

Abstract
Objective
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease in which glia are central mediators of motor neuron (MN) death. Since multiple cell types are involved in disease pathogenesis, the objective of this study was to determine the benefit of co‐targeting independent pathogenic mechanisms in a familial ALS mouse model.
Methods
Recently, our laboratory identified that ALS microglia induce MN death in an NF‐κB‐dependent mechanism. We also demonstrated that a single, post‐natal, intravenous injection of adeno‐associated viral vector serotype 9 encoding a shRNA against mutant SOD1 is able to traverse the blood–brain barrier of ALS mice and reduce SOD1‐expression in astrocytes and MNs. Reducing mutant SOD1 in MNs and astrocytes led to a robust increase in survival. To evaluate the benefit of co‐targeting multiple cell types in ALS, we combined microglial NF‐κB suppression with SOD1 reduction in astrocytes and MNs.
Results
Targeting both astrocytes and microglia resulted in an additive increase in survival and motor function by delaying both onset and progression. Strikingly, targeting all three cell types (astrocytes, motor neurons [MNs], and microglia) resulted in an additive increase in lifespan and motor function, with maximum survival reaching 204 days, 67 days longer than the mean survival of untreated control animals.
Interpretation
Our data suggest that a combinatorial approach co‐targeting different pathogenic mechanisms in independent cell types is a beneficial therapeutic strategy for ALS.
Introduction
Mounting evidence suggests multiple cell types are involved in the devastating loss of MNs in amyotrophic lateral sclerosis (ALS). Seminal studies utilizing chimeric mice containing a mixture of SOD1‐G93A and nontransgenic cells, demonstrate that animals with greater numbers of normal, nontransgenic cells develop a slower, less severe ALS phenotype and survive longer.1 Interestingly, nontransgenic MNs in close‐proximity to mutant SOD1‐expressing non‐neuronal cells exhibit neuronal pathology, suggesting that non‐neuronal cells can induce MN damage in a non‐cell autonomous manner.1
Conditional deletion of the mutant SOD1 gene in specific cell lineages provided evidence that individual cell‐types mediate different aspects of disease. For example, reducing mutant SOD1 in MNs in the slow‐progressing SOD1‐G37R mouse delays disease onset and early disease progression.2 However, reducing mutant SOD1 in glial fibrillary acidic protein (GFAP)‐expressing astrocytes or CD11b‐positive myeloid cells does not alter disease onset but slows disease progression.2, 3 Collectively, these data suggest that mutant SOD1 damage in MNs dictates onset and early progression, and astrocytes and microglia contribute to the rate at which disease progresses.4, 5
While these genetic studies provide a proof‐of‐principle for the cell types involved, the therapeutic potential of SOD1 reduction was not evaluated until recently. Our laboratory demonstrated that postnatal reduction of mutant SOD1 in MNs or astrocytes utilizing a shRNA targeting mutant SOD1 extends survival in both SOD1‐G93A and SOD1‐G37R mice.6 To deliver the shRNA construct, we performed single intravenous injections of adeno‐associated viral vector serotype 9 (AAV9), which has the unique ability to traverse the blood–brain barrier and induce transgene expression throughout the brain and spinal cord. Injection of AAV9 into neonates at postnatal day one (p1) results in robust transduction of the spinal cord with about 60% of ChAT‐positive MNs and 30% of GFAP‐positive astrocytes expressing the transgene.7 When AAV9 is injected intravenously into adult mice, the viral tropism shifts from neuronal to astrocytic transduction, with 90% of transgene‐positive cells co‐expressing astrocytic markers such as GFAP and EAAT2.8 Reducing mutant SOD1 predominantly in astrocytes by injecting AAV9‐shRNA‐SOD1 at p21 extends survival in the SOD1‐G93A mouse model by delaying disease progression. Delivering AAV‐shRNA‐SOD1 at p1 reduces mutant SOD1 predominantly in MNs and astrocytes and increases median lifespan by about 51 days.6
Despite the robust transduction of AAV9 and reduction of mutant SOD1 in MNs and astrocytes, it was not sufficient to completely hinder the progression in ALS mice. It is likely that other cell types that are not targeted efficiently with AAV9, such as microglia, are still able to induce MN death.
We previously demonstrated that microglia mediate MN death via NF‐κB in the SOD1‐G93A mouse model and dampening NF‐κB activation in myeloid cells delayed disease progression by 47%. To transgenically inhibit NF‐κB in microglia in SOD1‐G93A mice, we mated SOD1‐G93A mice to mice with conditional mutants of IKKβ (IKKβ flox/flox) that express cre recombinase under the c‐fms (CSF1R).10 In these mice, CSF‐1R is expressed throughout the mononuclear phagocyte system, but only microglia express CSF‐1R in the postnatal mouse brain.10, 11, 12, 13 Therefore, we hypothesized that a combinatorial approach involving viral‐mediated SOD1 suppression in MNs and astrocytes and transgenic NF‐κB inhibition in microglia would lead to a greater increase of survival in SOD1‐G93A mice.
Materials and Methods
Transgenic mice
All procedures were performed in accordance with the United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals, and the Institutional Animal Care and Use Committee of the Research Institute at Nationwide Children's Hospital approved these studies. Animals were housed under light:dark (12:12 h) cycle and provided with food and water ad libitum. Transgenic male B6SJ/L(SOD1‐G93A)1Gur/J mice (JacksonLaboratory, Bar Harbor, ME USA) were used for breeding with other transgenic lines. As previously described, homozygous IKKβ flox/wt; CSF1R‐cre+ mice display severe immune dysfunction, chronic eye infections, and enlarged spleens, so we evaluated heterozygous mice.10 SOD1‐G93A; IKKβ flox/wt; CSF‐1R‐icre were generated as previously described by breeding SOD1‐G93A mice to C57BL/6 CSF‐1R‐cre (Jackson) mice that had been bred to IKKβ flox/flox mice.9 SOD1‐G93A; IKKβ flox/wt; CSF1R‐cre+ and SOD1‐G93A; IKKβ flox/wt; CSF1R‐cre− mice are abbreviated CSF1R‐cre+ and CSF1R‐cre−, respectively. SOD1 transgene copy number was confirmed for all study animals by quantitative real‐time polymerase chain reaction.
Immunoblot analysis
Lumbar spinal cord tissues were homogenized in Tissue Protein Extraction Reagent (Pierce, Rockford, IL, USA) with EDTA, Complete protease inhibitor (Roche, Indianapolis, IN, USA) and Phospho‐STOP (Roche). The samples were run on NuPAGE Novex 4‐12% Bis‐Tris polyacrilamide gels and transferred to a PVDF membrane (Life Technologies, Carlsbad, CA, USA). Blots were blocked in 5% milk powder, 0.5% BSA in PBS‐Tween for 1 h, and then incubated for overnight at 4°C with primary antibody. Primary antibodies (all Cell Signaling, Danvers, MA, USA): Human SOD1 (1:750), phospho‐p65 (1:500), p65 (1:500), β‐Actin (1:1000). Bound primary antibody was detected by horseradish peroxidase conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) followed by chemiluminescence detection (ECL Western Blot Substrate, Pierce).
Immunohistochemistry
Animals were deeply anesthetized with a lethal dose of xylazene/ketamine and perfused transcardially with saline, then with 4% paraformaldehyde. Spinal cords were sectioned 40 μm thick using a vibrating blade microtome (Leica Microsystems, Wetzlar, Germany). Sections were incubated for 2 h at room temperature in TBS+ 1% Triton‐X + 10% donkey serum. Samples were incubated for 72 h at 4°C with primary antibodies, followed by 2 h incubation at RT with secondary antibodies. Primary antibodies: green fluorescent protein (GFP) (Abcam, 1:400), GFAP (Abcam, Cambridge, UK, 1:500), Iba‐1 (Wako, Richmond, VA, USA, 1:400), ChAT (Millipore,Darmstadt, Germany, 1:50). All images were captured on a Zeiss confocal microscope (Carl Zeiss Microscopy, Thornwood, NY). For quantification of transduced MNs and astrocytes, lumbar spinal cords were sectioned 40 μm thick from the end of thoracic level 14 to sacral level 1.
Viral vectors
The shRNA construct targeting human SOD1 was generated and obtained as previously described from the Life Technologies design tool.6 The shRNA sequence was cloned into pSilencer 3.1 (Genscript, Piscataway, NJ, USA) under the human H1 promoter and tested for human SOD1 reduction in vitro. The shRNA and H1 promoter was further cloned into an AAV vector with GFP under the chicken β‐actin promoter. Self‐complementary AAV9‐SOD1‐shRNA was produced by transient transfection procedures using a double‐stranded AAV2‐ITR‐based CB‐GFP vector, with a plasmid encoding the Rep2Cap9 sequence, along with an adenoviral helper plasmid pHelper (Stratagene, Santa Clara, CA) in HEK293 cells.
Injections
For neonatal mouse injections, postnatal day 1 (p1) SOD1‐G93A pups were used. Total volume of 50 μL containing 5 × 1011 (3.6 × 1014 vg/kg) DNase‐resistant viral particles of AAV9‐SOD1‐shRNA (Virapur LLC, San Diego, CA) was injected through temporal vein as previously described.8 A successful injection was verified by noting blanching of the vein. Pups were returned to their cage after the injection. For adult, postnatal day 21 (p21) tail vein injections, animals were placed in a restraint that positioned the tail in a lighted, heated groove. The tail was swabbed with alcohol and then injected intravenously with AAV9‐SOD1‐shRNA. SOD1‐G93A mice were injected with 200 μL viral solution containing 2 × 1012 DNase‐resistant viral particles, for an average dose of 1.7 × 1014 vg/kg. It is important to note that all cohorts of mice were generated simultaneously for comparison, and p1 and p21 data are plotted separately for clarity. Therefore, the uninjected control groups are the same in Figures 2, 3, 4 and 7, 8, 9.
Figure 2.
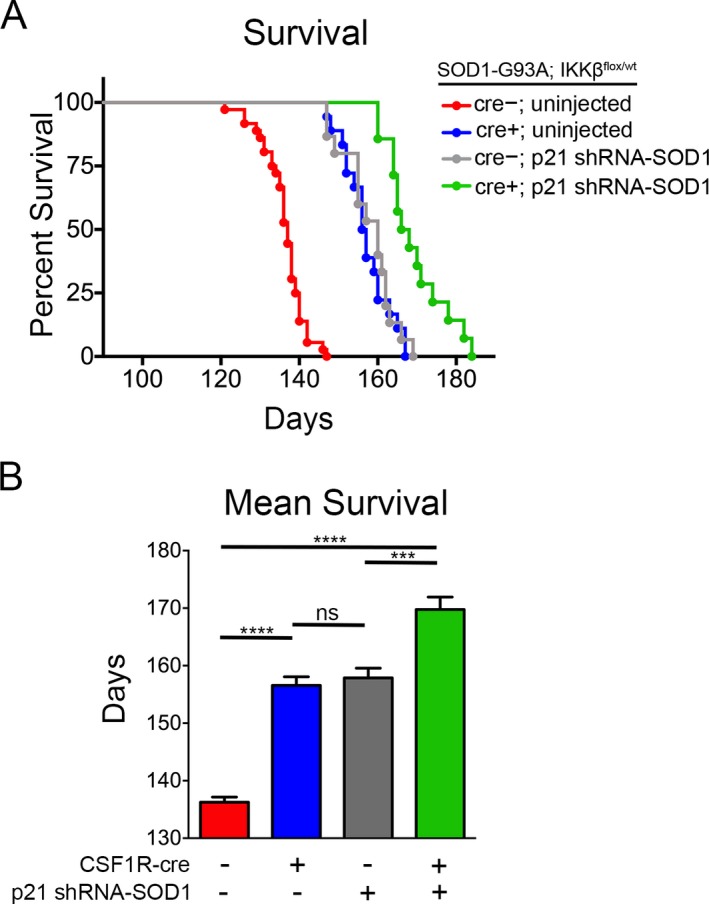
Targeting microglia and astrocytes leads to additive increase in survival in SOD1‐G93A mice. SOD1‐G93A; IKK β F/wt; CSF1R‐cre− mice and SOD1‐G93A; IKK β F/wt; CSF1R‐cre+ mice received a single intravenous injection of AAV9‐SOD1‐shRNA at postnatal day 21. Injected CSF1R‐cre− (gray) and CSF1R‐cre+ (green) mice and uninjected controls (red and blue) were monitored up to end‐stage of disease. (A) Kaplan–Meier survival analysis of the probability of survival as a function of age in SOD1‐G93A; IKK βF/wt; CSF1R‐cre− (shown in red, n = 33), SOD1‐G93A; IKK βF/wt; CSF‐1R‐cre+ mice (blue, n = 13), CSF1R‐cre− mice injected with SOD1‐shRNA at p21 (gray, n = 14), and CSF1R‐cre+ mice injected with AAV9‐SOD1‐shRNA at p21 (green, n = 13). Median survival: uninjected CSF1R‐cre− = 137 days, uninjected CSF‐1R‐cre+ = 157 days, CSF1R‐cre− p21 injected = 160 days, CSF1R‐cre+ p21 injected = 168 days. (B) Mean survival of mice from uninjected CSF1R‐cre− = 136.9 ± 0.9 days, uninjected CSF‐1R‐cre+ = 157.9 ± 1.8 days, CSF1R‐cre− p21 injected = 158.1 ± 1.8 days, CSF1R‐cre+ p21 injected = 169.8 ± 2.2 days. AAV9, adeno‐associated viral vector serotype 9.***p < 0.001; ****p < 0.0001.
Figure 3.
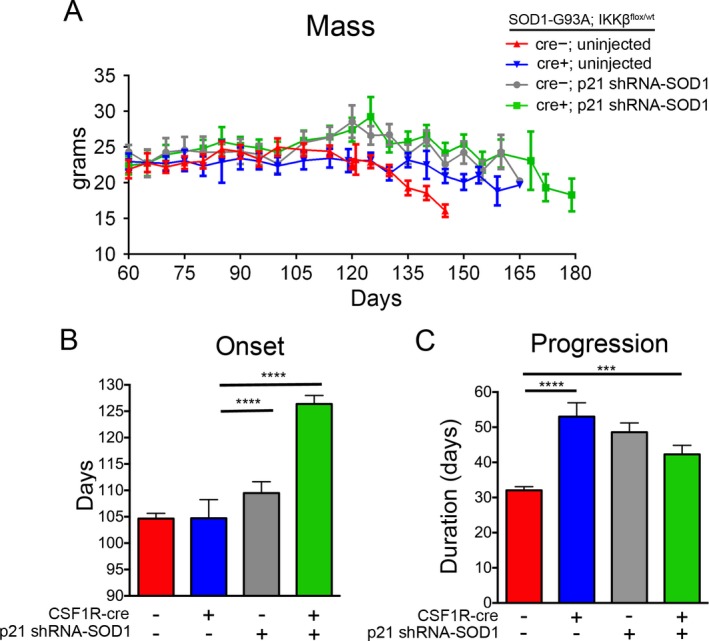
Disease onset and progression are delayed in SOD1‐G93A mice by targeting both astrocytes and microglia. (A) Uninjected CSF1R‐cre+ mice (blue) retained more body mass in late stages of disease compared to uninjected CSF1R‐cre− mice (red). P21‐treated mice, both CSF1R‐cre− (gray) and CSF1R‐cre+ (green), maintained body mass throughout their increased lifespan (B) Onset defined by peak weight is delayed in CSF1R‐cre+; p21 injected mice (green) compared to all uninjected (blue and green) and CSF1R‐cre−; p21 injected mice (gray). (C) Disease progression, defined by time from peak weight until death, is delayed in all mice with either microglia (blue), astrocytes (gray), or both (green) targeted, compared to untreated controls (red). ***p < 0.001; ****p < 0.0001.
Figure 4.

Motor function is improved in SOD1‐G93A mice by targeting microglia and astrocytes in concert. SOD1‐G93A; IKK flox/wt; CSF1R‐cre+; p1 injected mice (green) exhibit improved motor performance as demonstrated in accelerating rotarod testing (A) forelimb grip strength (B) and hindlimb grip strength (C). Motor performance was improved in all treated groups (blue, gray, and green) compared to untreated controls (red).
Figure 7.
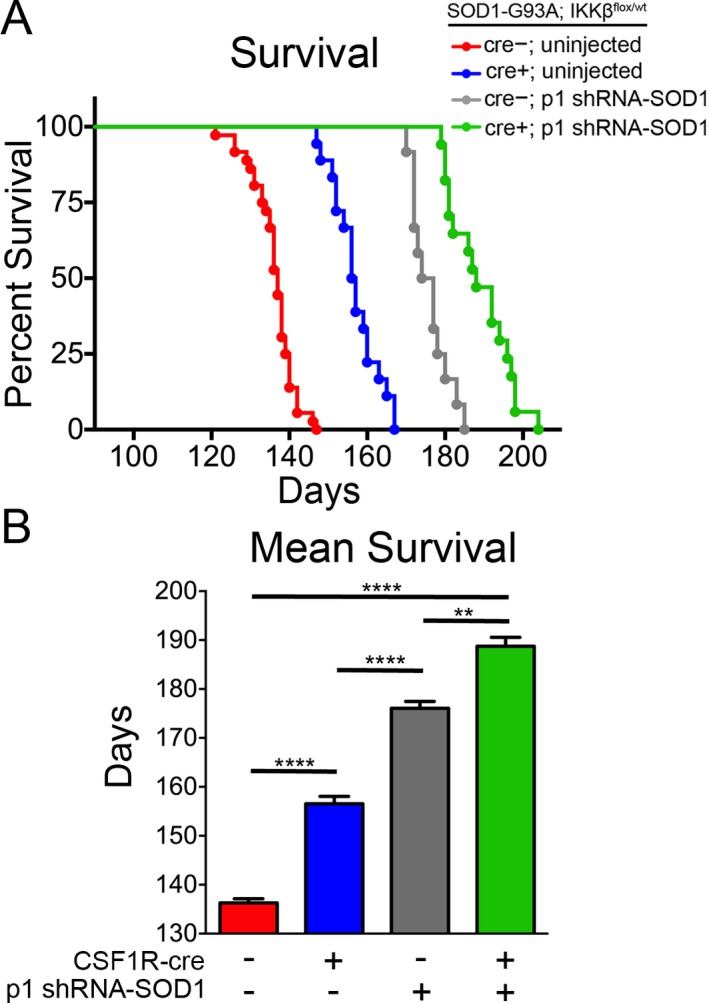
Targeting microglia, astrocytes, and motor neurons leads to additive increase in survival in SOD1‐G93A mice. SOD1‐G93A; IKK β F/wt; CSF1R‐cre− mice and SOD1‐G93A; IKK β F/wt; CSF1R‐cre+ mice received a single intravenous injection of AAV9‐SOD1‐shRNA at postnatal day 1. Injected CSF1R‐cre− (gray) and CSF1R‐cre+ (green) mice and uninjected controls (red and blue) were monitored up to end‐stage of disease. (A) Kaplan–Meier survival analysis of the probability of survival as a function of age in SOD1‐G93A; IKK βF/wt; CSF1R‐cre− (shown in red, n = 33), SOD1‐G93A; IKK βF/wt; CSF‐1R‐cre+ mice (blue, n = 13), CSF1R‐cre− mice injected with SOD1‐shRNA at p1 (gray, n = 8), and CSF1R‐cre+ mice injected with AAV9‐SOD1‐shRNA at p1 (green, n = 12). Median survival: uninjected CSF1R‐cre− = 137 days, uninjected CSF‐1R‐cre+ = 157 days, CSF1R‐cre− p1 injected = 177.5 days, CSF1R‐cre+ p1 injected = 185 days. (B) Mean survival of mice from uninjected CSF1R‐cre− = 136.9 ± 0.9 days, uninjected CSF‐1R‐cre+ = 157.9 ± 1.8 days, CSF1R‐cre− p1 injected = 177.4 ± 1.9 days, CSF1R‐cre+ p1 injected = 189.1 ± 2.7 days. **p < 0.01; ***p < 0.001; ****p < 0.0001.
Figure 8.
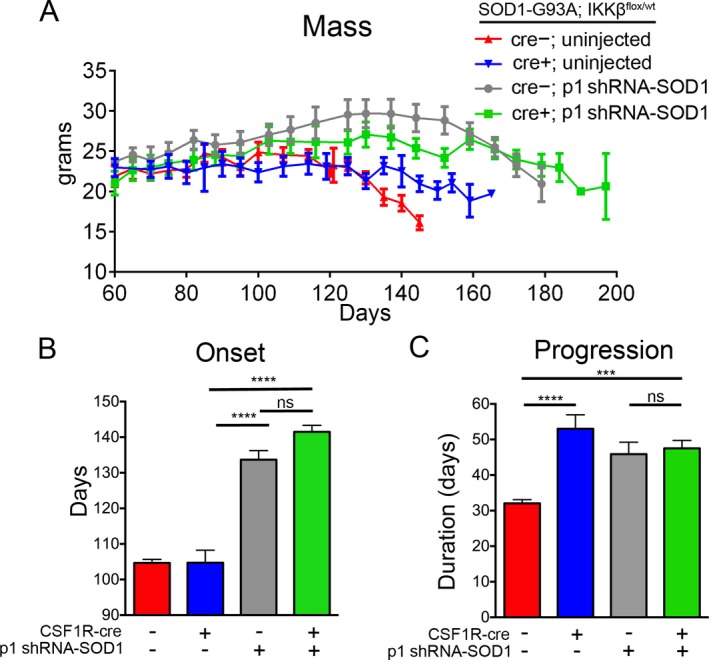
Targeting microglia, astrocytes, and motor neurons delays onset and disease progression in SOD1‐G93A mice. (A) Both CSF1R‐cre− (gray) and CSF1R‐cre+ (green) p1‐treated mice maintained body mass throughout their increased lifespan. Onset (B) and disease progression (C) was delayed in both CSF1R‐cre− and CSF1R‐cre+; p1 injected mice (gray and green) compared to all uninjected (blue and green) mice. ***p < 0.001; ****p < 0.0001.
Figure 9.
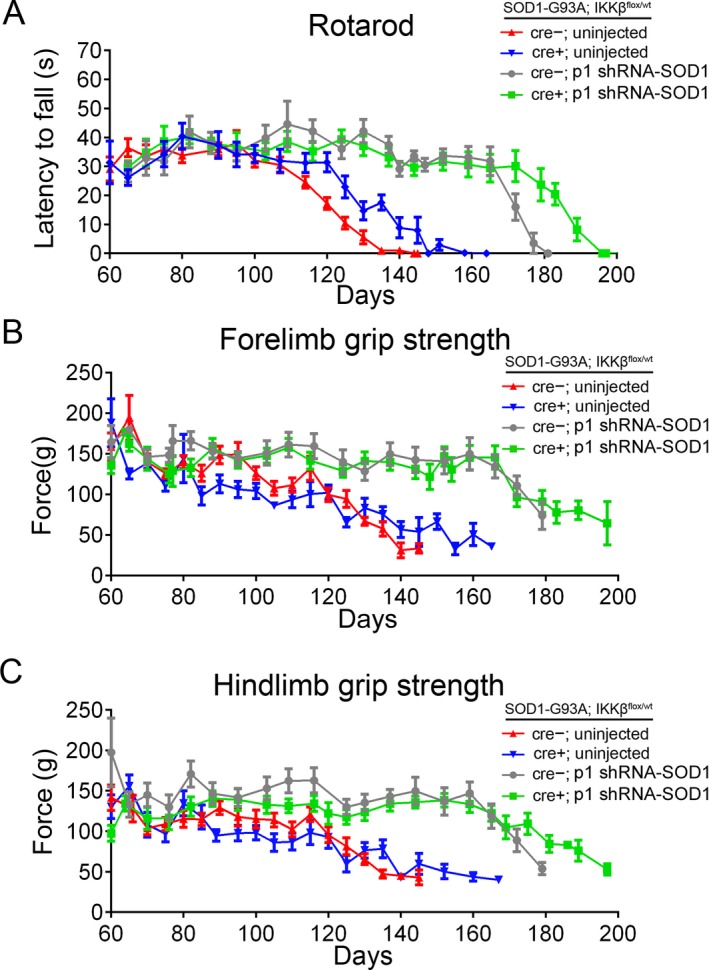
Motor function improves additively in SOD1‐G93A mice by targeting microglia, motor neurons, and astrocytes. SOD1‐G93A; IKK flox/wt; CSF1R‐cre+; p1‐injected mice (green) exhibit improved motor performance as demonstrated in accelerating rotarod testing (A) forelimb grip strength (B) and hindlimb grip strength (C). All treated groups (blue, gray, and green) showed improved motor performance over untreated controls (red).
Disease scoring and behavior analysis
Disease onset and progression was determined retrospectively from animal mass data. Onset is defined at the stage mice reach peak body mass. Progression is defined as the time from peak body mass until death. Testing of motor function using a rotarod device (Columbus Instruments, Columbus, OH) began at 60 days of age. Each session consisted of three trials that were averaged on the elevated accelerating rotarod beginning at 5 rpm/min measuring the time the mouse was able to remain on the rod. Grip strength measurements for hindlimb were tested weekly using a grip strength meter (Columbus Instruments). Each session consisted of three tests per animal and values were averaged.
Results
To target both astrocytes and microglia, we injected SOD1‐G93A; IKKβ flox/wt; CSF1R‐cre+ (abbreviated CSF1R‐cre+) mice intravenously with AAV9‐SOD1‐shRNA at postnatal day 21. Along with the SOD1‐shRNA, the AAV9 vector encodes contains GFP allowing us to visualize AAV9 transduction. Shown by immunohistochemistry (Fig. 1A), we observed that 90% of transduced cells were astrocytes in the lumbar spinal cord of 100‐day‐old mice, similar to levels we previously reported.6, 8 Quantification of GFP+ cells indicated the intravenous injection of AAV9‐shRNA‐SOD1 targeted about 70% of total GFAP‐positive astrocytes and 10% of ChAT+ MNs (Fig. 1B).
Figure 1.
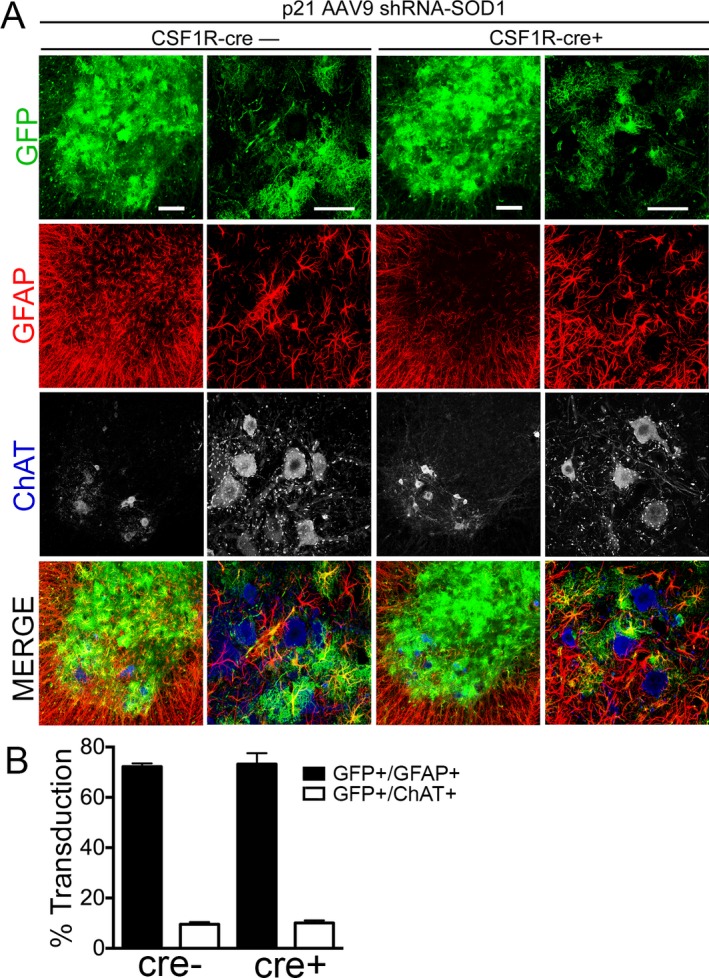
AAV9 transduces astrocytes when injected in adult CSF1R‐cre+ and cre− SOD1‐G93A mice. (A) Immunohistochemistry and quantification of lumbar spinal cords of SOD1‐G93A; IKK β F/wt; CSF1R‐cre− mice (left) and SOD1‐G93A; IKK β F/wt; CSF1R‐cre+ mice (right) injected at postnatal day 21 with AAV9‐SOD1‐shRNA expressing GFP. (B) Quantification of transduced (GFP+) cells at postnatal day 100 shows no significant difference in the mean percent of transduced astrocytes (GFP+/GFAP+) between CSF1R‐cre− (72.2 ± 0.7%) and CSF1R‐cre+ mice (73.2 ± 2.5%). Few motor neurons were transduced at p21 in both CSF1R‐cre− (9.6 ± 0.5%) and CSF1R‐cre+ mice (10.1 ± 0.6%), determined by counting transduced cells (GFP) co‐labeled ChAT+ motor neurons throughout the lumbar spinal cord. Lumbar spinal cords from three animals per group were analyzed. Scale bars indicate 100 μm (left) and 50 μm (right). AAV9, adeno‐associated viral vector serotype 9; GFP, green fluorescent protein.
As expected, median survival of uninjected CSF1R‐cre+ mice was 20 days longer than uninjected CSF1R‐cre− littermates. P21 injected CSF1R‐cre− mice had an increased median survival of 23 days compared to uninjected controls. Strikingly targeting both NF‐κB activation in microglia and reducing SOD1 in astrocytes, extended median survival to 167 days compared to 137 days in untreated, control mice (Fig. 2A). This amounted to a 25% increase in mean survival in p21 injected CSF‐1R‐cre+ mice (169.8 ± 2.2 days) compared to uninjected CSF‐1R‐cre− mice (136.3 ± 0.9 days) (Fig. 2B).
Consistent with the increase in lifespan, body mass was maintained longer in injected mice and uninjected CSF1R‐cre+ mice compared to uninjected CSF1R‐cre− mice (Fig. 3A). Disease onset, which is retrospectively defined as the age at which the mouse reaches peak weight, was not altered in uninjected CSF1R‐cre+ mice compared to uninjected controls (104.7 ± 2.5 and 104.2 ± 1.0 days, respectively). However, p21 injected CSF1R‐cre− mice exhibited a slight delay in disease onset (108.9 ± 2.1 days), most likely attributed to the small amount of MNs transduced. Surprisingly, onset was significantly delayed in p21 CSF1R‐cre+ mice (127.6 ± 1.9 days), about 23 days longer than uninjected controls (Fig. 3B). It is important to note that similar numbers of MNs (about 10% of total ChAT+ cells) were transduced with p21 injections of AAV9 in CSF1R‐cre+ and CSF1R‐cre− mice.
Disease progression, measured from time at which the animal reaches peak weight until death, was prolonged in all treated groups compared to uninjected CSF1R‐cre− mice (Fig. 3C). However, targeting both astrocytes and microglia did not result in an additive extension in progression. Disease progression of CSF1R‐cre+ p21 injected mice averaged 42.3 ± 2.6 days compared to 48.6 ± 2.6 days in p21 injected CSF1R‐cre− and 51.44 ± 2.7 days for uninjected CSF1R‐cre+ mice (Fig. 3C). We hypothesize the additive increase in survival was distributed between early and late phases of disease; thus we did not observe a preferential additive increase in disease onset or progression.
Motor performance measured by accelerating rotarod (Fig. 4A), forelimb (Fig. 4B), and hindlimb grip strength (Fig. 4C) was improved in all conditions in which astrocytes and/or microglia were targeted. CSF1R‐cre+ uninjected mice exhibited improved rotarod performance during disease progression compared to CSF1R‐cre− uninjected littermates (Fig. 3A). Similar to the late‐stage differences between uninjected CSF1R‐cre+ and CSF1R‐cre− mice, p21 injected groups (CSF1R‐cre+ and CSF1R‐cre−) exhibited improved motor performance until the late‐stage of disease (Fig. 4A–C). These data suggest targeting microglia and astrocytes leads to an additive increase in survival and motor function in SOD1‐G93A mouse model.
Since we observed an additive increase in median survival by targeting both astrocytes and microglia in the SOD1‐G93A mice, we hypothesized that reducing levels of mutant SOD1 in MNs and astrocytes would lead to an additional increase survival in CSF1R‐cre+ mice. To efficiently target MNs and astrocytes with AAV9, we injected a cohort of mice with AAV9‐SOD1‐shRNA intravenously at postnatal day 1. It is important to note that all cohorts of mice were generated simultaneously for comparison, and p1 and p21 data are plotted separately for clarity. As shown by immunohistochemistry, GFP can be detected in both astrocytes and MNs of p1‐injected animals (Fig. 5A). Quantification reveals about 60% of ChAT+ MNs and 40% of astrocytes are transduced when AAV9‐shRNA‐SOD1 is injected at p1 (Fig. 5B).
Figure 5.
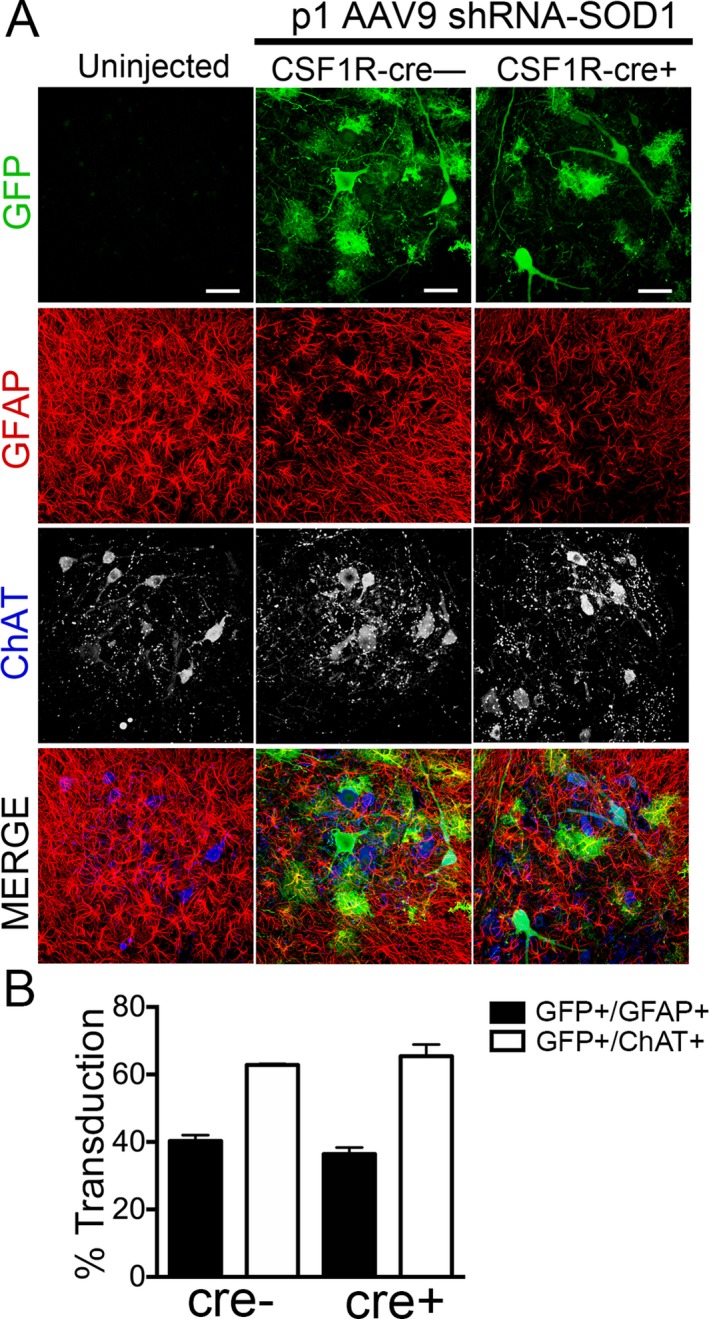
Injection of AAV9 at postnatal day 1 transduces motor neurons and astrocytes in CSF1R‐cre+ and cre− ALS mice. (A) Immunohistochemistry and quantification of lumbar spinal cords of SOD1‐G93A; IKK β F/wt; CSF1R‐cre− mice (left) and SOD1‐G93A; IKK β F/wt; CSF1R‐cre+ mice (right) injected at postnatal day 1 with AAV9‐SOD1‐shRNA expressing GFP. Scale bar indicates 50 μm. (B) Quantification of transduced (GFP+) cells at postnatal day 100 shows no significant difference in the mean percent of transduced astrocytes (GFP+/GFAP+) between CSF1R‐cre− (40.3 ± 1.7%) and CSF1R‐cre+ mice (36.4 ± 2.0%). Few motor neurons were transduced at p1 in both CSF1R‐cre− (62.8 ± 0.3%) and CSF1R‐cre+ mice (65.5 ± 3.4%), determined by counting transduced cells (GFP) co‐labeled ChAT+ motor neurons throughout the lumbar spinal cord. Lumbar spinal cords from three animals per group were analyzed. AAV9, adeno‐associated viral vector serotype 9; ALS, amyotrophic lateral sclerosis; GFP, green fluorescent protein.
To determine the level of SOD1 suppression between p21 and p1 injections, we performed immunoblot analyses of end‐stage whole‐lumbar spinal cord lysate. Mutant SOD1 was reduced by 50% at end‐stage when AAV9‐shRNA‐SOD1 was injected at p1 or p21, similar to our previously findings (Fig. 6A and B).6 No difference in mutant SOD1 suppression was observed between CSF1R‐cre+ and cre− injected mice (Fig. 6A). To confirm NF‐κB inhibition in our CSF1R‐cre+ mice, we performed immunoblot analysis in lumbar spinal cord homogenate for phosphorylated p65 (phospho‐p65). Phospho‐p65 was reduced in end‐stage lumbar spinal cord homogenate of CSF1R‐cre+ compared to cre− mice (Fig. 6C).
Figure 6.
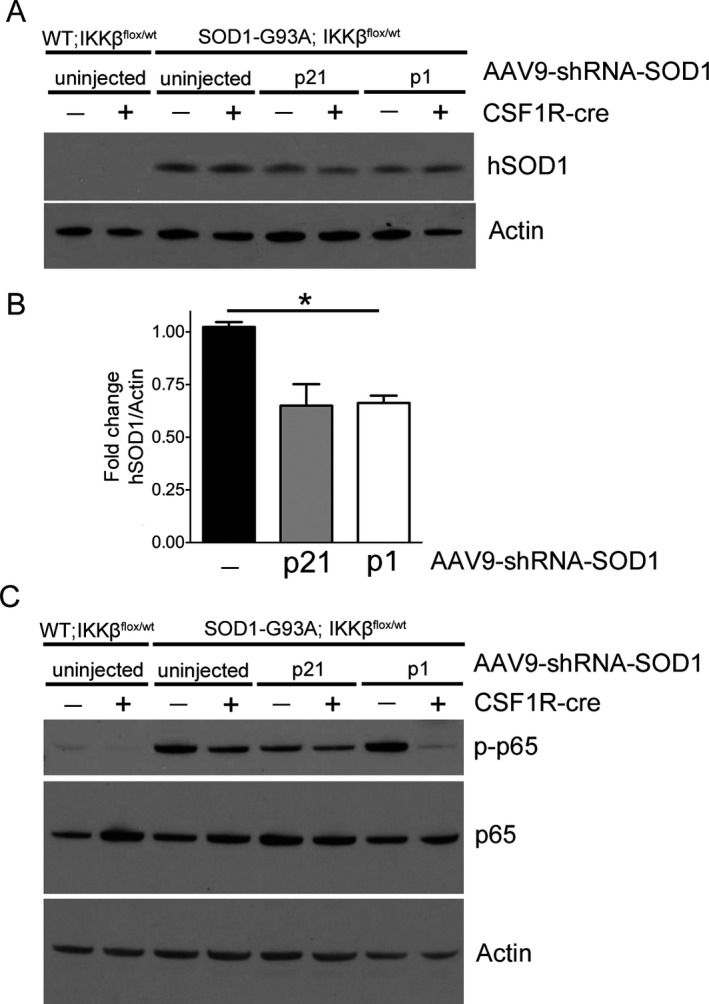
AAV9 shRNA‐SOD1 efficiently reduces mutant SOD1 protein in CSF‐1R‐cre− and CSF‐1R‐cre+ mice. (A) End‐stage spinal cord lysates of SOD1‐G93A; IKK β F/wt CSF1R‐cre− and CSF1R‐cre+ mice injected at either p21 or p1 with AAV9‐shRNA‐SOD1 were evaluated by immunoblot for levels of mutant human SOD1 protein. (B) Quantification of (A), hSOD1 normalized to actin. (C) End‐stage lumbar spinal cord lysates were evaluated for NF‐κB activation by probing for phosphorylated‐p65 (p‐p65). All CSF1R‐cre+ mice exhibited reduced p‐p65, indicative of decreased NF‐κB activity. AAV9, adeno‐associated viral vector serotype 9. *p < 0.05.
Strikingly targeting both NF‐κB activation in microglia and reducing SOD1 in MNs and astrocytes (CSF1R‐cre+, p1‐shRNA‐SOD1) extended median survival to 188 days compared to 137 days in CSF1R‐cre− uninjected mice (Fig. 7A). This amounted to a 38% increase in mean survival (188.7 ± 1.8 and 136.3 ± 0.9 days) (Fig. 7B). The longest‐lived mouse survived to 204 days of age, one of the longest extensions in survival ever reported for this fast‐progressing SOD1‐G93A mouse model. CSF1R‐cre− p1 injected mice had a median survival of 175.5 days, and had a 27% increase in mean survival compared to CSF1R‐cre− uninjected mice (Fig. 7B). These data demonstrate that heterozygous inhibition of NF‐κB signaling in microglia leads to an additive increase in survival when combined with SOD1 suppression in MNs and astrocytes.
Consistent with the increase in lifespan, body mass was maintained longer in injected mice compared to uninjected mice. Interestingly, less mass was lost overall in both CSF1R‐cre+ groups (uninjected and p1 injected) compared to both CSF1R‐cre− groups (Fig. 8A). Although not statistically significant, onset was delayed in CSF1R‐cre+; p1 injected mice compared to CSF1R‐cre−; p1 injected mice (134.9 ± 2.3 and 139.1 ± 1.8 days, respectively) (Fig. 8B). As predicted, disease progression was prolonged in CSF1R‐cre− p1 injected mice and CSF1R‐cre+ p1 injected mice (Fig. 8C). Thus, all conditions targeting microglia and/or MNs and astrocytes resulted in an extension in disease progression.
Motor performance measured by accelerating rotarod, forelimb and hind limb grip strength was substantially improved in p1‐injected mice compared to both uninjected groups (Fig. 9A–C). Similar to the late‐stage differences in uninjected CSF1R‐cre+ and CSF1R‐cre− mice, p1 injected groups (CSF1R‐cre+ and CSF1R‐cre−) exhibited similar motor performance until the late‐stage of disease, when CSF1R‐cre+ mice maintained motor function, which coincided with the longer lifespans. These data suggest a combinatorial approach to target microglia, MNs and astrocytes confers greatest benefit to motor function and survival of SOD1‐G93A mice.
One of the most striking disease hallmarks that is shared by patients and rodent models of ALS is gliosis, characterized by alterations in astrocyte and microglial morphology. NF‐kB inhibition in microglia and SOD1 suppression both independently decrease gliosis.6, 10 Therefore, to determine the impact of a combinatorial treatment on gliosis at disease onset, we analyzed lumbar spinal cord sections for levels of Iba‐1 as an indicator of microgliosis and GFAP for astrogliosis. We observed a robust decrease in Iba1 and GFAP levels at 100 days of age in CSF1R‐cre+ mice that were injected with AAV9‐shRNA‐SOD1 at either p1 or p21 (Fig. 10). Since gliosis is an indicator of homeostasis in the spinal cord, these data suggest disease pathogenesis is substantially delayed in combinatorial treated mice.
Figure 10.
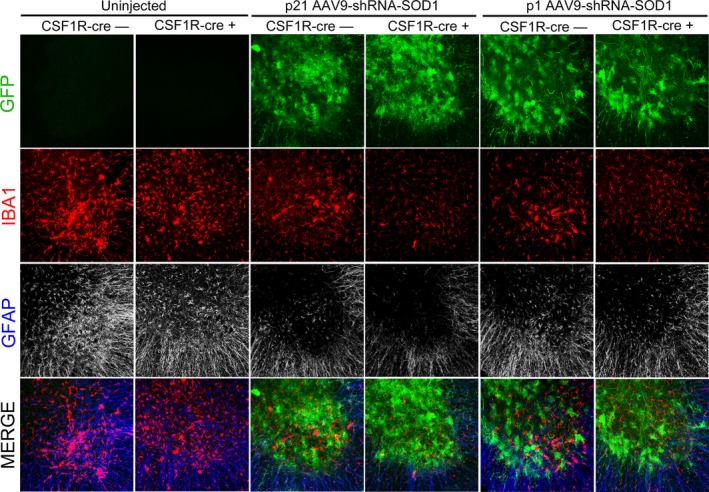
Astrogliosis and microgliosis is delayed in SOD1‐G93A mice when multiple pathogenic mechanisms are targeted. Immunohistochemistry of lumbar spinal cords at 100 days of age shows marked reduction in reactivity of microglia (Iba1, red) and astrocytes (GFAP, blue) in both p21‐ and p1‐injected mice. GFP transduction of AAV9‐shRNA‐SOD1 is shown in green. Three animals were analyzed for each group. Scale bar indicates 200 μm. GFP, green fluorescent protein; AAV9, adeno‐associated viral vector serotype 9.
Discussion
Here, we report that co‐targeting independent pathological mechanisms in different cell types in combination leads to one of the largest extensions in survival observed in the high copy number, fast‐progressing SOD1‐G93A mouse model of ALS. We previously demonstrated that NF‐κB is upregulated in SOD1‐G93A microglia with disease progression. Heterozygous inhibition of NF‐κB resulted in a 20‐day increase in median survival by delaying disease progression by 47%. We now show that suppressing NF‐κB activation in microglia and reducing mutant SOD1 levels in both astrocytes and MNs extended median lifespan to 188 days compared to 137 days in controls. This amounted to a 38% increase in median survival with maximum survival reaching up to 204 days. These animals displayed a striking improvement in motor function compared to untreated controls shown by the accelerating rotarod test, and forelimb and hindlimb grips strength. Rotarod performance did not decline in combinatorial treated mice until around 180 days of age, nearly 70 days after motor impairment was first detected in untreated controls.
By combining microglial NF‐κB inhibition with SOD1 suppression in astrocytes and MNs, we observed an additive increase in survival. This suggests that NF‐κB activation in microglia functions independently of SOD1 in astrocytes and MNs in the pathogenesis of ALS. Interestingly, we observed a surprising shift in disease onset when targeting predominantly astrocytes and microglia, but not MNs. Previous studies suggest that astrocytes and microglia are not involved in disease onset but contribute to the rate of disease progression.2, 3, 4 As expected, targeting either glial cell type independently corroborated previous studies and significantly extended disease progression but not onset.6, 10 Since reducing mutant SOD1 in MNs delays disease onset and early disease progression, it is important to note that a small percentage of MNs are transduced when AAV9 is injected at postnatal day 21.6, 8 This may account for a 5 day delay in mean disease onset in CSF1R‐cre‐negative mice injected with AAV9‐shRNA‐SOD1 at p21. However, the number of GFP‐positive MNs was not different between injected cre‐positive and cre‐negative mice. Thus, the delay in disease onset in CSF1R‐cre+, p21‐injected mice cannot be attributed to the small percentage of MNs targeted with AAV9‐shRNA‐SOD1.
Immunohistochemical analysis of lumbar spinal cords at 100 days (disease onset of uninjected mice), showed that all combinatorial‐targeted mice had reduced gliosis to near wild‐type levels. We previously demonstrated that microglial NF‐κB inhibition in SOD1‐G93A mice slowed astrogliosis and microglial activation during disease progression.10 In this study, targeting both astrocytes and microglia delayed activation from the point of disease onset. Since glial reactivity is an indicator of homeostasis in the spinal cord, targeting both astrocytes and microglia prolonged homeostasis compared to uninjected control animals. Thus, our data may reveal a more complicated, unknown interaction between astrocytes and microglia in early ALS pathogenesis and disease onset. Gene expression profiling of presymptomatic microglia corroborates immunohistochemical data, suggesting glial alterations occur early in SOD1‐G93A mice.14, 15 This raises the intriguing question of whether full correction of glial cell types can rescue or delay degeneration of diseased MNs.
While combinatorial targeting of microglia, astrocytes, and MNs resulted in a robust increase in survival, ultimately the mice still succumbed to disease. This can be attributed to several factors. First, we are not removing all mutant SOD1 from astrocytes and MNs or fully suppressing NF‐κB activity in microglia. Despite reducing mutant SOD1 levels by nearly 50% in the spinal cord, residual mutant SOD1 eventually leads to paralysis in this high copy number, aggressive SOD1‐G93A strain. We hypothesize that similar SOD1 reduction strategies will result in an even greater increase in survival in strains expressing lower levels of SOD1 initially, and even in SOD1 patients, considering patients only harbor one mutant allele. Second, targeting a more upstream pathogenic mechanism in microglia, such as mutant SOD1, will most likely confer greater benefit to survival. We hypothesize that synergistic epistasis between NF‐κB and SOD1 might only be observed if both are suppressed in microglia. Lastly, other cell types such as oligodendrocytes and NG2+ progenitors contribute to disease pathogenesis.16, 17 Recently, it was reported that removal of mutant SOD1 in oligodendrocyte progenitors delays disease onset and early disease progression in the slow‐progressing SOD1‐G37R mouse model of ALS.16 Interestingly, removing mutant SOD1 selectively from muscle or endothelia did not alter the disease course in ALS mouse models. However, it is possible that as treated mice live longer, more subtle cellular players or potentially new pathological mechanisms may emerge. Thus, the most efficacious therapeutic approach is likely to derive from co‐targeting multiple pathogenic mechanisms and cell types.
Cumulative evidence suggests microglial NF‐κB may be a promising therapeutic target for both familial and sporadic ALS. For example, several laboratories have confirmed NF‐κB is activated in glia in both familial and sporadic ALS patients by immunohistochemistry on postmortem tissue samples. Additionally, TDP‐43 and FUS have been shown to be co‐activators of NF‐κB, and NF‐κB inhibition in transgenic mice overexpressing mutant TDP‐43 ameliorated the disease phenotype.18, 19 Furthermore, several other ALS‐associated mutations such as OPTN, p62, and VCP are involved in regulating the NF‐κB pathway and could be involved in pathological NF‐κB activation in microglia23, 24, 25. Interestingly, the ALS‐causing nonsense and missense mutations in OPTN abolished the ability of OPTN to inhibit NF‐κB signaling.20, 21 One well‐characterized function of VCP is to assist in proteasomal degradation of the NF‐κB inhibitor, IκBα.22 Future studies should determine whether NF‐κB activation in microglia harboring these ALS mutations results in MN toxicity. However, the inability to easily study human microglia remains one of the biggest limitations in the field.
Whether SOD1 is a therapeutic target for sporadic ALS remains controversial. Recent studies have given rise to the hypothesis that wild‐type human SOD1 has the propensity to misfold, aggregate, and trigger pathogenic pathways similar to those induced by mutant SOD1.26, 27, 28, 29, 30 However, conflicting reports fail to detect misfolded SOD1 in postmortem sporadic tissue.31, 32, 33 While no consensus has emerged, the potential that some sporadic ALS patients could also benefit from AAV9‐mediated SOD1 reduction should be investigated. Furthermore, recent reports demonstrate that AAV9 can readily transduce MNs and astrocytes in non‐human primates.8, 34, 35 Thus, as new potential therapeutic targets emerge for sporadic ALS, AAV9 could be utilized as an efficient tool to manipulate these cell types.
Taken together, these data provide new insight and a proof‐of‐principle that co‐targeting multiple pathogenic mechanisms in independent cell types is a promising therapeutic strategy for ALS. A multipronged strategy has proved beneficial in treating other diseases and cancers and should be explored for neurodegenerative diseases such as ALS.36, 37, 38, 39 Thus, future efforts should focus on generating efficient and specific methods to target individual cell types in the brain and spinal cord.
Conflict of Interest
The authors report no conflict of interest regarding this work.
Acknowledgment
We thank Leah Schmelzer and Terri Schafer for technical assistance. This work was funded by the US National Institutes of Health (NIH) R01 NS644912, Project A.L.S. The ALS Association and Packard Center for ALS Research (P2ALS), and the Helping Link Foundation. A. F. is supported by NINDS T32NS077984 Training in Neuromuscular Disease.
References
- 1. Clement AM, Nguyen MD, Roberts EA, et al. Wild‐type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003;302:113–117. [DOI] [PubMed] [Google Scholar]
- 2. Boillee S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006;312:1389–1392. [DOI] [PubMed] [Google Scholar]
- 3. Yamanaka K, Chun SJ, Boillee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 2008;11:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boillee S, Vandevelde C, Cleveland D. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 2006;52:39–59. [DOI] [PubMed] [Google Scholar]
- 5. Beers DR, Henkel JS, Xiao Q, et al. Wild‐type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2006;103:16021–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foust KD, Salazar DL, Likhite S, et al. Therapeutic AAV9‐mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther 2013;21:2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol 2010;28:271–274. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Foust KD, Nurre E, Montgomery CL, et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 2008;27:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng L, Zhou J‐F, Sellers RS, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin‐dependent hyperproliferation of colonic epithelium to inflammation‐associated tumorigenesis. Am J Pathol 2010;176:952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frakes AE, Ferraiuolo L, Haidet‐Phillips AM, et al. Microglia induce motor neuron death via the classical NF‐κB pathway in amyotrophic lateral sclerosis. Neuron 2014;81:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erblich B, Zhu L, Etgen AM, et al. Absence of colony stimulation factor‐1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 2011;6:e26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasmono RT, Oceandy D, Pollard JW, et al. A macrophage colony‐stimulating factor receptor‐green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 2003;101:1155–1163. [DOI] [PubMed] [Google Scholar]
- 13. Sasmono RT, Williams E. Generation and characterization of MacGreen mice, the Cfs1r‐EGFP transgenic mice. Methods Mol Biol 2012;844:157–176. [DOI] [PubMed] [Google Scholar]
- 14. Butovsky O, Siddiqui S, Gabriely G, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest 2012;122:3063–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiu IM, Morimoto ETA, Goodarzi H, et al. A neurodegeneration‐specific gene‐expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 2013;4:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang SH, Li Y, Fukaya M, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 2013;16:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Philips T, Bento‐Abreu A, Nonneman A, et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013;136(Pt 2):471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Swarup V, Phaneuf D, Dupré N, et al. Deregulation of TDP‐43 in amyotrophic lateral sclerosis triggers nuclear factor κB‐mediated pathogenic pathways. J Exp Med 2011;208:2429–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uranishi H, Tetsuka T, Yamashita M, et al. Involvement of the pro‐oncoprotein TLS (translocated in liposarcoma) in nuclear factor‐kappa B p65‐mediated transcription as a coactivator. J Biol Chem 2001;276:13395–13401. [DOI] [PubMed] [Google Scholar]
- 20. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2011;465:223–226. [DOI] [PubMed] [Google Scholar]
- 21. Zhu G, Wu C‐J, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha‐ induced NF‐kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol 2007;17:1438–1443. [DOI] [PubMed] [Google Scholar]
- 22. Dai RM, Chen E, Longo DL, et al. Involvement of valosin‐containing protein, an ATPase co‐purified with IkappaBalpha and 26 S proteasome, in ubiquitin‐proteasome‐mediated degradation of IkappaBalpha. J Biol Chem 1998;273:3562–3573. [DOI] [PubMed] [Google Scholar]
- 23. Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF‐kappaB mediator in tumorigenesis. Cancer Cell 2008;13:343–354. [DOI] [PubMed] [Google Scholar]
- 24. Sanz L, Diaz‐Meco MT, Nakano H, Moscat J. The atypical PKC‐interacting protein p62 channels NF‐kappaB activation by the IL‐1‐TRAF6 pathway. EMBO J 2000;19:1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wooten MW, Geetha T, Seibenhener ML, et al. The p62 scaffold regulates nerve growth factor‐induced NF‐kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem 2005;280:35625–35629. [DOI] [PubMed] [Google Scholar]
- 26. Bosco DA, Morfini G, Karabacak NM, et al. Wild‐type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 2010;13:1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forsberg K, Jonsson PA, Andersen PM, et al. Novel antibodies reveal inclusions containing non‐native SOD1 in sporadic ALS patients. PLoS One 2010;5:e11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rakhit R, Robertson J, Vande Velde C, et al. An immunological epitope selective for pathological monomer‐misfolded SOD1 in ALS. Nat Med 2007;13:754–759. [DOI] [PubMed] [Google Scholar]
- 29. Pokrishevsky E, Grad LI, Yousefi M, et al. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 2012;7:e35050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haidet‐Phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 2011;29:824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kerman A, Liu H‐N, Croul S, et al. Amyotrophic lateral sclerosis is a non‐amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 2010;119:335–344. [DOI] [PubMed] [Google Scholar]
- 32. Liu H‐N, Sanelli T, Horne P, et al. Lack of evidence of monomer/misfolded superoxide dismutase‐1 in sporadic amyotrophic lateral sclerosis. Ann Neurol 2009;66:75–80. [DOI] [PubMed] [Google Scholar]
- 33. Brotherton TE, Li Y, Cooper D, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci USA 2012;109:5505–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bevan AK, Duque S, Foust KD, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther 2011;19:1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer K, Ferraiuolo L, Schmelzer L, et al. Improving single injection CSF delivery of AAV9‐mediated gene therapy for SMA: a dose‐response study in mice and nonhuman primates. Mol Ther 2015;23:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammer SM, Katzenstein DA, Hughes MD, et al. A trial comparing nucleoside monotherapy with combination therapy in HIV‐infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N Engl J Med 1996;335:1081–1090. [DOI] [PubMed] [Google Scholar]
- 37. Collier AC, Coombs RW, Schoenfeld DA, et al. Treatment of human immunodeficiency virus infection with saquinavir, zidovudine, and zalcitabine. AIDS Clinical Trials Group. N Engl J Med 1996;334:1011–1017. [DOI] [PubMed] [Google Scholar]
- 38. Chao MP, Alizadeh AA, Tang C, et al. Anti‐CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non‐Hodgkin lymphoma. Cell 2010;142:699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Conway D, Cohen JA. Combination therapy in multiple sclerosis. Lancet Neurol 2010;9:299–308. [DOI] [PubMed] [Google Scholar]