


Abstract
Sequential chromatin immunoprecipitation (SeqChIP) is a procedure in which formaldehyde-crosslinked, protein–DNA complexes from living cells are subjected to two sequential immunoprecipitations with antibodies of different specificity. SeqChIP has been used to address, in a qualitative manner, whether two proteins can simultaneously co-occupy a stretch of DNA in vivo. Here, we expand on our earlier work and describe theoretical and practical considerations for performing and interpreting SeqChIP experiments in a quantitative manner. We provide a detailed experimental procedure for designing and performing SeqChIP experiments as well as experimental examples of the three possible outcomes: full co-occupancy, no co-occupancy and partial co-occupancy. In some cases of partial co-occupancy, the order of immunoprecipitations in SeqChIP can strongly influence the outcome. We experimentally confirm a quantitative parameter that provides a measure of co-occupancy of two proteins on a given region of DNA and provide information on how to interpret the results of SeqChIP experiments. Our quantitative treatment of SeqChIP data substantially expands the usefulness of the technique for elucidating molecular mechanisms in vivo.
INTRODUCTION
Chromatin immunoprecipitation (ChIP) is a widely-used and powerful method for assaying protein–DNA interactions in vivo (1). In this technique, live cells are treated with formaldehyde to generate reversible crosslinks between protein molecules and DNA sequences located in close proximity on the chromatin template. Crosslinked chromatin is sheared by sonication to reduce DNA fragment size, and the resulting material is immunoprecipitated with an antibody against a desired protein, modified peptide (e.g. to detect acetylated, phosphorylated or methylated versions of a protein), or epitope (in situations where the protein of interest is epitope-tagged). DNA sequences that associate with a given protein (or modified variant) are selectively enriched in the immunoprecipitated, but not the input, sample. Typically, the amounts of specific genomic regions in control and immunoprecipitated samples are determined individually by quantitative PCR following the reversal of protein–DNA crosslinks, although other quantitative approaches have been employed. In addition, ChIP can be combined with microarray technology to identify the locations of specific proteins on a genome-wide basis (2–8). ChIP has been successfully used in a wide variety of organisms (e.g. bacteria, yeasts, flies, worms and mammalian cells) to analyze many different biological phenomena involving protein–DNA interactions.
Standard ChIP experiments provide quantitative information about the relative level of association of a given protein with different genomic regions. By comparing the results of multiple conventional ChIP experiments, the relative occupancy levels of different proteins at genomic regions can be determined. However, standard ChIP experiments do not address whether two proteins simultaneously occupy a given DNA sequence. The observation that two proteins associate with a given genomic region might reflect co-occupancy, but it also could indicate that the two proteins associate with different populations of DNA molecules. For example, if two proteins associate with a given DNA sequence in a mutually exclusive manner, standard ChIP experiments will nevertheless indicate that both proteins associate, perhaps even with a constant occupancy ratio over different binding sites. More generally, there are many potential situations in which it is critical to determine the extent to which two proteins co-occupy a given DNA sequence.
Sequential chromatin immunoprecipitation (SeqChIP; also referred to as Re-ChIP, ChDIP, double ChIP) has been used to ascertain whether two proteins can simultaneously associate with the same genomic region in vivo (9–16). In SeqChIP, protein–DNA complexes from the first immunoprecipitation are subjected to an additional immunoprecipitation with an antibody of a different specificity. The crosslinks of these doubly immunoprecipitated protein–DNA complexes are then reversed, and the DNAs are analyzed by quantitative PCR in an analogous manner to conventional ChIP samples. In general, SeqChIP has been used to qualitatively address whether two proteins co-occupy a given genomic region, but the results have not been interpreted in a quantitative fashion. In our previous work, we developed an initial approach for treating SeqChIP experiments in a quantitative manner, and used this approach to demonstrate that cellular stress alters the transcriptional properties of Mot1–TATA-box binding protein (TBP) complexes in yeast cells (16). Here, we expand on our earlier work to develop a comprehensive theoretical and practical method for measuring the co-occupancy of two proteins on a given region of DNA in a quantitative manner. Our quantitative treatment of SeqChIP data substantially expands the usefulness of the technique, particularly in elucidating molecular mechanisms involving multiple proteins that can associate with the same genomic region.
MATERIALS AND METHODS
Antibodies, peptides and oligonucleotides
Antibodies used in this work include those directed to the HA epitope (F-7; Santa Cruz Biotech), Myc epitope (06-549; Upstate Biotechnology), TFIIA and TFIIB (17), TBP-associated factors, TAF6 and TAF12 (kindly provided by Michael Green), and RNA Polymerase II (8WG16; Covance). Peptides encompassing the HA1 epitope (YPYDVPDYA) and Myc epitope (EQKLISEEDL) were synthesized and purified (≥95%) by reverse-phase high- performance liquid chromatography by American Peptide Company (www.americanpeptide.com).
Oligonucleotides were designed with Oligo 6.6 (www.oligo.net) in order to minimize primer dimers and other secondary structure concerns. Most primers were 22–28 bases in length and had calculated Tms between 53 and 56°C. Oligonucleotides were synthesized by Integrated DNA Technologies (www.idtdna.com) and were typically used without further purification. Amplification products were between 100–300 bases in length, and typically encompassed the TATA box/core promoter sequences. Primer sequences for individual genomic regions are available on request.
Cell growth, crosslinking and chromatin preparation
Cultures (400 ml) of LK25, a Saccharomyces cerevisiae strain containing a TBP allele tagged at the N-terminus with three copies of the HA1 epitope and a MOT1 allele fused to nine copies of the Myc epitope at the C-terminus (18), were grown at 30°C in casamino acids medium containing 2% dextrose to an optical density of 0.6 at 600 nm. Crosslinking was accomplished via the addition of 11 ml of 37% formaldehyde (1% final concentration) for 20 min at room temperature. The cultures were then quenched with 60 ml of 2.5 M glycine for 5 min at room temperature, and yeast cells were collected by centrifugation. The supernatant was discarded, and the cell pellet was then washed once with 250 ml ice-cold Tris-buffered saline and once with 10 ml FA lysis buffer (0.1 M NaCl; 50 mM HEPES–KOH, pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate and 0.1% SDS). Pellets were resuspended in 5 ml FA lysis buffer (0.1 M NaCl) containing 2 mM phenylmethylsulfonyl fluoride, and ∼1.2 ml aliquots were dispensed into 2.0 ml screw-cap tubes previously filled to three-fourths of the volume with 0.5 mm silica–zirconia beads (BioSpec). Special care was taken to minimize air bubbles in the tubes. The four tubes, each one containing ∼1 × 109 cells, were secured in Mini Bead Beater (Biospec) and disrupted with six three-minute cycles at the highest setting at 4°C. In between the cycles, samples were cooled for 1–2 min in an ice-water bath. The liquid containing cellular debris (∼5 ml) was split into three microfuge tubes and was centrifuged for 10 min (13 000 g, 4°C). The supernatant was discarded, 1 ml of FA lysis buffer (0.1 M NaCl) was added to each tube, and the samples were sonicated 3 times (30 s continuous pulses at 20% power in a Branson Sonifier equipped with a micro-tip probe) at 4°C, with 1 min cooling in an ice-water bath in between pulses. Sonicated samples were centrifuged for 15 min (13 000 × g, 4°C) to remove insoluble debris. The supernatants were pooled, diluted to ∼5 ml total volume with FA lysis buffer (0.1 M NaCl), and frozen on liquid nitrogen in 1 ml aliquots.
Single and sequential chromatin immunoprecipitation
Chromatin from ∼0.5 to 1 × 109 cells (1 ml chromatin aliquots as isolated above) was incubated with 5–20 μl of antibody and 20 μl of protein A-Sepharose for 90 min at room temperature on a rotating wheel. Protein complexes were either washed six times with FA lysis buffer (0.1 M NaCl; mild washes; F7 and 9E10 antibodies) or three times with FA lysis buffer (0.1 M NaCl), once with FA lysis buffer (0.5 M NaCl), once with ChIP wash buffer (10 mM Tris–HCl, pH 8.0, 250 mM LiCl, 1 mM EDTA, 0.5% Nonidet P-40 and 0.5% sodium deoxycholate) and once with TE (stringent washes; for all other antibodies). In the experiments described here, >50–75% of the protein in the original extract was immunoprecipitated, as determined by western blot analysis.
Precipitated complexes were eluted either by a 30 min incubation (at 30°C) with an epitope-specific peptide (100 μl of a 1 mg/ml solution in Tris-buffered saline; F7 and 9E10 antibodies) or via a 10 min incubation with 100 μl of ChIP elution buffer (50 mM Tris–HCl, pH 7.5, 10 mM EDTA and 1% SDS) at 68°C (all other antibodies). At this point, 10 μl was removed from each sample (except those not used for SeqChIP) for subsequent analysis of the first immunoprecipitation. Samples not used for SeqChIP were decrosslinked (2 h at 42°C followed by 6 h at 68°C) in 500 μl volume of 2-fold diluted ChIP elution buffer containing 0.8 mg/ml Pronase. After crosslink reversal, the samples were extracted once with phenol/chloroform and once with chloroform. DNAs were precipitated with ethanol in the presence of glycogen carrier, washed once with 70% ethanol, air-dried and resuspended in a final volume of 150 μl TE for use in subsequent analysis.
For SeqChIP, eluates (90 μl) were incubated (90 min at room temperature) in the presence of 25 μg/ml phage λ DNA, 5 mg/ml BSA(Fraction V; Sigma), 50 μg/ml Escherichia coli tRNA, 5–20 μl antibody and 20 μl protein A-Sepharose in a total volume of 1 ml FA lysis buffer (0.1 M NaCl). In an effort to make the second immunoprecipitation similar to the initial one, and to reduce background due to non-specific sticking to beads of the very limited material from the first immunoprecipitation, we experimented with a number of different carriers, including chromatin from other yeast species and various sources of carrier DNA. An advantage of chromatin from other yeast species is that one can monitor the amount of unrelated DNA through the procedure by using appropriate PCR primers. However, in this study we routinely used phage λ DNA, as it did not require the time-consuming preparation typically associated with non-S.cerevisiae yeast chromatin purification. The washes, elution and crosslink reversal following the second immunoprecipitation were carried out exactly as described above, with samples immunoprecipitated (the second time) with F7 or 9E10 antibodies receiving mild washes and peptide elution; all other samples were washed stringently and were eluted with heat in ChIP elution buffer. Eluted samples were extracted and precipitated exactly as described above. DNAs were resuspended in 150 μl TE for real-time quantitative PCR (QPCR) analysis.
Although epitope masking could theoretically affect the results of standard and SeqChIP experiments, there are few, if any examples in which epitope masking is locus-specific. Moreover, in many experiments including those presented here, immunoprecipitation is very efficient (>50–75%), and virtually all protein is crosslinked to DNA (this includes crosslinking to specific and non-specific genomic regions, and there is very little free protein. Epitope masking is unlikely to occur in experiments involving proteins containing multiple epitope tags that are unstructured and probably not in contact with protein or DNA that might occlude an antibody. Epitope masking is unlikely to have a significant effect in experiments using polyclonal antisera raised against full-length proteins or large protein domains, which typically contain a mixture of antibodies with specificities to different parts of protein. Finally, epitope masking can affect a SeqChIP experiment only in the specific, and very unlikely situation where both crosslinked proteins are required for antibody occlusion.
Quantitative PCR in real time
Quantitative PCR of the DNA samples described above was used to assess the extent of occupancy (or co-occupancy) at different genomic locations. Prior to PCR, input DNAs were 1:1000 diluted (relative to the first IPs) in TE. Each PCR reaction contained 2 μl of template DNA [either IP (∼1/75th of each immunoprecipitated sample) or input (∼1/75 000th of each immunoprecipitated sample, or ∼10 000 genome equivalents], 3 μl of primers (final concentration of 1 μM) and 5 μl of 2 × SYBR Green reaction mix (1). Quantitative PCR was performed on Applied Biosystems 7000 and 7700 instruments using a 10 min soak at 94°C followed by 40 cycles of 30 s at 94°C, 30 s at 53°C and 30 s at 72°C. Threshold cycles (Ct) were determined as recommended by the manufacturer's software and discussed in more detail elsewhere (1). For each amplification product, the NETCt was determined using the formula NETCt = Ct(IP) − Ct(Input). Fold occupancy (f) of any given region X over the POL1 control is given by f = 1.9[NETCt(POL1) − NETCt(X)]. The base value of 1.9 is an empirically derived average from hundreds of individual experiments and represents a remarkably consistent (±0.05) approximation of the amplification efficiency in this procedure. For short amplification products, this value is primer- and amplification sequence-independent. Occupancy values presented here represent an average of at least three independent experiments and have an error of approximately ±25%.
RESULTS AND DISCUSSION
Possible outcomes of SeqChIP experiments
Outcomes of SeqChIP experiments can fall into one of three categories: (i) complete co-occupancy, (ii) no co-occupancy or (iii) partial co-occupancy of the two factors (A and B) being tested (Figure 1). Complete co-occupancy occurs when two proteins always associate with the same DNA fragment, and neither factor is found on the DNA in the absence of the other. No co-occupancy occurs when A and B associate with the same genomic region in vivo but can do so only on mutually exclusive sub-populations of DNA fragments. In the case of partial co-occupancy, some DNA molecules have both A and B, while others have only A and/or only B.
Figure 1.

SeqChIP experiments have three different outcomes: complete co-occupancy, no co-occupancy, and partial co-occupancy. In full co-occupancy, proteins A and B always co-occupy promoters; neither one is found bound to a particular region without the other. No co-occupancy occurs when DNA binding of A and B is mutually exclusive. Partial co-occupancy has two distinct states: in the first, the binding of B is critically dependent on binding of A, but not vice versa. In the second state, both A and B can bind DNA independently, but may or may not be simultaneously bound on the same DNA fragments.
There are two types of partial co-occupancies (Figure 1). First, DNA binding of protein B always occurs in combination with protein A, whereas protein A can associate with DNA in the absence of protein B. For example, promoter binding by yeast TAFs depends on the TBP, yet TBP is found at many promoters that lack TAFs (17,19). Second, A and B can bind independently of one another to a given genomic region, but the proteins may co-occupy the same stretch of DNA in only a fraction of the cases. This second possibility may occur when two DNA-binding proteins do not interact with other and recognize different target sequences in an enhancer or other transcriptional regulatory region of a eukaryotic promoter.
Fold-enrichments and the nature of experimental background in SeqChIP experiments
The basic measurements in ChIP and SeqChIP experiments involve the fold-enrichments of ‘target’ genomic regions (e.g. an active promoter) to ‘non-target’ or control genomic regions (e.g. an intergenic region or coding region of an inactive gene). The experimental values for the control genomic regions are usually considered as experimental background. However, two separate components contribute to this experimental background.
One such experimental background can arise from the non-specific precipitation of DNA fragments that are either not crosslinked to proteins or are crosslinked with nuclear proteins not specifically targeted for immunoprecipitation. This background is frequently due to non-specific ‘purification’ by the antibody, protein A, or the agarose-based resin, and can occur even though those fragments are not associated with the protein being specifically precipitated. The extent to which this type of experimental background would be a problem largely depends on the avidity/affinity of the antibody toward the antigen and the conditions employed in the immunoprecipitation (e.g. stringencies of washes, elution conditions, etc.). Specific steps taken to minimize this type of background can include longer antibody–antigen incubation times, more stringent washes of bound complexes and optimized elution with epitope-containing peptides or protein fragments. Ideally, and often in practice, DNA fragments not crosslinked to the immunoprecipitated protein make a minor contribution to the overall experimental background.
A second experimental background in SeqChIP experiments can arise when the protein of interest gets crosslinked to genomic DNA in a non-sequence-specific manner. Such crosslinked material can arise from random collisions between the protein and DNA in the nucleus and/or true non-sequence-specific association of the protein. The extent to which non-specific DNA crosslinking contributes to the experimental background depends on both the average length of sheared DNA fragments and on the relative binding affinities to non-specific versus specific DNA sequences, which vary widely among proteins. Importantly, background due to non-specific protein–DNA crosslinking is an intrinsic feature of SeqChIP. It generally represents the major form of experimental background in SeqChIP, and it cannot be eliminated by varying the experimental procedure. As a consequence, meaningful information cannot be obtained from control experiments involving two sequential immunoprecipitations involving the same protein. In essence, the second immunoprecipitation largely re-purifies the same material (i.e. both crosslinked control and target regions), and it normally results in no change in fold-enrichments.
Theoretical predictions for fold-enrichments in SeqChIP experiments
The key concept for quantitating SeqChIP experiments is that the final fold-enrichment of the sequential ChIP should be equal to the product of the fold-enrichments of the individual ChIPs, if two proteins completely co-occupy DNA (12,16). In this regard, the immunopurifications involved in SeqChIP are analogous to sequential biochemical purification steps or fold-stimulation of a biochemical process by two independent events. Importantly, this concept relies only on the assumptions that in vivo formaldehyde crosslinking events are independent and inefficient.
Formaldehyde crosslinking of proteins to DNA is inefficient, with maximal crosslinking efficiencies for individual proteins typically ranging between 1 and 10% (1,20). As the physical crosslink of A to DNA is independent of the physical crosslink of B to DNA, most DNA molecules crosslinked to A will lack a similar physical link to protein B (and vice versa) even when proteins A and B always co-occupy a region of DNA. Hypothetically, extremely efficient protein–protein crosslinks between A and B (or crosslinks involving a common intermediary protein) could yield SeqChIP values that are significantly more than the product of the individual ChIPs. However, protein–protein contacts per se do not generate ChIP signals and as such play a secondary role relative to direct protein–DNA contacts. Furthermore, substantial experimental evidence presented here and elsewhere (16) validates these key concepts.
The theoretical results of a SeqChIP experiment are calculated as follows (Figure 2). There are three relevant classes of DNA molecules—those containing A alone, B alone, or A + B. ‘X’ is defined as the fraction of A-DNA molecules that also contain B, and ‘Y’ represents the fraction of B-DNA molecules that also contain A. X and Y are unrelated to each other because the occupancy characteristics of proteins A and B are different. Thus, if the fold-enrichments of the individual ChIP experiments are defined as A and B, then the expected fold-enrichments for the different classes of molecules are as follows:
Figure 2.

Predicted experimental outcomes for quantitative SeqChIP. Predicted full co-occupancy (A), no co-occupancy (B) and partial co-occupancy (C) values are expressed as functions of parameters X and Y, where X and Y represent fractions of A-DNA molecules that also contain B and fractions of B-DNA that also contain A, respectively. Note that SeqChIP order is not important for full co-occupancy or no co-occupancy (AB = BA), but is critical for cases of partial co-occupancy (AB ≠ BA).
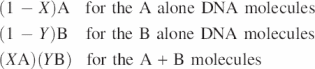
From the above equations it follows that:
![]()
![]()
For complete co-occupancy (X = 1 and Y = 1; Figure 2A), the fold-enrichment in a SeqChIP equals the product of the individual ChIPs, and fold-enrichment is not affected by the order of the individual ChIPs (AB = BA). For no co-occupancy (X = 0 and Y = 0; Figure 2B), the fold-enrichment in the SeqChIP is within experimental error of the fold-enrichment of the first IP, and is also independent of the actual order of the IPs (AB = BA = 0). Partial co-occupancy occurs when the fold-enrichment of a SeqChIP is significantly higher (in practical terms, this is often considered to be 2-fold) than the fold-enrichment of the first ChIP, but significantly lower than the product of the two ChIPs (Figure 2C). Unlike the examples above, the order of the individual IPs can matter in cases of partial co-occupancy (AB ≠ BA). In fact, partial co-occupancy will always be the result if two proteins do not completely co-occupy DNA, even though it might be experimentally observed only in one direction under special circumstances (see below).
In cases of partial co-occupancy, the order of individual ChIPs makes a difference (Figure 2C). Furthermore, partial co-occupancy will be observed in only one direction when X = 1 and Y is small; i.e. factor A always co-occupies DNA with factor B, but only a small proportion of B molecules co-occupy with A molecules (Figure 1C). When DNA-crosslinked A is immunoprecipitated first, most or all of those molecules will also have B associated and hence will benefit from the second IP. In contrast, when B is immunoprecipitated first, only a small percentage of molecules will contain A, so that the second IP will not add much fold-enrichment to the already highly enriched pool of B-DNA molecules.
To illustrate the importance of the order of individual ChIPs in cases of partial co-occupancy, consider an example in which X = 1, Y = 0.1, A = 10, B = 50. In this case, the predicted fold-enrichments are 50 when the A ChIP is first and 95 when the B ChIP is first. When the A ChIP is first, the SeqChIP value of 50 represents a 5-fold enrichment over the A ChIP, which indicates partial co-occupancy as it is well below the theoretically predicted maximal co-occupancy value of 500. However, when the B ChIP is first, the SeqChIP value of 95 is <2-fold enrichment over the individual B ChIP, and hence is within experimental error, making it impossible to distinguish partial co-occupancy from experimental background. In contrast, in situations where both X and Y are both significant (e.g. each being 0.5), the predicted SeqChIP values are 130 if the A ChIP is first and 150 if the B ChIP is first, indicating that partial co-occupancy should be observed in both directions. Even though partial and full co-occupancy results are definitive in one direction, the examples above underscore the importance of performing SeqChIP in both directions (e.g. A first then B and vice versa), as lack of co-occupancy in one direction is not necessarily indicative of no co-occupancy.
A quantitative metric of protein co-occupancy
We define a measure of SeqChIP efficiency (C; in percent) that is related to the extent of partial co-occupancy between two proteins. A C-value of 100 is defined as complete co-occupancy, a value of 0 is defined as no co-occupancy, and intermediate C-values represent partial co-occupancy. Specifically, C = 100(AB − A)/(A · B − A), where AB represents the fold-enrichment for the sequential ChIP and A and B represent the fold-enrichments for the individual ChIPs. The value A is subtracted from both AB (SeqChIP result) and A · B (product of the individual ChIP results), because A represents the contribution of the first IP and as such does not represent sequential IP enrichment per se. When the order of IPs is reversed (B ChIP is first, A ChIP is second), C is calculated according to the formula C = 100(BA − B)/(B · A − B).
In cases of partial occupancy, efficiencies are dependent on the order of individual IPs (as AB ≠ BA), which in turn means that typically CAB ≠ CBA. From the above example where X = 1, Y = 0.1, A = 10, and B = 50, CAB ≈ 8 (CBA is undefined SeqChIP enrichment over the first ChIP is within experimental error), a low partial co-occupancy value. When the X and Y values are 0.5 each, then CAB ≈ 24 and CBA ≈ 29, indicating substantial partial co-occupancy between factors A and B. Thus, aside from providing information on the extent to which two proteins occupy a given genomic region, this quantitative metric can be used to estimate values of X and Y and hence additional information about the nature of co-occupancy.
Considerations related to the relative locations of the two proteins associated with DNA
Our analysis of co-occupancy is best suited for two proteins with binding sites that are spaced closely together (<100–200 bases apart). As sonication of crosslinked chromatin produces a randomized population of fragments that average 400–500 bases in length, inter-site spacing of significantly >200 bases will result in a substantial proportion of DNA fragments that contain one binding site but not the other. For proteins that always co-occupy a given DNA region, but whose sites are separated significantly >200 bases, the outcome from a standard SeqChIP experiment will appear to be partial co-occupancy, because the percentage of fragments containing both sites will be significantly lower than if the sites are close together. To investigate co-occupancy of two proteins whose binding sites are separated by several hundred bases, sonication times should be decreased so that the average fragment length after sonication is 1.5–2 kb, and PCR primers should be designed so that amplification products encompass both binding sites if possible. However, these modifications will result in higher background as well as lowered signal. These considerations do not reflect any inadequacy of the theoretical treatment of SeqChIP experiments described above, but rather the fact that proteins that associate with DNA sequences too far apart from one another will not often be found on the same DNA fragments generated by sonication or other fragmentation methods.
Experimental validation
To validate the above theoretical treatment of SeqChIP data, we performed co-occupancy experiments for situations related to the RNA polymerase II transcription machinery for which detailed biochemical and structural information is available. It has been strongly suggested that basic components of the Pol II machinery completely co-occupy promoters (17,20,21). In accord with this prediction, SeqChIP experiments involving pairwise combinations of TBP, TFIIA, TFIIB, and Pol II at the PGK1 promoter show complete co-occupancy in all cases tested, with an average C-value remarkably close to 100 (Figure 3A). As expected, TBP and TFIIB do not co-occupy a tRNA promoter transcribed by RNA Polymerase III (Pol III) since TFIIB binding to this promoter is <2-fold above background. On the other hand, no co-occupancy of Mot1 and TFIIA is observed at the PYK1 promoter when the SeqChIP is performed in either direction (Figure 3B; C = 0), even though both Mot1 and TFIIA exhibit considerable binding to this promoter individually. The lack of co-occupancy by Mot1 and TFIIA is in accord with their functional antagonism in vitro (22–24) and their competitive binding to the solvent-exposed surface of TBP (25). Finally, we observe varying degrees of partial co-occupancy between TBP and TAFs depending on whether the promoter is TAF-dependent or TAF-independent (17,19). C-values for TAF6–TBP and TAF12–TBP co-occupancies at TAF-dependent promoters range from 33 to 50, whereas C-values for TAF-independent promoters are 2- to 3-fold lower (Figure 3C). Partial co-occupancy between TBP and TAFs is expected, because distinct TBP complexes lacking TAFs can associate with promoters (18,26–28). Furthermore, the difference in TBP–TAF co-occupancy values at TAF-dependent and TAF-independent promoters is consistent with previous genetic and molecular observations (17,19), and it provides strong evidence for a direct correlation between the magnitude of C and extent of co-occupancy. More extensive SeqChIP analysis of the co-occupancy behaviors of TBP, TFIIB, TFIIA, Pol II, TAFs and Mot1 demonstrate that cellular stress alters the transcriptional properties of promoter-bound Mot1–TBP complexes, and are presented elsewhere (16).
Figure 3.

The three predicted outcomes of SeqChIP: full co-occupancy, no co-occupancy and partial co-occupancy. (A) Full co-occupancy of TFIIA, TFIIB, TBP and Pol II at the PGK1 promoter. (B) No co-occupancy of Mot1 and TFIIA at the PYK1 promoter irrespective of IP order, and no co-occupancy of TBP and TFIIB at a Pol III-transcribed tRNA gene (C) Partial co-occupancy of TAF6–TBP and TAF12–TBP at the TAF-dependent RPL9A and RPS9B promoters. C-values are shown in parenthesis below individual experiments. Data for Mot1/TFIIA co-occupancy shown in part (B) is taken from (16).
Practical experimental considerations
To apply SeqChIP in a quantitative manner as described above, it is essential that the individual immunoprecipitations are efficient, and that the fold-enrichment for a given protein is equivalent when the immunoprecipitation is first or second. Ideally, both individual immunoprecipitations should yield ≥5-fold enrichment of target sites over non-target sites in order to unambiguously resolve instances of partial co-occupancy from cases of full (or no) co-occupancy. It is highly recommended to perform SeqChIP experiments in both forward (A-B) and reverse (B-A) directions. Although technically unnecessary in the cases of complete co-occupancy, performing SeqChIP in both directions will provide compelling evidence of complete co-occupancy, and will permit unambiguous differentiation between states of partial and no co-occupancy in the event A-B do not fully co-occupancy the DNA fragment being studied.
To demonstrate that the first and second immunoprecipitations are equivalent, it is critical to have a positive control involving two proteins that are known or strongly suspected to completely co-occupy genomic regions and hence give C-values = 100 in the control experiment. It is best, although not always possible (or practical), to individually design positive control experiments for every protein analyzed by SeqChIP. In the examples described above, the positive controls for the partial co-occupancy of TBP and TAF6 are the complete TBP–TFIIB and TAF6–TAF12 co-occupancies at the same genomic sequences. While the actual positive controls are determined on a case-by-case basis, tightly associated polypeptides or subunits of multiprotein complexes as determined by biochemical studies will often be good choices. It is also possible to use some of the protein combinations described here and elsewhere (16) as general controls for the SeqChIP procedure, depending on the availability of antibodies or epitope-tagged strains. As discussed in the section on the nature of experimental background, control experiments involving sequential immunoprecipitations involving the same protein are inappropriate, because re-purification of the same material normally does not change the fold-enrichment. In contrast, sequential immunoprecipitations involving two subunits of a multi-protein complex results in increased fold-enrichments described by the theoretical treatment above, because the two purification steps involve different classes of DNA molecules due to the low crosslinking efficiency of the two proteins.
CONCLUSION
Quantitative SeqChIP is a powerful technique for examining co-occupancy of two proteins in vivo. It has three distinct outcomes that are now both theoretically and experimentally defined: no co-occupancy, partial co-occupancy and complete co-occupancy. SeqChIP efficiency provides a quantitative measure of the extent of partial co-occupancy between two factors at any given sequence. With recent advancement in microarray technology, it should be possible to apply quantitative SeqChIP on a genome-wide scale to study a wide variety of biological phenomena.
REFERENCES
- 1.Aparicio O.M., Geisberg,J.V. and Struhl,K. (2004) Chromatin immunoprecipitation for determining the association of proteins with specific genomic locations in vivo. In Ausubel,F.A., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (eds), Current Protocols in Molecular Biology. John Wiley & Sons, New York, NY, pp. 21.3.1–21.3.17. [Google Scholar]
- 2.Ren B., Robert,F., Wyrick,J.J., Aparicio,O., Jennings,E.G., Simon,I., Zeitlinger,J., Schreiber,J., Hannett,N., Kanin,E. et al. (2000) Genome-wide location and function of DNA binding proteins. Science, 290, 2306–2309. [DOI] [PubMed] [Google Scholar]
- 3.Iyer V.R., Horak,C.E., Scafe,C.S., Botstein,D., Snyder,M. and Brown,P.O. (2001) Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature, 409, 533–538. [DOI] [PubMed] [Google Scholar]
- 4.Weinmann A.S., Yan,P.S., Oberley,M.J., Huang,T.H. and Farnham,P.J. (2002) Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev., 16, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren B., Cam,H., Takahashi,Y., Volkert,T., Terragni,J., Young,R.A. and Dynlacht,B. (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev., 16, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martone R., Euskirchen,G., Bertone,P., Hartman,S., Royce,T.E., Luscombe,N.M., Rinn,J.L., Nelson,F.K., Miller,P., Gerstein,M. et al. (2003) Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl Acad. Sci. USA, 100, 12247–12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cawley S., Bekiranov,S., Ng,H.H., Kapranov,P., Sekinger,E.A., Kampa,D., Piccolboni,A., Smentchenko,V., Cheng,J., Williams,A.J. et al. (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of non-coding RNAs. Cell, 116, 499–509. [DOI] [PubMed] [Google Scholar]
- 8.Odom D.T., Zizlsperger,N., Gordon,D.B., Bell,G.W., Rinaldi,N.J., Murray,H.L., Volkert,T.L., Schreiber,J., Rolfe,P.A., Gifford,D.K. et al. (2004) Control of pancreas and liver gene expression by HNF transcription factors. Science, 303, 1378–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scully K.M., Jacobson,E.M., Jepsen,K., Lunyak,V., Viadiu,H., Carriere,C., Rose,D.W., Hooshmand,F., Aggarwal,A.K. and Rosenfeld,M.G. (2000) Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science, 290, 1127–1131. [DOI] [PubMed] [Google Scholar]
- 10.Chaya D., Hayamizu,T., Bustin,M. and Zaret,K.S. (2001) Transcription factor FoxA (HNF3) on a nucleosome at an enhancer complex in liver chromatin. J. Biol. Chem., 276, 44385–44389. [DOI] [PubMed] [Google Scholar]
- 11.Soutoglou E. and Talianidis,I. (2002) Coordination of PIC assembly and chromatin remodeling during differentiation-induced gene activation. Science, 295, 1901–1904. [DOI] [PubMed] [Google Scholar]
- 12.Proft M. and Struhl,K. (2002) Hog1 kinase converts the Sko1-Cyc8-Tup1 repressor complex into an activator that recruits SAGA and SWI/SNF in response to osmotic stress. Mol. Cell, 9, 1307–1317. [DOI] [PubMed] [Google Scholar]
- 13.Henry K.W., Wyce,A., Lo,W.S., Duggan,L.J., Emre,N.C., Kao,C.F., Pillus,L., Shilatifard,A., Osley,M.A. and Berger,S.L. (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev., 17, 2648–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metivier R., Penot,G., Hubner,M.R., Reid,G., Brand,H., Kos,M. and Gannon,F. (2003) Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell, 115, 751–763. [DOI] [PubMed] [Google Scholar]
- 15.Ijpenberg A., Tan,N.S., Gelman,L., Kersten,S., Seydoux,J., Xu,J., Metzger,D., Canaple,L., Chambon,P., Wahli,W. et al. (2004) In vivo activation of PPAR target genes by RXR homodimers. EMBO J., 23, 2083–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geisberg J.V. and Struhl,K. (2004) Cellular stress alters the transcriptional properties of promoter-bound Mot1-TBP complexes. Mol. Cell, 14, 479–489. [DOI] [PubMed] [Google Scholar]
- 17.Kuras L., Kosa,P., Mencia,M. and Struhl,K. (2000) TAF-containing and TAF-independent forms of transcriptionally active TBP in vivo. Science, 288, 1244–1248. [DOI] [PubMed] [Google Scholar]
- 18.Geisberg J.V., Moqtaderi,Z., Kuras,L. and Struhl,K. (2002) Mot1 associates with transcriptionally active promoters and inhibits the association of NC2 in yeast. Mol. Cell. Biol., 22, 8122–8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X.-Y., Bhaumik,S.R. and Green,M.R. (2000) Distinct classes of yeast promoters revealed by differential TAF recruitment. Science, 288, 1242–1244. [DOI] [PubMed] [Google Scholar]
- 20.Kuras L. and Struhl,K. (1999) Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature, 399, 609–612. [DOI] [PubMed] [Google Scholar]
- 21.Li Y.F., Le Gourierrec,J., Torki,M., Kim,Y.J., Guerineau,F. and Zhou,D.X. (1999) Characterization and functional analysis of Arabidopsis TFIIA reveal that the evolutionary unconserved region of the large subunit has a transcription activation domain. Plant Mol. Biol., 39, 515–525. [DOI] [PubMed] [Google Scholar]
- 22.Auble D.T. and Hahn,S. (1993) An ATP-dependent inhibitor of TBP binding to DNA. Genes Dev., 7, 844–856. [DOI] [PubMed] [Google Scholar]
- 23.Auble D.T., Hansen,K.E., Mueller,C.G.F., Lane,W.S., Thorner,J. and Hahn,S. (1994) Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev., 8, 1920–1934. [DOI] [PubMed] [Google Scholar]
- 24.Chicca J.J., Auble,D.T. and Pugh,B.F. (1998) Cloning and biochemical characterization of TAF-172, a human homolog of yeast Mot1. Mol. Cell. Biol., 18, 1701–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cang V., Auble,D.T. and Prelich,G. (1999) A new regulatory domain on the TATA-binding protein. EMBO J., 18, 6662–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geisberg J.V., Holstege,F.C., Young,R.A. and Struhl,K. (2001) Yeast NC2 associates with the RNA polymerase II preinitiation complex and selectively affects transcription in vivo. Mol. Cell. Biol., 21, 2736–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dasgupta A., Darst,R.P., Martin,K.J., Afshari,C.A. and Auble,D.T. (2002) Mot1 activates and represses transcription by direct, ATPase-dependent mechanisms. Proc. Natl Acad. Sci. USA, 99, 2666–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creton S., Svejstsrup,J.Q. and Collart,M.A. (2002) The NC2 alpha and beta subunits display different roles in vivo. Genes Dev., 16, 3265–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]