% • Paternal duplications of 6q24 (majority submicroscopic)- 32% • Idiopathic LOM at maternal TNDM1 DMR- 28%. 14 |
• MS-MLPA at 6q24 will detect all mechanisms. • If MS-MLPA is without CNV positive, UPD analysis test to delineate molecular mechanism. • If no UPD or CNV detected, ZFP57 sequencing. |
MID found in ∼60% of TNDM1 patients with primary TNDM1 DMR LOM. More than half of MID cases are caused by homozygous or compound heterozygous ZFP57 mutation. 19 |
| Temple syndrome/UPD maternal 14 OMIM #616222 Unknown (rare) |
14q32 | Prenatal and postnatal growth restriction, neonatal hypotonia, feeding difficulty, and early puberty. In infancy can resemble PWS 82 or RSS. 66 |
Reduced expression of paternally expressed genes DLK1 and RTL1 . | • UPD(14)mat −70–80% • Paternal deletions 14q32.2∼10% • LOM at IG-DMR and/or MEG3 DMR ∼12% • Paternal microdeletions of IG-DMR ≤2% 42 |
• MS-MLPA testing at 14q32. • If MS-MLPA is positive without CNV, UPD analysis to delineate molecular mechanism |
Not reported |
| Kagami-Ogata Syndrome(KOS)/UPD paternal 14 OMIM #608149 Unknown (rare) |
14q32 | Facial abnormality, small bell-shaped thorax, abdominal wall defects, placentomegaly, and polyhydraminos. |
Overexpression of paternally expressed gene
RTL1.
Maternally expressed RTL1 antisense transcript (RTL1-as) is a micro-RNA down-regulating RTL 1 expression in trans, thus maternal deletions involving RTL1 and RTL1- as result in mild KOS phenotype even if RTL1 is present in single copy. 83 |
• UPD(14) pat-65–70% • Maternal deletions 14q32.2–15–20% • GOM at IG-DMR and/or MEG3 DMR 10–15% • Maternal ICR microdeletions (IG-DMR and/or MEG3 DMR) < 5% 42 84 Note: MEG3 DMR is unmethylated in placenta and cannot be used for prenatal diagnosis in CVS. |
• MS-MLPA testing at 14q32. • If MS-MLPA is positive without CNV, UPD analysis to delineate molecular mechanism. |
Not reported |
| Pseudo-hypoparathyroidism type 1a (PHP1a) OMIM #103580 1:150,000 for PHP1a, PHP1b and PPHP (AHO without PHO) |
20q13.3 | AHO: short stature, round face, subcutaneous calcification or ossifications, brachymetacarpia, and cognitive impairment. + End-organ resistance to PTH, TSH, and gonadotropins. |
AHO features are caused by constitutional 50% reduction of stimulatory G-protein subunit Gsα activity. Hormone resistance is caused by loss of function of Gsα in endocrine tissues (pituitary, thyroid, and ovary) with paternal imprinting (silencing) of Gsα. |
• Loss of function mutations on maternal allele of Gsα (exons 1–13 of
GNA
S) – majority of cases.
• Microdeletions of maternal alleles of Gsα- rare 85 Inactivating mutations on paternal allele of Gsα cause AHO without hormone resistance. |
• Sequencing of
GNAS
locus.
• If negative, CNV analysis of GNAS . |
Not applicable |
| Pseudo-hypoparathyroidism type 1b (PHP1b) OMIM #603233 |
20q13.3 | End-organ resistance to PTH, TSH, and gonadotropins. Some PHP1b cases (based on molecular diagnosis) can manifest mild features of AHO. 86 |
Loss of expression of Gsα in endocrine tissues (pituitary, thyroid, and ovary) with paternal imprinting (silencing) of Gsα. |
• LOM at maternal
GNAS1A
DMR (sometimes accompanied by LOM at
GNASXL/NESPAS
and/or GOM at
NESP55
DMRs)—majority of cases
• UPD(20) pat –rare • In familial cases, LOM is associated with maternal deletions in cis at STX16 or NESP55 . 87 |
• MS-MLPA testing at 20q13.3. • If MS-MLPA is positive without CNV, UPD analysis to delineate molecular mechanism. 88 • Somatic mosaicism for methylation defect was observed, 88 thus testing of another cell type can be considered in case of negative result. |
∼12% of PHP1b cases with idiopathic methylation defects at GNAS locus exhibit MID. 15 |
Abbreviations: AHO, albright hereditary osteodystrophy; ART, assisted reproductive technologies; BMI, body mass index; BP, break point; CMA, chromosomal microarray; CNV, copy number variant; CVS, chorionic villus samples; DMR, differentially methylated region; GOM, gain of methylation; ICR, imprinting control region; IUGR, intrauterine growth restriction; LOM, loss of methylation; MID, multiple imprinting defect (methylation alterations at multiple ICRs); MS-MLPA, methylation-specific multiplex ligation-dependent probe amplification; PCNA, proliferating cell nuclear antigen; PTH, parathyroid hormone; snoRNA, small noncoding RNAs; SRO, smallest region of overlap; TSH, thyroid-stimulating hormone; UPD, uniparental disomy.
Fig. 1.
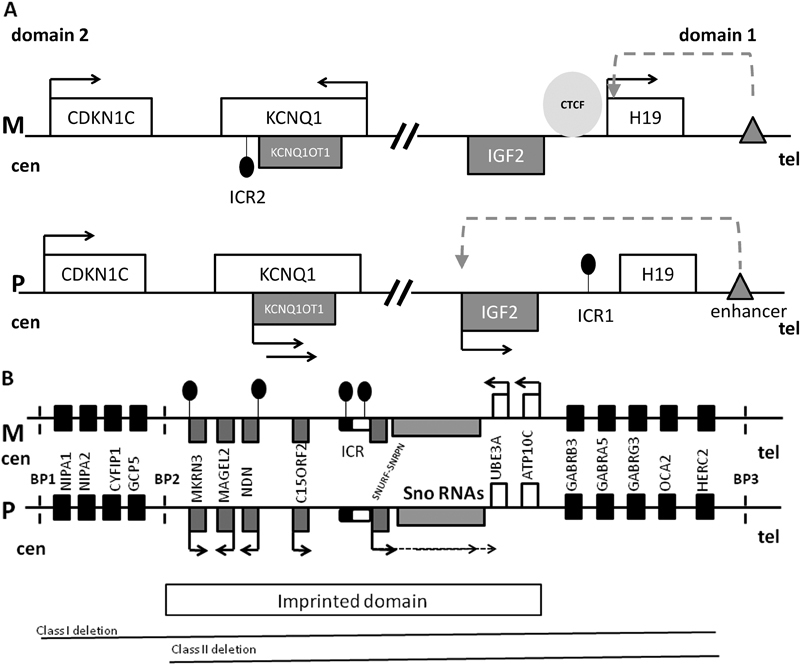
( A ) The map of chromosome 11p15.5 imprinted cluster consisting of domain 1 and 2. M is maternal chromosome, P is paternal chromosome, cen is centromere, tel is telomere, white rectangles are maternally expressed genes, gray rectangles are paternally expressed genes, black arrows indicate gene expression, black circles indicate DNA methylation, gray triangle is enhancer, dashed arrow indicates accessibility of IGF2 promoter to the enhancer, and light gray circle is CCCTC-binding factor (CTCF). In domain 1, imprinting control region 1 (ICR1) is located upstream of H19 noncoding RNA and is methylated on the paternal allele. Methylation prevents CTCF binding to the paternal ICR1, permitting access of the IGF2 promoter to the downstream enhancer. Thus, IGF2 is expressed, whereas H19 is silenced. On the maternal allele, CTCF binds to the unmethylated ICR1, blocking IGF2 promoter access to the enhancer. Thus, IGF2 is silenced and H19 uses the enhancer and is transcribed. In domain 2, ICR2 is located at the 5′ end of the noncoding RNA KCNQ1OT1 (KCNQ1 opposite strand transcript) and is methylated on the maternal allele. When ICR2 is unmethylated, KCNQOT1 is transcribed, KCNQ1 and CDKN1C are silenced in cis . Methylation of ICR2 results in KCNQOT1 silencing and expression of KCNQ1 and CDKN1C. IGF2 promotes growth, and CDKN1C suppresses growth. Overexpression of IGF2 and/or loss of expression of CDKN1C is associated with Beckwith–Wiedemann syndrome (overgrowth), whereas overexpression of CDKN1C and/or loss of expression of IGF2 is associated with Russell–Silver syndrome (growth restriction). ( B ) Map of human chromosomal region 15q11-q13 involved in Prader-Willi (PWS), Angelman (AS), and 15q11q13 maternal duplication syndromes, containing an imprinted domain as well as biallelically expressed genes. M is maternal chromosome, P is paternal chromosome, cen is centromere, tel is telomere, white rectangles are maternally expressed genes, gray rectangles are paternally expressed genes, black arrows indicate gene expression in a parent of origin-specific manner, and black rectangles are biallelically expressed genes. Imprinted control region (ICR) is shown as rounded rectangle with the black half being the PWS critical element and the white part the AS critical element; black circles indicate DNA methylation. SNURF/SNRPN gene expression is regulated by ICR methylation and it is expressed in multiple splice forms, including the UBE3A antisense transcript which is paternally expressed in brain and silences paternal UBE3A transcription in brain. Clusters of C/D box small nucleolar RNAs (snoRNAs) are encoded within introns of SNURF-SNRPN and are regulated by its promoter. Promoters of MKRN3 and NDN are methylated on the maternal allele. Dashed vertical lines show the regions of common break points (BP) located within regions of low copy repeats. The most common causes of PWS and AS are paternal and maternal deletions, respectively, occurring between BP1 and BP3 or between BP2 and BP3. It is known that the critical gene for AS is UBE3A which is expressed from the maternal allele in human brain and biallelically in other tissues. The relative contributions of single genes to PWS and maternal 15q11-q13 duplication syndrome remain unknown. The drawing is not to scale.
In addition to ICRs there are also secondary or somatic DMRs (sDMRs) found in the promoters of some protein-coding imprinted genes which also exhibit parent of origin allele-specific DNA methylation acquired in somatic cells. 11 However, the promoters of the majority of protein-coding imprinted genes are unmethylated on both parental alleles. 11 The complexity of imprinted gene regulation is exemplified by two imprinting domains located at 11p15.5 and 15q11-q13 ( Fig. 1 ).
Genomic Imprinting and Human Disease
The consequence of genomic imprinting is that only one of the two parental alleles is active. Thus, loss of function of an active allele of an imprinted gene cannot be compensated by the silent allele resulting in a disease phenotype. On the other hand, a mutation in the inactive allele of an imprinted gene does not have phenotypic consequences. 7
There are several human pediatric conditions, known as imprinting disorders (IDs), which are caused by abnormal expression of imprinted genes at seven imprinted domains on six human chromosomes ( Fig. 2 , Table 1 ). These disorders can be caused by several types of molecular alterations: (1) copy number variation (CNV, loss or gain) overlapping imprinted genes; (2) uniparental disomy (UPD)— inheritance of both chromosomes in a pair from the same parent; (3) point mutation of the active allele; (4) epimutation defined as a heritable change in gene activity that is not associated with an alteration of DNA sequence but rather with gain or loss of DNA methylation at ICR; (5) microdeletion or microduplication restricted to ICR, causing loss or gain of DNA methylation; and (6) structural chromosome rearrangements ( Fig. 3 , Table 1 ).
Fig. 2.
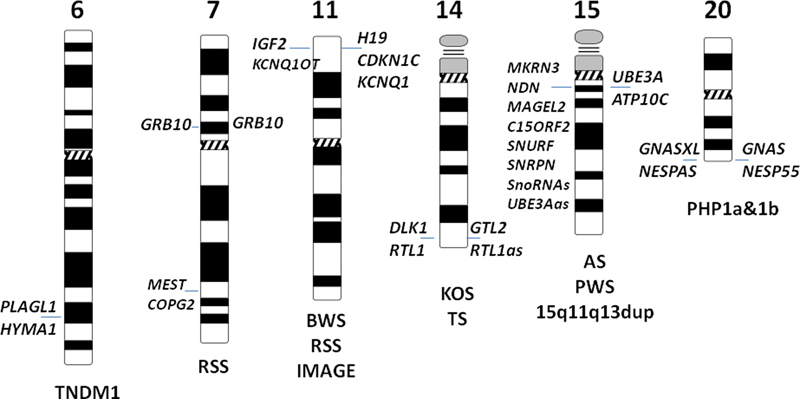
Map of human imprinted domains known to be associated with disease. Paternally expressed genes are shown on the left side of the chromosome and maternally expressed genes are shown on the right side of the chromosome (parent of origin-specific expression of GRB10 is different in different tissues). Associated conditions are shown under the chromosome. TNDM1 is transient neonatal diabetes mellitus (TNDM1), RSS is Russell–Silver syndrome, BWS is Beckwith–Wiedemann syndrome, KOS is Kagami-Ogata syndrome, TS is Temple syndrome, AS is Angelman syndrome, PWS is Prader-Willi syndrome, and PHP1a&b are pseudo-hypoparathyroidism types 1a&b.
Fig. 3.
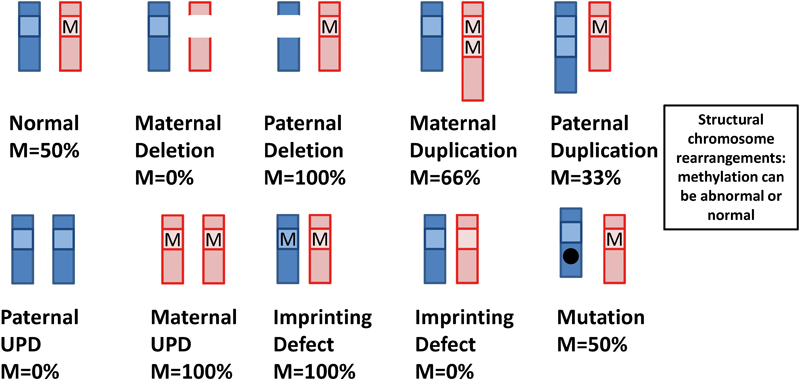
Schematic drawing of types of molecular alterations observed in imprinting disorders and their effect on DNA methylation at imprinting control regions (ICRs) in a scenario when methylation occurs on the maternal allele. The paternal chromosome is shown in light gray, and the maternal chromosome is shown in dark gray. M- is DNA methylation at the ICR. Imprinting defect can result either from idiopathic DNA methylation change or microdeletion/microduplication at ICR.
The frequency of underlying defects varies among IDs. For example, in Beckwith–Wiedemann syndrome (BWS), Russell–Silver syndrome (RSS), and transient neonatal diabetes mellitus type 1 (TNDM1), idiopathic epimutation is the most frequent underlying cause. In contrast, for Prader–Willi syndrome (PWS) and Angelman syndrome (AS), the most frequent cause is de novo deletions, due to the presence low copy number repeats in 15q11-q13 region making this region susceptible to nonhomologous recombination. For pseudo-hypoparathyroidism type 1a (PHP1a) and IMAGE syndrome, point mutations are the only known cause. In other IDs, point mutations are rare and are usually found in familial cases ( Table 1 ).
Inheritance of Imprinting Disorders
The recurrence risk of ID depends on the underlying molecular alteration. UPD and idiopathic loss of DNA methylation usually occur sporadically and recurrence risk is low. In contrast, CNVs or point mutations can either occur de novo or be inherited from the parent, which could be affected or unaffected, depending which grandparent transmits the mutant allele. Whether a specific ID is predominantly sporadic or there is a substantial proportion of familial cases depends on the reproductive fitness of this disorder. For example, in severe developmental disorders, such a AS and PWS, the genetic alterations almost exclusively occur de novo , and recurrence risk is low, with the exception of point mutations of UBE3A which cause AS and are associated with 50% recurrence risk when are inherited from the unaffected mother, and are not associated with a phenotype when inherited from the father. 12 In contrast, patients with BWS have generally favorable outcomes as adults and approximately 15% of BWS cases are familial. 13 In these families, the disorder is inherited as a dominant trait with parent of origin-specific effect and a recurrence risk of up to 50% when transmitted from the parent contributing the expressed allele. 13 TNDM1 can be inherited as a recessive trait when caused by homozygous or compound heterozygous mutations in the ZFP57 gene with a recurrence risk of 25% 14 (see Multiple Imprinting Defect section).
Multiple Imprinting Defects
Recent research studies have shown that the methylation alterations in IDs are not always restricted to one imprinted domain. In some IDs, methylation alterations can be found at multiple ICRs. Several terms have been used in literature to describe this phenomenon, namely multiple imprinting defect (MID), multilocus methylation defect (MLMD), hypomethylation of imprinting (HIL), and hypomethylation syndrome (HMS). To date, cases of MID have been reported in four IDs where primary epimutation is one of the most frequent causes of the disorder: TNDM1, BWS, RSS, and PHP1b ( Table 1 ). 15 Aberrant DNA methylation (mostly loss of maternal methylation) at multiple imprinted loci is frequently found to be mosaic, suggesting that the mechanism is failure of postfertilization maintenance of DNA methylation imprints. 16 In TNDM1, the MID has been associated with mutations in ZFP57 gene, 17 encoding Kruppel-associated box domain zinc finger protein, involved in maintenance of DNA methylation at imprinted loci. 18 It is estimated that ∼60% of TNDM1 MID cases carry homozygous/compound heterozygous mutation in ZFP57 , making it ∼5% of all TNDM1 cases. 19 Individuals with ZFP57 mutations, in addition to loss of methylation at TNDM1 DMR, always exhibit loss of methylation at the PEG3 DMR (chr19q13.43) and GRB10 DMR (7p12.1) and sometimes loss of methylation at the MEST DMR (7q32.2), ICR2 (11p15.5), and the GNASXL/NESPAS DMR (20q13.3). 19
In addition to ZFP57 , other genes implicated in MID to date, include the NLRP2 , NLRP5, NLRP7 , and KHDC3L genes. Recessive maternal effect mutations of NLRP7 and KHDC3L are found in familial cases with recurrent biparental hydatiform moles where loss of methylation is observed at all maternal DMRs. 20 21 In addition, maternal effect homozygous mutations in NLRP2 were identified in one family with BWS/MID. 22 Sequencing of these four genes as well as other candidate genes ( DNMT3L and TRIM28 ) involved in the regulation of genomic imprinting failed to identify causative mutations in BWS, SRS, and PHP1b MID cases. 23 24 25 Recent exome sequencing of a large cohort of families with children affected with MID has identified mutations in NLRP5 gene in both heterozygous and compound heterozygous forms in five mothers of children with various forms of MID, including BWS, SRS, and MID with unspecific neurodevelopmental presentations, these mothers also exhibited higher than usual rates of miscarriages. 26 An environmental factor that has been suggested to predispose to MID in BWS and TNDM1 is the conception with the help of assisted reproductive technologies. 17 27 28
Clear genotype/epigenotype/phenotype correlations are not well established for MID disorders, with some patients not presenting any additional clinical features compared with isolated epimutation associated with the primary diagnosis, whereas other patients may present more severe phenotypes. For example, patients with TNDM1 MID caused by ZFP57 mutation may present with various combinations of congenital anomalies and developmental delay, in addition to classical features of TNDM1. 19 24 29 For BWS, significantly lower birth weight, decreased frequency of nevus flammeus and hemihyperplasia, 30 and significant increase of mild-to-moderate developmental delay 31 in BWS-MID compared with isolated ICR2 epimutation were reported in two studies, whereas others do not report any significant phenotypic differences. 27 32 No significant differences were found for RSS MID 31 32 33 34 35 and PHP1b MID 24 36 37 versus respective single epimutation phenotypes.
The variability of clinical MID phenotypes can potentially be explained by mosaic methylation patterns in different tissues, that is, the methylation pattern in blood leukocytes, the tissue most frequently used for diagnostic testing, is different from the tissue important for the development of specific disease phenotypes. There is also a possibility of contribution of abnormal methylation at other loci not tested by current assays or other genomic determinants that have not been identified. In addition, long-term follow-up in these cases might reveal as yet unknown adult-onset disorders that can be associated with MID.
To date, routine practice in genetic diagnostic laboratories is testing for epimutations at imprinted loci specific for ID in question. Testing for methylation alterations at multiple loci is usually performed through research laboratories, experienced in multilocus analysis of DNA methylation. For TNDM1, the current recommendation is that all patients with an epimutation at the TNDM1 DMR without an underlying genomic alteration (UPD or duplication) are tested for ZFP57 mutation. This allows estimation of recurrence risk (25% for ZFP57 mutation). 14 However, given the clinical variability of phenotype, counseling of families with ZFP57 mutations may be challenging. 19
Genetic Testing of Imprinting Disorders
This section will focus on description of molecular genetics and cytogenetics methods used to diagnose IDs.
DNA Methylation Analysis
Abnormal DNA methylation at ICRs is a feature of multiple molecular alterations observed in IDs, including epimutation, CNV, and UPD ( Fig. 3 ); therefore, for majority of IDs, DNA methylation analysis is used as first-tier diagnostic test ( Fig. 4 ).
Fig. 4.
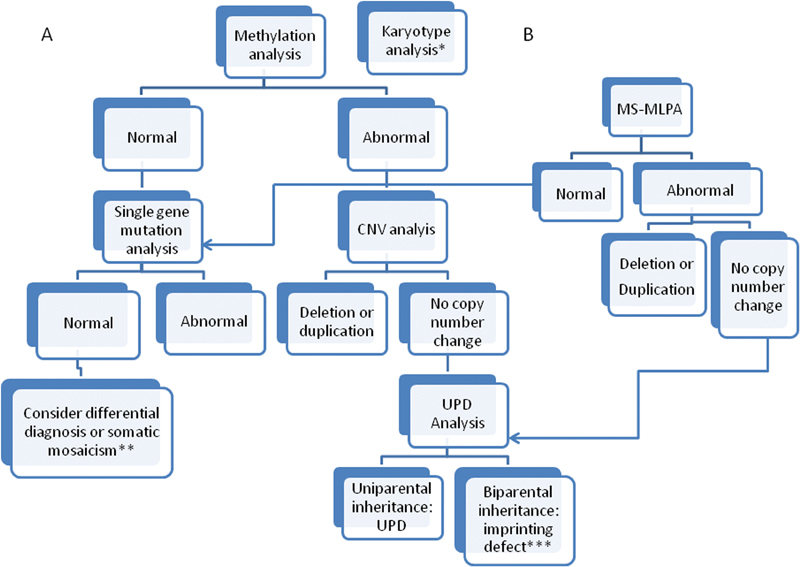
Testing strategies for imprinting disorders (ID) based on initial methylation analysis ( A ) or simultaneous methylation and CNV analysis by MS-MLPA ( B ). *Karyotype analysis to test for structural chromosome rearrangements can be initiated at the same time as methylation analysis if this is a known cause for an ID. **If all testing is negative, but the phenotype is highly suggestive of a specific ID, testing of another cell type (skin fibroblasts, buccal cells) can be considered to detect somatic mosaicism. ***In the majority of cases, an ID will be due to an idiopathic methylathion change, however depending on the resolution of previous CNV analysis, testing for ICR microdeletion/microduplication can be performed to rule out inherited genomic alterations.
There are a variety of molecular techniques that can be used to assess DNA methylation. 38 However, many of them are too labor intensive/time consuming to be used in routine clinical diagnostic laboratories. Here, we will focus on techniques that have been used in diagnostic laboratories and also some of the new technologies that are currently used in the research setting but have potential in future to be used for diagnostics.
DNA methylation analysis techniques can be based either on restriction enzyme digestion or sodium bisulfite conversion. Some restriction enzymes are sensitive to methylation of cytosine in CpG dinucleotides within the restriction site. Such sites can be used for analysis of methylation status, that is, unmethylated DNA molecules will be cut at this site, whereas methylated molecules remain undigested. As a control, a restriction enzyme cutting at the same sequence but not sensitive to methylation is often used. The generated restriction pattern can be analyzed by downstream techniques such as Southern blot hybridization or methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA).
Sodium bisulfite conversion methods are based on chemical conversion of unmethylated cytosine to uracil; methylated C is resistant to this conversion. The ratio of DNA molecules with methylated C to unmethylated C (T in amplified DNA) can then be assessed in qualitative and quantitative assays including methylation-specific PCR (MS-PCR), methylation-specific single nucleotide primer extension (MS-SNuPe), MALDI-TOF mass spectrometry (Sequenom), sequencing methods including Sanger, pyrosequencing, and next-generation sequencing, as well as methylation BeadChip microarrays using Infinium chemistry. 38
These techniques differ by the number and location of CpG sites they are capable of detecting, for example, Southern blot and MS-PCR target one CpG, whereas Sanger, pyrosequencing, and MALDI-TOF mass spectrometry can analyze multiple consecutive CpGs within the same amplification product. The other techniques can be mutiplexed and multiple independent CpGs can be analyzed within one assay, for example, about a dozen of CpGs can be assayed using MS-MLPA or MS-SNuPE and up to several hundred thousand can be assayed using the methylation BeadChip microarrays (Illumina).
Until recently, Southern blot and MS-PCR were the most frequently used techniques in diagnostic laboratories. Their main advantage is that they do not require special equipment. However, their major disadvantage is that they provide only qualitative data and assess only one CpG site in one assay. In addition, a Southern blot is labor intensive, requires large quantities of DNA and the use of radioactive labeling. In past years, a semiquantitative technique, MS-MLPA, has become a method of choice in many diagnostic laboratories. MS-MPLA can simultaneously detect methylation status and copy number change for several dozens of selected sequences. It is based on hybridization of two adjacent locus-specific probes, ligation and simultaneous digestion with methylation-sensitive enzymes and further amplification of target sequences using universal primers, the fragment length of which is further analyzed using an automated sequencer. If the probes are not ligated due to deletion of target sequences or digestion of umethylated CpG sites, no amplification product is formed. The copy number change and relative percent of methylation can be estimated by comparing peak heights of test probes to control probes with normal copy number and fully methylated loci. 39 In the case of IDs with CNVs, the conclusive molecular diagnosis can be achieved using MS-MLPA, which is particularly useful for AS and PWS, where deletions are the most frequent causal mechanism. Another advantage of MS-MLPA is the option of testing several independent CpG sites within an ICR, reducing false-positive results due to SNPs in the probe-binding site.
Copy Number Variation Analysis
Copy number analysis is usually used simultaneously with methylation test (MS-MLPA) or as follow-up testing for abnormal methylation result ( Fig. 4 ).
Copy number change can be detected by a variety of methods including quantitative PCR, chromosomal microarray (CMA), MLPA, and fluorescent in situ hybridization (FISH). FISH is only suitable for identification of relatively large deletions (> 50 kb); other methods can identify both large copy number variants and microdeletions/microduplicatons within ICRs if the probes/primers are located within this region. As microdeletion/microduplication restricted to ICR can be associated with a 50% recurrence risk, it is important to differentiate DNA methylation changes associated with genomic alterations from an idiopathic DNA methylation defect.
Currently, CMA is a first-tier diagnostic test for individuals with developmental delay, intellectual disability, autism, and congenital anomalies. 40 Using CMA, infants and young children with AS and PWS caused by deletions can be identified prior to the time when the clinical phenotype has become fully evident and a disorder specific methylation-based diagnostic test is considered. However, CMA will not distinguish paternal versus maternal deletion and if a deletion of AS/PWS critical region was identified, a methylation test is then recommended to confirm the diagnosis and verify the parent of origin of the deletion.
Uniparental Disomy
In the course of a molecular diagnosis of ID, once a methylation alteration is identified and copy number alteration is ruled out, testing for uniparental disomy (UPD) is recommended. UPD testing is also recommended if structural or numerical abnormalities involving imprinted chromosome regions are identified in prenatal diagnosis (see section Prenatal Diagnosis of Imprinting Disorders).
UPD is defined as the inheritance of both homologous chromosomes of a pair from one parent and no copy from the second parent in a diploid genome. 41 Depending on parent of origin, UPD can be paternal or maternal. In addition, UPD can be either isodisomic or heterodisomic. Isodisomy refers to the inheritance of two identical chromosomes from the same parent; this is associated with loss of heterozygosity. Heterodisomy occurs when two homologous chromosomes are inherited from the same parent. Due to meiotic recombination, uniparentally inherited chromosomes are frequently a mixture of isodisomic and heterodisomic segments. In addition to UPD for an entire chromosome, segmental UPD for a region of chromosome and biparental inheritance of the rest of the chromosome can also occur.
The mechanisms of generating different types of UPD are reviewed in 42 43 44 and are shown in Fig. 5 . UPD for the majority of human chromosomes is without any adverse phenotypic consequences, with the exception of situations of isodisomic inheritance of a mutant allele for an autosomal recessive condition from a carrier parent. However, UPD in a chromosomal region carrying imprinted domain ( Table 1 , Fig. 2 ) can result in imprinted gene disorder depending on the parent of origin.
Fig. 5.
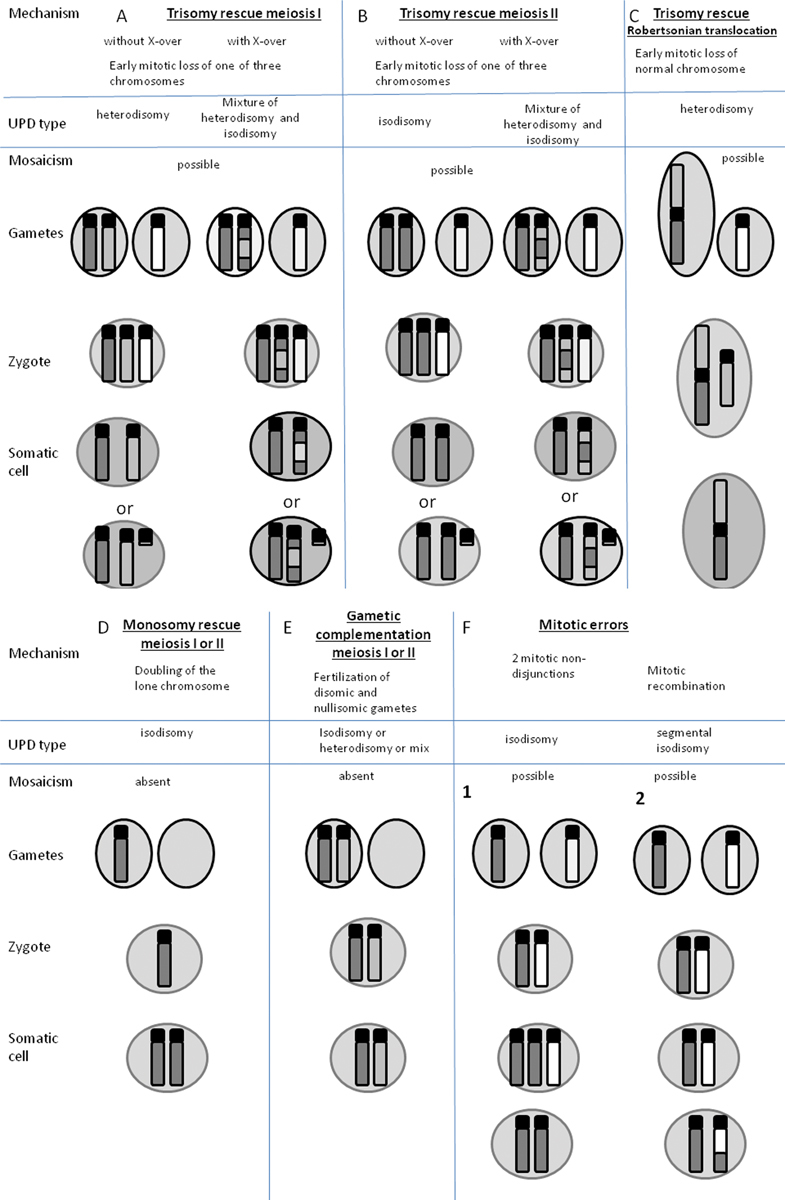
Possible mechanisms of uniparental disomy (UPD) formation. Dark gray and light gray are maternal nonhomologous chromosomes, white is paternal chromosomes. The examples of UPD are shown for maternal chromosomes, however paternal UPD can occur by the same mechanism. Where mosaicism is shown as a possible outcome, there is a possibility of the presence of trisomic cells in the placenta or the fetus for ( A ), ( B ), ( C ), and ( F1) , in the case of segmental isodisomy ( F2 ), mosaicism is present for a biparental cell line. ( A , B , C ) Trisomy can result from meiosis I or II nondisjunction in a parent with a normal karyotype. Rescue can occur by complete loss of one of the parental chromosomes or formation of a residual small supernumary marker chromosome. Trisomy rescue and UPD can also occur if the egg or sperm carries a balanced translocation and nondisjunction occurs. ( C ) Oocyte carriers of a homologous Robertsonian translocation or isochromosome. ( D ) Monosomic fertilized zygote has to undergo immediate endoreduplication of monosomic chromosomes, otherwise the embryo won't survive. ( E ) Gamete complementation is a very rare event as it assumes nondisjunction of the same chromosome in both oocyte and sperm. ( F1 ) Two sequential mitotic nondisjunction events can lead to isodisomy. ( F2 ) Segmental UPD can result from homologous chromatid exchange during mitosis, resulting in segmental loss of heterozygosity and mosaicism for a UPD cell line and a biparental cell line. (This figure was adapted with modifications from Yamazawa K, Ogata T, Ferguson-Smith AC. 2010). 63
Routinely, UPD testing is performed by genotyping of microsatellite markers distributed along the length of the chromosome in question in the proband and both parents (trio analysis). The American College of Medical Genetics guidelines requires a minimum of two informative markers to establish uniparental or biparental inheritance of an entire chromosome. If UPD is identified, it is also recommended that at least one other chromosome is tested to demonstrate biparental inheritance and verify paternity. 45 There are no established guidelines for diagnosing segmental UPD. If microsatellite analysis reveals uniparental origin of the tested chromosome, a diagnosis of UPD can be made. If the inheritance of the tested chromosome is biparental, it is presumed that the ID is caused by an imprinting defect. In this situation, if the ICR was not previously assessed for microdeletion/microduplication by MS-MLPA or targeted CNV analysis, it is important to perform this testing at this point to better define the recurrence risk.
The UPDs occurring due to mitotic errors are often mosaic, which can complicate the establishment of the diagnosis. For example, the majority of individuals with BWS and UPD for chromosome 11 have segmental paternal mosaic UPD of 11p15. If a low level of UPD mosaicism occurs in the tissue tested (usually peripheral blood leukocytes), the methylation-based test could be negative. Thus, if there is a strong clinical suspicion of BWS, but test results in peripheral blood are negative, testing in additional cell types (e.g., skin fibroblasts, tumor biopsy, buccal epithelial cells) is clinically indicated. 13 In addition, the majority of currently used CMAs also contain probes for single nucleotide polymorphisms (SNPs), and SNP-based CMA has been shown to be capable of detecting mosaicism as low as 5%. 46 Thus, SNP-CMA testing of the proband and parents can be considered if mosaic UPD is suspected.
Also, SNP-based CMA could suggest UPD in children with IDs, before specific clinical features become apparent as CMA is used as a first-tier diagnostic test for infants or young children with developmental delay/congenital anomalies. 40 Long contiguous stretches of homozygosity identified by SNP-based CMA are suggestive of either consanguinity/origin from isolated population if distributed throughout the genome or could be a hallmark of UPD if found for a single chromosome. 43 Long contiguous stretches of homozygosity, however, are not diagnostic for UPD and follow-up testing using either methylation assays and/or microsatellite analysis in trios should be performed.
In addition, for cases with UPD of one chromosome, there are also rare cases with genome-wide mosaic uniparental disomies characterized by somatic mosaicism of a normal cell line and another cell line with two sets of chromosomes of uniparental origin. More than 10 live-born cases of paternal mosaic genome-wide UPD, presenting with complex variable phenotypes, including features of BWS, AS, TNDM1, 47 48 49 and three live-born cases of mosaic genome-wide maternal UPD with phenotypic features of RSS have been reported to date. 44 50 51
Cytogenetic Analysis
Rare cases of IDs can be associated with microscopically visible chromosomal abnormalities: including balanced rearrangements disrupting expression of genes within imprinted domains but not resulting in methylation alterations. For example, for BWS, balanced inversions and translocations of 11p15.5 or unbalanced rearrangements resulting in copy number changes at imprinting domains can occur. 52 In ID cases caused by UPD, karyotype abnormalities can sometimes also be observed, for example, Robertsonian translocations involving chromosome 14 or 15 and small supernumerary marker chromosomes (SMCs) ( Fig. 5 ). 42 53 There is no consensus regarding the need for routine karyotype analysis for diagnosis of IDs as structural chromosomal abnormalities are relatively rarely observed in IDs (< 1% of cases, Table 1 ), and these analyses are costly and labor intensive. However, the importance of karyotype analysis is that in some situations parents can be carriers of balanced rearrangements with potential risks for UPD or unbalanced karyotypes in future offspring. Thus, the need for karyotype analysis should be assessed on an individual basis. If a chromosome rearrangement is identified in a proband, parental karyotypes should be also assessed to define the future risks to offspring for chromosomal abnormalities.
Sequence Analysis
In two IDs, PHP1A and IMAGE syndrome point mutations are the main disease-causing mechanism: PHP1a is caused by loss of function mutations on the maternal allele of GNAS , and IMAGE syndrome is caused by gain of function mutations on the maternal allele of CDKN1C ( Table 1 ). For these two disorders, Sanger sequencing can be undertaken as a first-tier diagnostic test. There are also several IDs (BWS, TNDM1, AS, and PWS) where point mutations are found in a minority of cases ( Table 1 ). Thus, Sanger sequencing is usually performed if first-tier methylation testing is negative in sporadic cases. In familial cases, sequencing can be performed as first-tier diagnostic test or in parallel with ICR microdeletion/microduplication analysis. Currently, next-generation sequencing panels designed for simultaneous testing of a wider spectrum of patient phenotypes, such as overgrowth, intellectual disability, or neonatal diabetes ( www.genetests.org ) may include genes that have been implicated in causing IDs.
Diagnostic Algorithms for Imprinting Disorders
For all IDs except for 15q11-q13 duplication, PHP1a and IMAGE syndrome, the diagnostic algorithm is similar, starting with a DNA methylation test, and if positive followed by copy number and/or UPD tests. If the DNA methylation test is negative, sequencing of imprinted genes can be undertaken ( Fig. 4 , Table 1 ). An abnormal DNA methylation test is reflective of one of the following molecular alterations: CNV, including large CNVs of part or entire imprinted domains or ICR microdeletions/microduplications, UPD, or an idiopathic methylation defect. However, with the exception MS-MLPA, DNA methylation testing alone will not provide information regarding the underlying molecular mechanism. Establishing the correct molecular mechanism of the ID is important for several reasons. First, differentiating heritable and nonheritable abnormalities is important for estimating recurrence risk, that is, risk for offspring and other family members. For example, a recent study estimating CNV rates in BWS using MS-MLPA revealed a higher frequency of CNVs (8.4%) than previously anticipated (2–6%), with one third of these CNVs being inherited. 54 Second, different molecular subtypes of IDs may be associated with phenotypic differences. Therefore, knowledge of molecular subtype could provide phenotype/genotype correlations. This is relevant to patient management in that BWS patients with paternal UPD 11 or gain of methylation at ICR1 have a higher embryonal tumor risk than patients with ICR2 loss of methylation or CDKN1C mutations. 13 In PWS, the risk of autism and psychosis is significantly elevated in patients with maternal UPD 15 versus other molecular subtypes. 12 Third, idiopathic DNA methylation defects without underlying genomic alterations in cis can be associated with MID, which has further implications for patient management/genetic counseling (see MID section for discussion).
Prenatal Diagnosis of Imprinting Disorders
Prenatal testing is possible for future pregnancies in families in which an ID diagnosis in proband has been confirmed, the molecular mechanism of ID has been defined, and the recurrence risk has been estimated. For cases with inherited mutations or genomic alterations, prenatal diagnosis should be offered as recurrence risk is high, that is, 50% for the majority of IDs and 25% for TNDM1 caused by ZFP57 mutation. In cases with de novo mutations or genomic alterations, the recurrence risk is low; however, the possibility of germ-line mosaicism exists, 55 56 thus, prenatal testing could be offered for reassurance. UPD with normal karyotype is considered a sporadic event and invasive prenatal testing is not indicated. 57 However, if a parent is a carrier of Robertsonian translocation involving chromosomes 14 and 15, he/she is at risk of having a child with UPD for these chromosomes and prenatal karyotype analysis and UPD testing should be offered. 58 For IDs caused by idiopathic methylation defects, recurrence risk is also very low. However, a recurrence cannot be completely excluded (e.g., unknown mutation affecting methylation at ICR in cis or trans ) and prenatal diagnosis could be offered for reassurance. However, due to the uncertainty of the exact timing of the establishment of stable DNA methylation patterns in the developing fetus, and because some DMRs can exhibit different methylation patterns in placental versus fetal cells, 42 a methylation analysis for prenatal diagnosis of an ID should only be used if validated for specific DMRs in the appropriate cell type (chorionic villus samples [CVS] and amniocytes). For example, methylation at the MEG3 sDMR is routinely used for diagnosis of Temple and Kagami–Ogata syndromes in peripheral blood cells. However, it is unmethylated on both alleles in normal placenta and thus cannot be used for prenatal diagnosis in CVS. 42 53
Ultrasound findings can also be indicative of IDs and trigger prenatal testing for specific IDs. A small thorax with ribs resembling coat hangers could be a sign of Kagami–Ogata syndrome (paternal UPD 14), which has a poor prognosis; prenatal testing should be offered. 57 59 Omphalocele, overgrowth, and macroglossia are suggestive of BWS, and prenatal testing should be offered to enable better perinatal management of these patients. 59 Asymmetrical growth restriction could be a sign of RSS, and prenatal testing is also possible, but may be not pursued due to the relatively mild phenotype and generally benign perinatal course. 59
A question of an ID can arise in a pregnancy without a previously affected child or abnormal ultrasound findings, if UPD is suspected. This may occur if a prenatal karyotype or microarray is performed for advanced maternal age or another indication and numerical or structural chromosome abnormalities involving imprinted chromosomal regions are detected. For example, prenatal testing for UPD is recommended in cases of confined placental mosaicism for trisomies involving clinically relevant imprinted chromosomes as the risk for UPD is estimated to be 11 to 25%. 45 59 The presence of a nonhomologous Robertsonian translocation is associated with a 0.6 to 0.8% risk for UPD, and the risk is very high (60%) for homologous Robertsonian translocations; UPD testing is indicated if chromosomes 14 or 15 are involved. 58 59 An SMC 15 not containing euchromatic material has been reported in cases of paternal or maternal UPD 15 and a risk of 5% for UPD has been estimated. Thus, testing for UPD is indicated if SMC (15) or (14) is detected on prenatal karyotype. 58 59
Future Directions
In the past few years, there has been a lot of discussion in the literature regarding the need for multilocus testing in IDs associated with MID to elucidate the clinical spectrum of the multiple MIDs. 15 16 19 31 In addition, it would be more efficient to utilize platforms that support testing of imprinted DMRs genome wide. Various methylation array platforms capable for simultaneous analysis of thousands of CpG have been used in epigenetic research; however, the majority of them are not suitable for routine diagnostic use as they are labor intensive and require sophisticated bioinformatics tools for analysis. 38 A promising array platform that could be used in molecular diagnostics is the Methylation BeadChip microarrays using Infinium chemistry (Illumina), which have single CpG resolution of up to several hundred thousand sites genome wide. Despite some methodological challenges due to the presence of two types of probes 60 and cross-reactivity of some of the probes, 54 it represents a robust, high throughput method with the capability to run 96 samples simultaneously. Various versions of this arrays are widely used in epigenetic research including the discovery of new imprinted genes 61 and the characterization of methylation profiles in patients with MID. 24 34 47 62
It is also possible that in parallel to the development of next-generation sequencing in genetic diagnostics, next-generation bisulfite sequencing could be implemented for molecular diagnosis of imprinting and other epigenetic disorders in certain diagnostic laboratories, thereby providing simultaneous detection of methylation patterns, copy number changes, and genomic sequence variants.
References
- 1.Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet. 2014;15(08):517–530. doi: 10.1038/nrg3766. [DOI] [PubMed] [Google Scholar]
- 2.Kaneda M, Okano M, Hata Ket al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting Nature 2004429(6994):900–903. [DOI] [PubMed] [Google Scholar]
- 3.Bourc'his D, Xu G L, Lin C S, Bollman B, Bestor T H.Dnmt3L and the establishment of maternal genomic imprints Science 2001294(5551):2536–2539. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Ito M, Zhou F et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15(04):547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Messerschmidt D M, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles B B.Trim28 is required for epigenetic stability during mouse oocyte to embryo transition Science 2012335(6075):1499–1502. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura T, Arai Y, Umehara H et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9(01):64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 7.Horsthemke B. In brief: genomic imprinting and imprinting diseases. J Pathol. 2014;232(05):485–487. doi: 10.1002/path.4326. [DOI] [PubMed] [Google Scholar]
- 8.Plasschaert R N, Bartolomei M S. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141(09):1805–1813. doi: 10.1242/dev.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barlow D P, Bartolomei M S.Genomic imprinting in mammals Cold Spring Harb Perspect Biol 2014602a018382. Doi: 10.1101/cshperspect.a018382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choufani S, Shuman C, Weksberg R. Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2013;163C(02):131–140. doi: 10.1002/ajmg.c.31363. [DOI] [PubMed] [Google Scholar]
- 11.Barlow D P. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 12.Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(03):365–376. doi: 10.1002/ajmg.c.30273. [DOI] [PubMed] [Google Scholar]
- 13.Weksberg R, Shuman C, Beckwith J B. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18(01):8–14. doi: 10.1038/ejhg.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Temple I K, Mackay D JG, Docherty L E. Seattle, WA: University of Washington; 1993. Diabetes Mellitus, 6q24-Related Transient Neonatal. [PubMed] [Google Scholar]
- 15.Eggermann T, Heilsberg A K, Bens S et al. Additional molecular findings in 11p15-associated imprinting disorders: an urgent need for multi-locus testing. J Mol Med (Berl) 2014;92(07):769–777. doi: 10.1007/s00109-014-1141-6. [DOI] [PubMed] [Google Scholar]
- 16.Eggermann T, Elbracht M, Schröder C et al. Congenital imprinting disorders: a novel mechanism linking seemingly unrelated disorders. J Pediatr. 2013;163(04):1202–1207. doi: 10.1016/j.jpeds.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 17.Mackay D J, Callaway J L, Marks S M et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(08):949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 18.Zuo X, Sheng J, Lau H T et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem. 2012;287(03):2107–2118. doi: 10.1074/jbc.M111.322644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boonen S E, Mackay D J, Hahnemann J M et al. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up. Diabetes Care. 2013;36(03):505–512. doi: 10.2337/dc12-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murdoch S, Djuric U, Mazhar B et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38(03):300–302. doi: 10.1038/ng1740. [DOI] [PubMed] [Google Scholar]
- 21.Parry D A, Logan C V, Hayward B E et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am J Hum Genet. 2011;89(03):451–458. doi: 10.1016/j.ajhg.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer E, Lim D, Pasha Set al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome) PLoS Genet 2009503e1000423. Doi: 10.1371/journal.pgen.1000423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boonen S E, Hahnemann J M, Mackay D et al. No evidence for pathogenic variants or maternal effect of ZFP57 as the cause of Beckwith-Wiedemann Syndrome. Eur J Hum Genet. 2012;20(01):119–121. doi: 10.1038/ejhg.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Court F, Martin-Trujillo A, Romanelli V et al. Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum Mutat. 2013;34(04):595–602. doi: 10.1002/humu.22276. [DOI] [PubMed] [Google Scholar]
- 25.Spengler S, Gogiel M, Schönherr N, Binder G, Eggermann T. Screening for genomic variants in ZFP57 in Silver-Russell syndrome patients with 11p15 epimutations. Eur J Med Genet. 2009;52(06):415–416. doi: 10.1016/j.ejmg.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Docherty L E, Rezwan F I, Poole R Let al. Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans Nat Commun 201568086. Doi: 10.1038/ncomms9086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossignol S, Steunou V, Chalas C et al. The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J Med Genet. 2006;43(12):902–907. doi: 10.1136/jmg.2006.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tee L, Lim D H, Dias R Pet al. Epimutation profiling in Beckwith-Wiedemann syndrome: relationship with assisted reproductive technology Clin Epigenetics 201350123. Doi: 10.1186/1868-7083-5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackay D J, Boonen S E, Clayton-Smith J et al. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet. 2006;120(02):262–269. doi: 10.1007/s00439-006-0205-2. [DOI] [PubMed] [Google Scholar]
- 30.Bliek J, Verde G, Callaway J et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2009;17(05):611–619. doi: 10.1038/ejhg.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poole R L, Docherty L E, Al Sayegh A et al. Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am J Med Genet A. 2013;161A(09):2174–2182. doi: 10.1002/ajmg.a.36049. [DOI] [PubMed] [Google Scholar]
- 32.Azzi S, Rossignol S, Steunou V et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet. 2009;18(24):4724–4733. doi: 10.1093/hmg/ddp435. [DOI] [PubMed] [Google Scholar]
- 33.Begemann M, Spengler S, Kanber D et al. Silver-Russell patients showing a broad range of ICR1 and ICR2 hypomethylation in different tissues. Clin Genet. 2011;80(01):83–88. doi: 10.1111/j.1399-0004.2010.01514.x. [DOI] [PubMed] [Google Scholar]
- 34.Kannenberg K, Urban C, Binder G. Increased incidence of aberrant DNA methylation within diverse imprinted gene loci outside of IGF2/H19 in Silver-Russell syndrome. Clin Genet. 2012;81(04):366–377. doi: 10.1111/j.1399-0004.2012.01844.x. [DOI] [PubMed] [Google Scholar]
- 35.Turner C L, Mackay D M, Callaway J L et al. Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur J Hum Genet. 2010;18(06):648–655. doi: 10.1038/ejhg.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maupetit-Méhouas S, Azzi S, Steunou V et al. Simultaneous hyper- and hypomethylation at imprinted loci in a subset of patients with GNAS epimutations underlies a complex and different mechanism of multilocus methylation defect in pseudohypoparathyroidism type 1b. Hum Mutat. 2013;34(08):1172–1180. doi: 10.1002/humu.22352. [DOI] [PubMed] [Google Scholar]
- 37.Perez-Nanclares G, Romanelli V, Mayo S et al. Detection of hypomethylation syndrome among patients with epigenetic alterations at the GNAS locus. J Clin Endocrinol Metab. 2012;97(06):E1060–E1067. doi: 10.1210/jc.2012-1081. [DOI] [PubMed] [Google Scholar]
- 38.Ammerpohl O, Martín-Subero J I, Richter J, Vater I, Siebert R. Hunting for the 5th base: techniques for analyzing DNA methylation. Biochim Biophys Acta. 2009;1790(09):847–862. doi: 10.1016/j.bbagen.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Nygren A O, Ameziane N, Duarte H Met al. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences Nucleic Acids Res 20053314e128. Doi: 10.1093/nar/gni127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller D T, Adam M P, Aradhya S et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(05):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet. 1980;6(02):137–143. doi: 10.1002/ajmg.1320060207. [DOI] [PubMed] [Google Scholar]
- 42.Hoffmann K, Heller R. Uniparental disomies 7 and 14. Best Pract Res Clin Endocrinol Metab. 2011;25(01):77–100. doi: 10.1016/j.beem.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Kearney H M, Kearney J B, Conlin L K.Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations Clin Lab Med 20113104595–613, ix.ix [DOI] [PubMed] [Google Scholar]
- 44.Yamazawa K, Nakabayashi K, Kagami M et al. Parthenogenetic chimaerism/mosaicism with a Silver-Russell syndrome-like phenotype. J Med Genet. 2010;47(11):782–785. doi: 10.1136/jmg.2010.079343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaffer L G, Agan N, Goldberg J D, Ledbetter D H, Longshore J W, Cassidy S B. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet Med. 2001;3(03):206–211. doi: 10.1097/00125817-200105000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalish J M, Conlin L K, Mostoufi-Moab S et al. Bilateral pheochromocytomas, hemihyperplasia, and subtle somatic mosaicism: the importance of detecting low-level uniparental disomy. Am J Med Genet A. 2013;161A(05):993–1001. doi: 10.1002/ajmg.a.35831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inbar-Feigenberg M, Choufani S, Cytrynbaum C et al. Mosaicism for genome-wide paternal uniparental disomy with features of multiple imprinting disorders: diagnostic and management issues. Am J Med Genet A. 2013;161A(01):13–20. doi: 10.1002/ajmg.a.35651. [DOI] [PubMed] [Google Scholar]
- 48.Johnson J P, Waterson J, Schwanke C, Schoof J. Genome-wide androgenetic mosaicism. Clin Genet. 2014;85(03):282–285. doi: 10.1111/cge.12146. [DOI] [PubMed] [Google Scholar]
- 49.Kalish J M, Conlin L K, Bhatti T R et al. Clinical features of three girls with mosaic genome-wide paternal uniparental isodisomy. Am J Med Genet A. 2013;161A(08):1929–1939. doi: 10.1002/ajmg.a.36045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strain L, Warner J P, Johnston T, Bonthron D T. A human parthenogenetic chimaera. Nat Genet. 1995;11(02):164–169. doi: 10.1038/ng1095-164. [DOI] [PubMed] [Google Scholar]
- 51.Horike S, Ferreira J C, Meguro-Horike M et al. Screening of DNA methylation at the H19 promoter or the distal region of its ICR1 ensures efficient detection of chromosome 11p15 epimutations in Russell-Silver syndrome. Am J Med Genet A. 2009;149A(11):2415–2423. doi: 10.1002/ajmg.a.33065. [DOI] [PubMed] [Google Scholar]
- 52.Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(03):343–354. doi: 10.1002/ajmg.c.30267. [DOI] [PubMed] [Google Scholar]
- 53.Liehr T, Ewers E, Hamid A B et al. Small supernumerary marker chromosomes and uniparental disomy have a story to tell. J Histochem Cytochem. 2011;59(09):842–848. doi: 10.1369/0022155411412780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baskin B, Choufani S, Chen Y A et al. High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum Genet. 2014;133(03):321–330. doi: 10.1007/s00439-013-1379-z. [DOI] [PubMed] [Google Scholar]
- 55.Fernández-Novoa M C, Vargas M T, Vizmanos J L et al. [Prader-Willi syndrome large deletion on two brothers. Is this the exception that confirm the rule?] Rev Neurol. 2001;32(10):935–938. [PubMed] [Google Scholar]
- 56.Kokkonen H, Leisti J. An unexpected recurrence of Angelman syndrome suggestive of maternal germ-line mosaicism of del(15)(q11q13) in a Finnish family. Hum Genet. 2000;107(01):83–85. doi: 10.1007/s004390000336. [DOI] [PubMed] [Google Scholar]
- 57.Aypar U, Brodersen P R, Lundquist P A, Dawson D B, Thorland E C, Hoppman N. Does parent of origin matter? Methylation studies should be performed on patients with multiple copies of the Prader-Willi/Angelman syndrome critical region. Am J Med Genet A. 2014;164A(10):2514–2520. doi: 10.1002/ajmg.a.36663. [DOI] [PubMed] [Google Scholar]
- 58.Shaffer L G. Risk estimates for uniparental disomy following prenatal detection of a nonhomologous Robertsonian translocation. Prenat Diagn. 2006;26(04):303–307. doi: 10.1002/pd.1384. [DOI] [PubMed] [Google Scholar]
- 59.Kotzot D. Prenatal testing for uniparental disomy: indications and clinical relevance. Ultrasound Obstet Gynecol. 2008;31(01):100–105. doi: 10.1002/uog.5133. [DOI] [PubMed] [Google Scholar]
- 60.Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium methylation 450K technology. Epigenomics. 2011;3(06):771–784. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- 61.Choufani S, Shapiro J S, Susiarjo M et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 2011;21(03):465–476. doi: 10.1101/gr.111922.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Docherty L E, Rezwan F I, Poole R L et al. Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J Med Genet. 2014;51(04):229–238. doi: 10.1136/jmedgenet-2013-102116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamazawa K, Ogata T, Ferguson-Smith A C. Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet. 2010;154C(03):329–334. doi: 10.1002/ajmg.c.30270. [DOI] [PubMed] [Google Scholar]
- 64.Azzi S, Salem J, Thibaud N et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015;52(07):446–453. doi: 10.1136/jmedgenet-2014-102979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Crescenzo A, Citro V, Freschi A et al. A splicing mutation of the HMGA2 gene is associated with Silver-Russell syndrome phenotype. J Hum Genet. 2015;60(06):287–293. doi: 10.1038/jhg.2015.29. [DOI] [PubMed] [Google Scholar]
- 66.Kagami M, Mizuno S, Matsubara K et al. Epimutations of the IG-DMR and the MEG3-DMR at the 14q32.2 imprinted region in two patients with Silver-Russell syndrome-compatible phenotype. Eur J Hum Genet. 2015;23(08):1062–1067. doi: 10.1038/ejhg.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prickett A R, Ishida M, Böhm S et al. Genome-wide methylation analysis in Silver-Russell syndrome patients. Hum Genet. 2015;134(03):317–332. doi: 10.1007/s00439-014-1526-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brioude F, Oliver-Petit I, Blaise A et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet. 2013;50(12):823–830. doi: 10.1136/jmedgenet-2013-101691. [DOI] [PubMed] [Google Scholar]
- 69.Eggermann T, Begemann M, Binder G, Spengler S.Silver-Russell syndrome: genetic basis and molecular genetic testing Orphanet J Rare Dis 2010519. Doi: 10.1186/1750-1172-5-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arboleda V A, Lee H, Parnaik R et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. 2012;44(07):788–792. doi: 10.1038/ng.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hamajima N, Johmura Y, Suzuki S, Nakanishi M, Saitoh S.Increased protein stability of CDKN1C causes a gain-of-function phenotype in patients with IMAGe syndrome PLoS One 2013809e75137. Doi: 10.1371/journal.pone.0075137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Smith A J, Purmann C, Walters R G et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18(17):3257–3265. doi: 10.1093/hmg/ddp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sahoo T, del Gaudio D, German J R et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40(06):719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schaaf C P, Gonzalez-Garay M L, Xia F et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat Genet. 2013;45(11):1405–1408. doi: 10.1038/ng.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim S J, Miller J L, Kuipers P J et al. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012;20(03):283–290. doi: 10.1038/ejhg.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Driscoll D J, Miller J L, Schwartz S, Cassidy S B. Seattle, WA: University of Washington; 1993. Prader-Willi syndrome. [PubMed] [Google Scholar]
- 77.Baple E L, Poole R L, Mansour S et al. An atypical case of hypomethylation at multiple imprinted loci. Eur J Hum Genet. 2011;19(03):360–362. doi: 10.1038/ejhg.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sahoo T, Bacino C A, German J R et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array-CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet. 2007;15(09):943–949. doi: 10.1038/sj.ejhg.5201859. [DOI] [PubMed] [Google Scholar]
- 79.Dagli A I, Williams C A. Seattle, WA: University of Washington; 1993. Angelman syndrome. [PubMed] [Google Scholar]
- 80.Ramsden S C, Clayton-Smith J, Birch R, Buiting K.Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes BMC Med Genet 20101170. Doi: 10.1186/1471-2350-11-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buiting K, Gross S, Lich C, Gillessen-Kaesbach G, el-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72(03):571–577. doi: 10.1086/367926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hosoki K, Kagami M, Tanaka T et al. Maternal uniparental disomy 14 syndrome demonstrates Prader-Willi syndrome-like phenotype. J Pediatr. 2009;155(06):900–9030. doi: 10.1016/j.jpeds.2009.06.045. [DOI] [PubMed] [Google Scholar]
- 83.Davis E, Caiment F, Tordoir X et al. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol. 2005;15(08):743–749. doi: 10.1016/j.cub.2005.02.060. [DOI] [PubMed] [Google Scholar]
- 84.Kagami M, Kato F, Matsubara K, Sato T, Nishimura G, Ogata T. Relative frequency of underlying genetic causes for the development of UPD(14)pat-like phenotype. Eur J Hum Genet. 2012;20(09):928–932. doi: 10.1038/ejhg.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mitsui T, Nagasaki K, Takagi M, Narumi S, Ishii T, Hasegawa T. A family of pseudohypoparathyroidism type Ia with an 850-kb submicroscopic deletion encompassing the whole GNAS locus. Am J Med Genet A. 2012;158A(01):261–264. doi: 10.1002/ajmg.a.34393. [DOI] [PubMed] [Google Scholar]
- 86.Mantovani G, de Sanctis L, Barbieri A M et al. Pseudohypoparathyroidism and GNAS epigenetic defects: clinical evaluation of albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab. 2010;95(02):651–658. doi: 10.1210/jc.2009-0176. [DOI] [PubMed] [Google Scholar]
- 87.Kelsey G. Imprinting on chromosome 20: tissue-specific imprinting and imprinting mutations in the GNAS locus. Am J Med Genet C Semin Med Genet. 2010;154C(03):377–386. doi: 10.1002/ajmg.c.30271. [DOI] [PubMed] [Google Scholar]
- 88.Garin I, Mantovani G, Aguirre U et al. European guidance for the molecular diagnosis of pseudohypoparathyroidism not caused by point genetic variants at GNAS: an EQA study. Eur J Hum Genet. 2015;23(04):438–444. doi: 10.1038/ejhg.2014.127. [DOI] [PMC free article] [PubMed] [Google Scholar]