

ABSTRACT
High extracellular osmolarity results in a switch from an adaptive to an inflammatory gene expression program. We show that hyperosmotic stress activates the protein kinase R (PKR) independently of its RNA-binding domain. In turn, PKR stimulates nuclear accumulation of nuclear factor κB (NF-κB) p65 species phosphorylated at serine-536, which is paralleled by the induction of a subset of inflammatory NF-κB p65-responsive genes, including inducible nitric oxide synthase (iNOS), interleukin-6 (IL-6), and IL-1β. The PKR-mediated hyperinduction of iNOS decreases cell survival in mouse embryonic fibroblasts via mechanisms involving nitric oxide (NO) synthesis and posttranslational modification of proteins. Moreover, we demonstrate that the PKR inhibitor C16 ameliorates both iNOS amplification and disease-induced phenotypic breakdown of the intestinal epithelial barrier caused by an increase in extracellular osmolarity induced by dextran sodium sulfate (DSS) in vivo. Collectively, these findings indicate that PKR activation is an essential part of the molecular switch from adaptation to inflammation in response to hyperosmotic stress.
KEYWORDS: DSS, PKR, hyperosmotic stress, iNOS
INTRODUCTION
The intracellular response to increased extracellular osmolarity (hyperosmotic stress) involves unique stress-induced signaling pathways that are distinct from other cellular stress conditions. After initial detection of hyperosmotic conditions and ionic perturbations, the cell either begins transcriptional and translational recovery programs or commits to apoptosis (1). Phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) increases with hyperosmotic stress duration and intensity (2), which contributes to the global inhibition of protein synthesis by sequestering eIF2 in an inactive complex (3). Other investigators have shown previously that the commitment to apoptosis is a result of prolonged shutdown of translation of mRNAs encoding antiapoptotic proteins (4, 5). However, we have shown that apoptotic determination in severe stress occurs independently of decreased protein synthesis and correlates with phosphorylation of eIF2α (2). eIF2α phosphorylation is only one mechanism by which protein synthesis is decreased. In cells bearing a phosphorylation-incompetent eIF2α (serine-51–alanine), protein synthesis is still inhibited to the same degree as in wild-type (WT) cells, pointing to redundant mechanisms of protein synthesis inhibition. Previous studies have shown that apoptosis can be regulated during hyperosmotic stress via mechanisms either dependent on eIF2α phosphorylation, such as the cytoplasmic localization of hnRNP A1 and translational inhibition of antiapoptotic mRNAs (2), or independent of eIF2α phosphorylation, such as angiogenin-mediated cleavage of tRNAs and inhibition of the apoptosome (6). Much of the cellular signaling surrounding eIF2α phosphorylation remains to be elucidated, including additional mechanisms that contribute to apoptotic cell death.
One possible mechanism of cell death downstream of hyperosmotic stress is via the initiation of a subsequent inflammatory response. It has been shown previously that proinflammatory gene and cytokine induction, as well as invasion of immune cells, is observed in tissues susceptible to hyperosmotic changes in the extracellular environment, such as the kidney (7), cornea, liver, and gastrointestinal tract (8). Several inflammatory diseases have been linked to a hyperosmotic stress response, such as diabetes (9), dry eye syndrome (10, 11), and inflammatory bowel disease (IBD) (12, 13).
Many genes are expressed in the inflammatory response, with the combined effect of a prosurvival or proapoptotic outcome. Signaling molecules, including cytokines (such as interleukin-6 [IL-6]) and chemokines (such as Mcl-1) can play context-dependent roles in determining cell fate (14, 15). These signaling molecules exert effects in trans, initiating responses in other cells that lead to a proteome that promotes survival or apoptosis. A well-accepted marker of inflammation in both IBD and in dry eye syndrome is the induction of inducible nitric oxide synthase (iNOS). Although the enzymatic product of iNOS, the signaling molecule nitric oxide (NO), has been shown to be protective in small amounts and for short durations (16–18), upregulation of iNOS can lead to prolonged and increased NO production. NO at high levels can be detrimental as it reacts with superoxide to form peroxynitrite, posttranslationally modify proteins, and alter or interfere with their function (18–20). iNOS and NO have both been found to be increased in relevant tissues of patients diagnosed with IBD and dry eye syndrome (21), as well as positively correlated with cellular damage (22) and disease onset (23).
The regulation of iNOS is primarily under transcriptional control (24). The transcription factor nuclear factor κB (NF-κB) induces transcription of the iNOS gene under conditions such as ethanol treatment (25), Toll-like receptor (TLR) activation (26), and hyperosmotic stress (27). NF-κB is a dimeric transcription factor that is held inactive in the cytoplasm but is released upon signal-induced phosphorylation and degradation of the inhibitory binding partner (IκBα) to enter the nucleus (28). There are over 500 genes known to be regulated by NF-κB, including cytokine and chemokine genes, immunoreceptor genes, and stress response genes (for a full list, see http://www.bu.edu/nf-kb/gene-resources/target-genes/). The combination of genes actually induced is highly cell type and stimulus specific (29). One way in which the appropriate pattern of gene expression is regulated is via the pattern of modifications to each subunit of the NF-κB dimer, known as the barcode hypothesis (30, 31). As apparent from the list of responsive genes, NF-κB has been shown to be active both in inflammation (32, 33) and under hyperosmotic stress conditions (34, 35).
Although NF-κB activation and hyperosmotic stress are associated with the inflammatory response, the molecular mechanisms linking the hyperosmotic stress response, NF-κB activation, and the initiation of inflammation remain poorly defined. Here, we investigate the involvement of protein kinase R (PKR) in both the hyperosmotic stress response and the induction of proinflammatory signaling molecules, including iNOS. PKR is considered activated when it becomes phosphorylated, is in an open conformation, and homodimerizes (36). This classically occurs when PKR binds double-stranded RNA (dsRNA), such as in the event of viral infection (37). However, PKR can also become activated independently of dsRNA binding when it is bound to PKR-activating protein (PACT), whose binding disrupts the closed inactive conformation of PKR (36). PKR is also known to be involved in the initiation of inflammation in response to viral infection via the NF-κB signaling pathway (38). We use mouse embryonic fibroblasts (MEFs) to elucidate a PKR-amplified proinflammatory signaling pathway that is initiated by the exposure of cells to hyperosmotic medium, that progresses via nuclear localization of a subset of NF-κB subunit p65 species phosphorylated at serine-536, and that leads to iNOS induction and subsequently to apoptosis. We show that dextran sodium sulfate (DSS) increases both the extracellular osmolarity and similar proinflammatory signaling in a cell culture model. We also show evidence of the therapeutic potential of PKR inhibition using a small-molecule inhibitor of PKR in vivo in a mouse model of IBD.
RESULTS
PKR is activated under hyperosmotic conditions and amplifies p65-mediated iNOS gene expression.
PKR is a serine/threonine kinase that homodimerizes and autophosphorylates, becoming activated classically in response to dsRNA (37). PKR is known to phosphorylate eIF2α at serine-51 in response to viral infection and is essential in the NF-κB-mediated inflammatory response to this event (39). We hypothesized that as PKR is known to be involved in the inflammatory response and can phosphorylate eIF2α, it may represent a molecular link between these signaling events in the hyperosmotic stress response. In order to determine the role of PKR in hyperosmotic stress, we treated MEF cells with 600 mosM sucrose in Dulbecco's modified Eagle's medium (DMEM) for increasing durations. Western blot analysis showed that PKR is activated, as indicated by phosphorylation at threonine-451 upon treatment with hyperosmotic medium (Fig. 1A). The use of a small-molecule inhibitor of PKR kinase activity 6,8-dihydro-8-(1H-imidazol-5-ylmethylene)-7H-pyrrolo[2,3-g]benzothiazol-7-one (C16; designated PKRi) inhibited phosphorylation and activation of PKR under this stress condition. PKR activation correlated with increased cleavage of caspase-3, hinting at a proapoptotic role for PKR in hyperosmotic stress signaling. Short hairpin RNA (shRNA)-mediated suppression of PKR protein (shPKR) decreased hyperosmotic stress-induced caspase-3 cleavage, confirming a role for PKR in this process (Fig. 1B).
FIG 1.
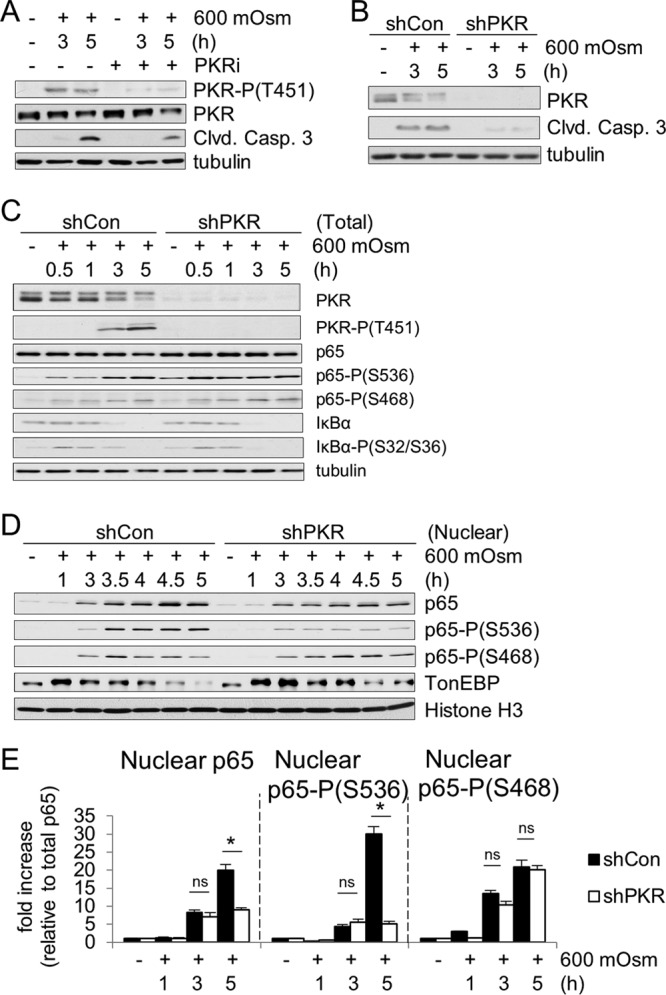
PKR is activated under hyperosmotic stress conditions and affects NF-κB phosphorylation and nuclear localization. (A and B) MEFs were treated with the PKR inhibitor (PKRi; 1.25 μM) for 1 h and then with the addition of hyperosmotic medium (A) or selected for lentivirus infection of control (shCon) or shPKR cells before treatment with hyperosmotic medium (B) for the indicated durations. Lysates were analyzed via immunoblotting for the indicated proteins. (C and D) Total cell lysates and nuclear fractions from control and shPKR MEFs treated with hyperosmotic medium for the indicated durations. Hyperosmotic medium was a final osmolarity of 600 mosM (sucrose). Proteins were detected at the following masses: PKR at 55 kDa; PKR phosphorylated at T451 [PKR-P(T451)] at ∼60 kDa; cleaved (Clvd) caspase-3 at 15 kDa; tubulin at 55 kDa; p65, p65-P(S536), and p65-P(S468) at 65 kDa; IκBα and IκBα-P(S32/S36) at 35 kDa; TonEBP at ∼200 kDa; and histone H3 at ∼20 kDa. (E) Quantification of NF-κB p65, p65-P(S536), and p65-P(S468) translocated to the nucleus (n = 3 biological replicates). Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
As NF-κB and its phosphorylation status have been demonstrated to play a role in inflammatory signaling (40), we investigated the involvement of PKR in phosphorylation of the p65 subunit of NF-κB. We hypothesized that hyperosmotic stress involves PKR-dependent and -independent mechanisms of activation of NF-κB subunit p65 and the subsequent transcriptional induction of target genes. The PKR-independent mechanisms should involve the release of NF-κB subunit p65 from its inhibitory cytoplasmic complex with IκBα via its phosphorylation and degradation (40). This hypothesis is in agreement with the observation that increased extracellular osmolarity caused a decrease in levels and an increase in phosphorylation of IκBα (Fig. 1C). Suppression of PKR did not change the levels and phosphorylation pattern of IκBα during hyperosmotic stress, suggesting that PKR is not essential for classical NF-κB p65 activation during hyperosmotic stress by the IκB kinase α/β (IKKα/β)-mediated phosphorylation of IκBα (41). We next hypothesized that PKR might influence the site-specific posttranslational phosphorylation of NF-κB p65, thus influencing the induction of target genes which are strictly dependent on specific NF-κB p65 phosphorylation events for their activation (30, 42). We focused on phosphorylation at serine-536 and serine-468, two sites involved in target gene-specific regulation (31). Hyperosmotic stress caused the phosphorylation of p65 at serine-536 and serine-468, suggesting enhanced transactivation potential (Fig. 1C). Interestingly, increased phosphorylation of p65 at serine-536 was observed in total cell lysates even prior to addition of hyperosmotic medium, suggesting a role for PKR in controlling basal NF-κB p65 phosphorylation. Moreover, stress-induced NF-κB p65 phosphorylation was found to be increased with hastened kinetics in shPKR cells. However, despite this greater signal of p65 phosphorylated at serine-536 in total lysate, the relative nuclear localizations of total p65 and of p65 phosphorylated at this residue were unchanged at 1 h and reduced in shPKR cells after 3 and 5 h of hyperosmotic treatment, corresponding to the absence, initiation, and maximal phosphorylation, respectively, of PKR (Fig. 1D and E). The nuclear localization of p65 phosphorylated at serine-468 was unchanged. These data support the idea that both the phosphorylation and the nuclear localization of serine-536 phosphorylated-NF-κB p65 involve a PKR-dependent mechanism. The functional outcome of this mechanism during hyperosmotic stress is expected to be the differential regulation of NF-κB subunit p65 target genes, a theme previously described as the NF-κB barcode hypothesis (30, 31). Given the prosurvival and proapoptotic functions of NF-κB p65 signaling (43, 44), we expect activation of the PKR/NF-κB p65 axis to contribute to cell fate during hyperosmotic stress. It should be mentioned that shPKR did not alter the nuclear localization of the tonicity enhancer binding protein (TonEBP) (Fig. 1D), further supporting the specificity of PKR in modulating the nuclear levels of NF-κB p65 phosphorylated at serine-536.
We hypothesized that PKR/NF-κB-mediated signaling may contribute to increased apoptosis in response to hyperosmotic stress based on the increase in caspase-3 cleavage (Fig. 1A and B). To test this hypothesis, first we performed reverse transcription-quantitative PCR (RT-qPCR) on mRNAs for a number of NF-κB target genes under hyperosmotic stress conditions. Particular attention was given to the regulation of iNOS expression that was previously described to be positively induced by NF-κB p65 phosphorylated at serine-536 and serine-468 (31, 42). Induction of iNOS mRNA was completely abrogated by loss of NF-κB p65 (Fig. 2A), indicating that induced expression of this gene is dependent on NF-κB p65 in this cell type and stress condition. This increase in iNOS mRNA correlated with an increase in iNOS protein accumulation under hyperosmotic stress conditions (Fig. 2B). As NF-κB p65 and its phosphorylation at serine-536 and serine-468 have been linked to iNOS mRNA induction via the intermediary kinase IKKε (31), we also measured the effects of inhibition of this protein kinase. Unlike IKKα/β, IKKε may not result in degradation of IκBα but has been shown to be necessary for downstream target gene induction via direct phosphorylation of NF-κB p65 (31). Treatment with a small-molecule inhibitor directed to the active site of IKKε was sufficient to inhibit the induction of hyperosmotic stress-induced iNOS mRNA (Fig. 2C). To confirm the importance of IKKε in iNOS regulation, we suppressed IKKε protein via shRNA and observed similar results (Fig. 2D). NF-κB p65 phosphorylation and nuclear localization were not absent in shIKKε MEFs (data not shown), suggesting that IKKε may be involved as an intermediary in a PKR/p65/IKKε axis mediating the regulation of a subset of NF-κB p65 target genes. Other genes showed a similar dependence on NF-κB p65 expression, including proinflammatory cytokine IL-6, the chemoattractant Mcl-1, and the NF-κB inhibitory molecule IκBα (Fig. 2E). This indicates that NF-κB p65 is necessary for transcriptional induction of several p65 target genes under hyperosmotic conditions. Other proteins involved in the unfolded protein response (UPR) including transcription factor ATF4, phosphatase subunit GADD34, and endoplasmic reticulum (ER) stress response genes Xbp1s, CHOP, BiP, and eIF2α kinase PKR-like endoplasmic reticulum kinase (PERK) were not activated or upregulated under hyperosmotic conditions (Fig. 2F).
FIG 2.
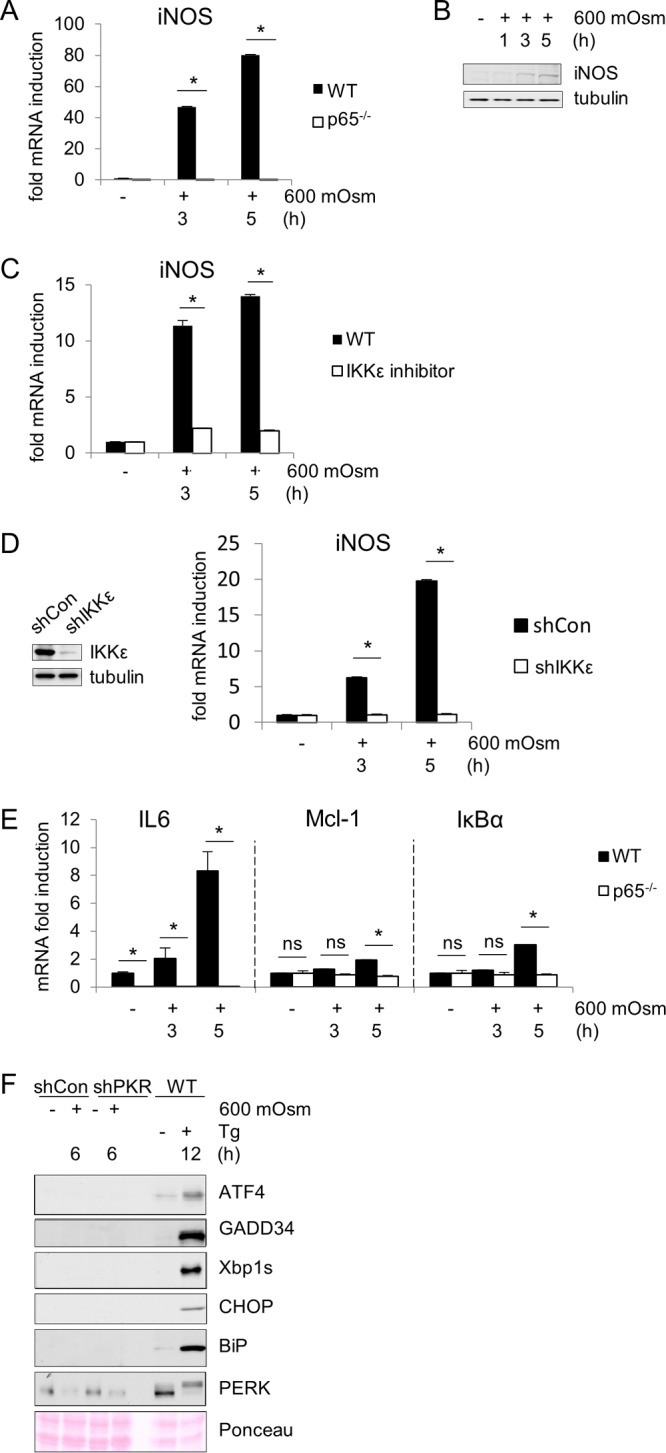
NF-κB p65 is necessary for full induction of iNOS and other mRNAs in response to hyperosmotic treatment. (A) RT-qPCR analysis of mRNA isolated from wild-type and p65-deficient MEFs treated with 600 mosM sucrose. (B) Western blot analysis of wild type MEFs treated with 600 mosM sucrose. (C) RT-qPCR analysis of mRNA isolated from MEFs cotreated with the IKKε inhibitor (100 nM) and hyperosmotic medium. (D) MEFs treated with shIKKε before treatment with hyperosmotic medium for the indicated durations. IKKε depletion was verified by Western blotting. (E) RT-qPCR analysis of mRNA isolated from wild-type and p65-deficient MEFs treated with 600 mosM sucrose. (F) Control and shPKR MEFs were treated with DSS medium, and wild type MEFs were treated with Tg for the indicated durations. Cell lysates were analyzed by immunoblotting. IKKε protein was detected at ∼75 kDa, tubulin was detected at 55 kDa, iNOS was detected at 130 kDa, ATF4 was detected at ∼48 kDa, GADD34 was detected at ∼110 kDa, Xbp1s was detected at 54 kDa, CHOP was detected at 27 kDa, BiP was detected at 75 kDa, and PERK was detected at ~140 to 160 kDa. mRNA levels in panels A to E were normalized to the level of GAPDH. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
To determine if PKR has any effect on NF-κB p65-mediated iNOS gene expression, we measured iNOS mRNA induction by RT-qPCR in MEFs in which PKR expression was suppressed. We found that induction of iNOS mRNA was greatly diminished in shPKR cells compared to that in wild-type cells (Fig. 3A). The attenuation of maximal induction was confirmed with the use of the PKR inhibitor (Fig. 3B). Similar attenuation of proinflammatory gene induction was found in PKR-deficient MEFs reconstituted with a kinase-deficient PKR (data not shown), supporting the necessity of PKR catalytic activity for maximal response. To further verify that maximal transcriptional induction of iNOS is both NF-κB and PKR dependent, we studied iNOS promoter-induced transcription in a luciferase reporter system (Fig. 3C). When MEFs were treated with hyperosmotic medium, luciferase activity was increased, indicating transcriptional control via the iNOS promoter (Fig. 3D). In addition, when the known κB elements in the promoter were mutated, this stress dependent-increase in activity was lost. Luciferase activity was diminished compared to that in control MEFs when the wild-type reporter plasmid was transfected into shPKR MEFs (Fig. 3E), confirming that iNOS is under transcriptional control of a PKR/NF-κB signaling axis. Similar to results with the NF-κB p65-deficient MEFs, the induction of one additional NF-κB p65 target (IL-6) was found to be inhibited by the loss of PKR, while other NF-κB p65 targets (Mcl-1 and IκBα) were unaffected (Fig. 3F); it is likely that other NF-κB gene targets may display similar patterns of PKR-dependent and -independent induction. This supports the hypothesis that only a subset of genes is coordinately regulated by both PKR and NF-κB p65 along this signaling axis under hyperosmotic stress conditions. Another class of stress response genes known to be activated only in adaptation, or osmoadaptive genes, were unaffected by loss of PKR, as exemplified by mRNA levels of aldose reductase (AR) (Fig. 3G). The trend in gene induction was consistent between wild-type and shRNA-transfected control MEFs. Taken together, these data suggest activation of a signaling pathway connecting the kinase ability of PKR to the phosphorylation and nuclear localization of NF-κB subunit p65 during the hyperosmotic stress response, leading to the induction of proinflammatory genes such as iNOS (Fig. 3H).
FIG 3.

PKR is necessary for maximal induction of iNOS mRNA. RT-qPCR analysis was performed of RNA isolated from control and shPKR MEFs treated with 600 mosM sucrose (A, F, and G) or from MEFs treated with PKRi for 1 h prior to addition of hyperosmotic medium (B) for the indicated durations. (C) Schematic representation of wild-type and κB-site mutant plasmid constructs of the iNOS promoter fragments fused to the luciferase reporter gene. (D) Luciferase activities were normalized to Renilla luciferase in MEFs transfected with the constructs shown in panel C and then treated with hyperosmotic medium for the indicated durations. (E) Control and shPKR MEFs were transfected with the wild-type luciferase reporter and treated with hyperosmotic medium for the indicated durations. Luciferase activity was normalized to that of the transfected Renilla luciferase reporter. (H) A model of iNOS mRNA amplification under hyperosmotic stress via PKR activation and NF-κB signaling. In all panels mRNA levels were normalized to the level of GAPDH. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
Amplification of iNOS has proapoptotic effects during hyperosmotic stress.
To determine the ultimate function of iNOS in hyperosmotic stress, we first measured global NO production. NO was detected at higher levels under hyperosmotic stress conditions, and this increase was reduced by inhibition of PKR kinase activity (Fig. 4A). As nitric oxide is produced from l-arginine, cells must have increased uptake of the amino acid l-arginine, the iNOS substrate (45). An amino acid uptake assay verified increased l-arginine uptake from extracellular medium during hyperosmotic exposure (Fig. 4B). In order to determine that the levels of nitric oxide are toxic and play an important role in apoptosis induction in hyperosmotic stress, we used a nitric oxide scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO). Use of this scavenger reduced caspase-3 cleavage during hyperosmotic stress (Fig. 4C), indicating that nitric oxide production plays a key role in hyperosmotic stress-induced cell death. The use of the PKR inhibitor also dramatically increased cell survival (Fig. 4D), suggesting a positive correlation between PKR activation, nitric oxide production, and apoptosis. This reduction in apoptosis was confirmed using a caspase-3 activity assay in both shPKR- and PKRi-treated MEF cells (Fig. 4E). As a previous report demonstrated that apoptosis was independent of global protein synthesis inhibition (2), we confirmed that protein synthesis levels were not a differentiating factor in PKRi-treated and untreated MEFs (Fig. 4F).
FIG 4.

NO produced during hyperosmotic stress is proapoptotic. (A) Cell lysates were assayed for nitric oxide from wild-type MEFs treated with hyperosmotic medium and PKRi or control and shPKR MEFs for the indicated durations. (B) Cell lysates were assayed for arginine uptake from wild-type MEFs treated with hyperosmotic medium. (C) Caspase-3 cleavage was determined by quantification of immunoblot signals of cell extracts from control and shPKR MEFs treated with hyperosmotic medium and the nitric oxide scavenger cPTIO (100 μM) for the indicated durations. Proteins were detected at the following masses: PKR at 55 kDa, cleaved caspase-3 at 15 kDa, and tubulin at 55 kDa. (D) Cell survival was quantified using an MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay of MEFs treated with hyperosmotic medium and PKRi for the indicated durations. (E) Caspase-3 activities were measured in cell lysates from control and PKRi-treated MEFs as well as from control and shPKR MEFs treated with hyperosmotic medium for the indicated durations. (F) Protein synthesis levels were measured by [35S]Met-Cys incorporation in MEFs treated with PKRi and hyperosmotic medium for the indicated durations. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
One mechanism by which nitric oxide alters cell function is via the nitrosylation of proteins. Although many proteins can be nitrosylated, we chose to examine a well-defined example as a proof of concept. The glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) can be nitrosylated, the effect of which is to alter its subcellular localization, which correlates positively with apoptosis (46). Using an established method to detect protein nitrosylation (47), it was shown that GAPDH was nitrosylated under conditions of hyperosmotic stress, as seen by a relative increase of signal in the ascorbate-treated sample after stress treatment (Fig. 5A). In addition, the nitrosylation of GAPDH was shown to decrease its catalytic activity during hyperosmotic stress as despite constant levels of protein (Fig. 5B), activity decreased over the duration of hyperosmotic treatment (Fig. 5C). Transfection of hemagglutinin (HA)-tagged GAPDH plasmid into MEF cells (Fig. 5D) showed that after hyperosmotic stress treatment, levels of GAPDH in the nucleus were increased (Fig. 5E). Using a plasmid bearing a substitution mutation that alters a single cysteine residue to a serine residue (C150S) (46), it was determined that the increase in nuclear localization was a result of modification at this cysteine-150 residue. In addition, inhibition of PKR led to the reversal of this nuclear localization of GAPDH, linking the effects of PKR activity to the nitrosylation of GAPDH protein and its subsequent nuclear localization (Fig. 5E). Taken together, these data support the hypothesis that during hyperosmotic stress, PKR becomes activated and contributes to increased nuclear levels of NF-κB p65 phosphorylated at serine-536. This accumulation of activated NF-κB contributes to the amplification of iNOS expression, which is translated to active iNOS protein, which produces nitric oxide. Nitric oxide modifies proteins to alter their functions and correlates with apoptotic cell death.
FIG 5.
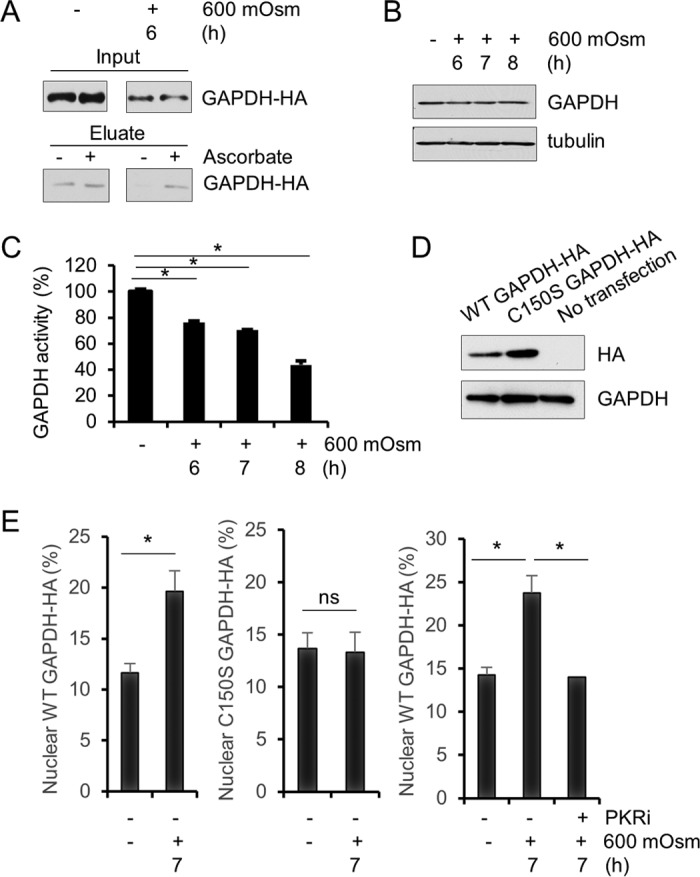
Nitrosylation of GAPDH occurs in hyperosmotic stress. (A) Cell lysates from MEFs treated with hyperosmotic medium for the indicated durations were analyzed for S-nitrosylation in the presence or absence of ascorbate. Eluates were analyzed via immunoblotting. GAPDH protein was detected at ∼35 kDa. (B) Cell lysates from MEFs treated with hyperosmotic medium for the indicated durations were analyzed via immunoblotting for the indicated proteins. (C) GAPDH activity was measured in cell lysates from MEFs treated with hyperosmotic medium for the indicated durations. (D) HA-tagged wild-type and mutant (C150S) GAPDH were expressed in MEFs as verified by Western blotting. HA and GAPDH were detected at ∼35 kDa. (E) Nuclear localization of GAPDH-HA was measured by immunofluorescence and quantified (n = 10) from MEFs transfected with wild-type or mutant GAPDH-HA and treated with hyperosmotic medium and PKRi for the indicated durations. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
PKR amplifies induction of iNOS gene expression in IECs and promotes a DSS-induced colitis phenotype.
In order to test whether PKR affects amplification of iNOS expression and cell fate in a tissue susceptible to inflammation, we turned to the colon in the mouse model of inflammatory bowel disease, DSS-induced colitis. DSS-induced colitis is a well-established, reproducible model of intestinal inflammation that mimics ulcerative colitis in the form of disruption of the integrity of the intestinal epithelial barrier (48). DSS-induced colitis has also been characterized as having increased expression of iNOS mRNA in colon tissue, which is similar to what occurs in human patients diagnosed with ulcerative colitis (49). In addition, increased osmolarity of colonic fluid has been reported in DSS-induced colitis (50). Although only the rectally located portion of the colon shows histological signs of inflammation in the DSS-induced colitis model (51), RT-qPCR analysis revealed an increase in iNOS mRNA in both distal (from the rectum) and rectal colon after DSS treatment (Fig. 6A). In addition, proinflammatory cytokine IL-22 increased only in the rectal colon, while its anti-inflammatory decoy receptor IL22ra2 was present at high levels in the distal colon under control conditions and decreased upon DSS treatment. IL-22 is expressed in a subset of T cells and innate lymphocytes (52). This exemplifies the regional difference in gene induction in this disease model as colitis is observed primarily in the rectal colon (51). Proinflammatory cytokine IL-6 increased in the distal colon as well and to a greater extent than in the rectal colon. There was no significant loss of epithelial cells as levels of villin mRNA did not dramatically decrease. Protein lysates extracted from the rectal colon revealed that both iNOS and cleaved caspase-3 proteins accumulate over the course of DSS treatment although TonEBP is not detectable (Fig. 6B). These data indicate that proinflammatory mRNAs and apoptotic markers colocalize to the region of the colon displaying the disease phenotype. It should be noted that the intestinal tissue gene expression (Fig. 6A) and protein levels (Fig. 6B) measured consist of many cell types. Because DSS-induced colitis involves disruption of the intestinal epithelial barrier, we next determined the expression of proinflammatory genes in intestinal epithelial cells (IECs). It has been previously shown that the colitis phenotype occurs upon DSS treatment in Rag1−/− and SCID mice (48), indicating that proinflammatory cytokine induction leading to loss of mucosal integrity is independent of T cells. In order to determine that the mRNA signals that we were investigating came from IECs themselves and not from invading immune cells, IECs were isolated, and mRNA was extracted from this cell subpopulation. RT-qPCR analysis revealed that proinflammatory iNOS, IL-6, and Cox2 mRNAs were induced in IECs (Fig. 6C). Other osmoadaptive mRNAs, known to be induced in the hyperosmotic stress response by TonEBP (SNAT2 and AR) but not in the inflammatory response, were not increased in IECs.
FIG 6.

PKR inhibition ameliorates inflammatory gene expression in a mouse model of IBD. (A) RT-qPCR of the indicated mRNAs, isolated from dissected rectal and distal sections of the colon from C57BL/6 mice given water containing DSS (2.5%) for 5 days. (B) Western blot analysis of rectal sections of the colon from DSS-treated mice and wild-type MEFs. Proteins were detected at the following masses: TonEBP at ∼200 kDa, cleaved caspase-3 at 15 kDa, iNOS at 130 kDa, and tubulin at 55 kDa. ¥ represents an artifact band at ∼125 kDa. (C) RT-qPCR analysis of mRNA isolated from colonic epithelial cells from DSS-treated mice for the indicated times. Values were normalized to villin mRNA. (D) Immunoblots for the indicated proteins from cell lysates isolated from colonic epithelial cells of mice given water containing DSS for 5 days. Injections of the PKRi (500 μg/kg) or DMSO were given intraperitoneally from 1 day prior to DSS treatment until day 5 of the DSS treatment. Proteins were detected at the following masses: PKR-P(T451) at ∼60 kDa, PKR at 55 kDa, p65-P(S536) at 65 kDa, p65 at 65 kDa, IKKε-P(S172) at ∼80 kDa, IKKε at 80 kDa, and villin at ∼90 kDa. (E) RT-qPCR analysis for the indicated mRNAs of RNA isolated from colonic epithelial cells from mice treated as described for panel C. Values were normalized to villin mRNA. (F) Hematoxylin and eosin stains of colons from mice given water containing DSS for 5 days. Injections of PKRi or DMSO were as described for panel C. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
In order to determine whether PKR activation played a role in the induction of these proinflammatory genes, mice were injected with the PKR inhibitor for the duration of the DSS treatment. IECs isolated from these mice showed that DSS treatment alone was sufficient to induce phosphorylation of PKR at threonine-451, of NF-κB p65 at serine-536, and of IKKε at serine-172; however, the PKR inhibitor was sufficient to reduce phosphorylation at all of these sites (Fig. 6D). In addition, the PKR inhibitor was able to attenuate hyperinduction of mRNAs for the proinflammatory iNOS, IL-6, and IL-1β genes in IECs after DSS treatment (Fig. 6E). The induction of tumor necrosis factor alpha (TNF-α) mRNA was unaffected, indicating that its gene is not among the subset of genes that are affected by PKR signaling and that other factors may contribute to the induction of other mRNAs associated with the inflammatory response. Histology of colon tissue upon treatment with DSS and the PKR inhibitor corroborated this evidence as loss of crypt morphology and macrophage invasion into the lamina propria, both present in the DSS-treated tissue, were profoundly reduced or even absent in tissue cotreated with the PKR inhibitor (Fig. 6F).
These data suggest that PKR has a proapoptotic and disease-facilitating role in DSS-induced colitis, which correlates with the induction of iNOS. However, the signaling pathway leading from DSS treatment to iNOS induction via PKR may not be the same as detailed above in the hyperosmotic stress response. Although DSS has been suggested to increase the extracellular osmolarity (50, 53), the molecular mechanisms of the cellular response to DSS are not well studied. In order to investigate the hypothesis that DSS induces a hyperosmotic stress response sufficient to cause colitis, we used a culture model of MEF cells treated directly with DSS as a hyperosmotic agent. It was first determined that addition of DSS in tissue culture medium increases the osmolarity of the medium in the absence of cells, as determined by an osmometer (Fig. 7A). Treating cells with increasing concentrations of DSS in the medium correlated with increased caspase-3 cleavage and activity levels (Fig. 7B). The caspase-3 activity levels in control cells are once again greater than in shPKR cells. Western blot analysis showed an increase in NF-κB p65 phosphorylation at serine-536 reminiscent of the hyperosmotic stress response induced by sucrose (Fig. 1C). The master transcription factor of the osmotic response, TonEBP—itself a member of the Rel family and capable of heterodimerizing with NF-κB p65 to induce target genes (54, 55)—is activated under hyperosmotic stress equally in control and shPKR cells, as indicated by the shift in molecular weight (Fig. 7C). The nuclear levels of NF-κB p65 phosphorylated at serine-536 are greater in control cells than in PKR-suppressed cells (Fig. 7D). At high concentrations of DSS, there is a PKR-dependent increase in proinflammatory iNOS (Fig. 7E) and IL-6 mRNA levels but not in the TNF-α level (Fig. 7F). The induction of iNOS was NF-κB p65 dependent (Fig. 7G), just as observed previously (Fig. 2A). The lower concentration of DSS (5%) was insufficient to induce apoptosis but did demonstrate the induction of osmoadaptive genes, exemplified by SNAT2 and AR mRNA induction (Fig. 7H). These data suggest that DSS in the extracellular medium induces a cellular response to hyperosmolarity. This response is adaptive in low concentrations of DSS and proinflammatory in higher concentrations. The mechanism that switches off the adaptive response in higher concentrations of DSS is not known.
FIG 7.

DSS induces hyperosmotic stress in MEFs. (A) Osmolarity was measured in medium containing DSS (5% or 10%, wt/vol). (B) Caspase-3 activities were determined at 6 h of DSS medium treatment. Immunoblot analysis of control and shPKR MEFs was performed. (C) Immunoblot analysis of the indicated proteins from cell extracts from control and shPKR MEFs treated with DSS medium for the indicated durations. Proteins were detected at the same molecular weights indicated previously. (D) Immunoblot analysis for the indicated proteins of nuclear extracts isolated from control and shPKR MEFs treated with DSS medium for the indicated durations. (E to H) RT-qPCR analysis for the indicated mRNAs isolated from control and shPKR MEFs (E and F), control and p65-knockout (KO) MEFs (G), and MEFs (H) treated with 5% or 10% DSS medium for the indicated times or 3 h (E, F, and H). mRNA levels were normalized to the level of GAPDH. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
An important question remains as to how PKR is activated in response to increased extracellular osmolarity. No reports exist in the literature to suggest that dsRNA is present under this stress condition, so we hypothesized an alternative method of PKR activation. Previous studies have elegantly shown the dependence of PKR activation by poly(I·C) in MEF cells genetically deficient in PKR and stably reconstituted with either wild-type human PKR (hPKR) or mutant human PKR (Mut hPKR) (56). We obtained cells in which residues lysine-64 in the dsRNA-binding motif (dsRBM) I and lysine-154 in the dsRNA-binding domain II were mutated to glutamines, impairing the binding of double-stranded RNA. The mutations were confirmed by sequencing of cDNA generated from cells expressing the WT or the mutant proteins (Fig. 8A). Levels of WT hPKR reconstitution were consistent with endogenous PKR expression in wild-type MEFs and slightly higher in Mut hPKR-reconstituted MEFs (Fig. 8B). When these cells were subjected to hyperosmotic stress, phosphorylation of PKR was observed in both the wild-type (WT hPKR) and mutant (Mut hPKR) hPKR-reconstituted cells (Fig. 8C and D). Interestingly, the basal levels of PKR activation/autophosphorylation also increased in the absence of stress (Fig. 8C). This may suggest that increased levels of the protein correspond to increased activation as PKR phosphorylation is increased in the more highly expressed mutant PKR-reconstituted cells. In addition, levels of PKR-dependent (iNOS and IL-6) and -independent (IκBα and Mcl-1) proinflammatory and osmoadaptive (AR) mRNAs isolated from these cells are unchanged upon the mutation of PKR (Fig. 8E). These mutations significantly decreased eIF2α phosphorylation in cells transfected with poly(I·C) (Fig. 8F). This indicates that the activation of PKR is not dependent on the function of its dsRNA binding under this stress condition.
FIG 8.

PKR activation is independent of dsRNA binding during hyperosmotic stress. (A) Schematic representation of the WT and mutant PKR constructs (confirmed by sequencing of cDNA isolated from transfected cells), introduced stably into MEFs deficient in PKR. (B) Immunoblot analysis of FLAG-PKR expression in reconstituted MEFs. (C) Immunoblot analysis for the indicated proteins in cell extracts isolated from MEFs reconstituted with constructs shown in panel A and treated with hyperosmotic medium for the indicated durations. (D) Quantification of PKR-P(T451) bands normalized to total PKR bands from the experiment shown in panel C. (E) RT-qPCR analysis for the indicated mRNAs of RNA isolated from reconstituted MEFs and treated with hyperosmotic medium for the indicated durations. (F) Immunoblot analysis for the indicated proteins isolated from wild-type or mutant FLAG-PKR-reconstituted MEFs transfected with poly(I·C) for 6 h. (G) Immunoblot analysis for the indicated proteins isolated from either immunoprecipitations of FLAG-PKR from the indicated MEFs (IP) or from cell extracts before immunoprecipitations (input). All MEFs were treated with 600 mosM medium for 3 h. An antibody against PKR was used for the immunoblotting. (H) In vitro kinase activity assays on immunoprecipitated FLAG-PKR from cells treated as described for panel G. An equal volume of immunoprecipitated protein was used for the kinase assays. The kinase assay was performed in the presence or absence of the substrate GST-eIF2α, as indicated. FLAG-PKR autophosphorylation and the substrate GST-eIF2α1–180 phosphorylation are shown at 60 kDa and 41 kDa, respectively. (I) In vitro kinase activity assays on immunoprecipitated FLAG-PKR from the indicated MEFs treated for 6 h with poly(I·C) (top) and autophosphorylated FLAG-PKR (bottom). Kinase assays were in the absence of substrate. Error bars represent standard errors of the means. *, P < 0.05; ns, not significant (Student's t test).
To further support this argument, we performed an in vitro kinase assay on PKR immunoprecipitated from WT hPKR- and Mut hPKR-expressing cells after 3 h of treatment with 600 mosM medium (Fig. 8G and H). We used recombinant glutathione S-transferase (GST)–eIF2α residues 1 to 180 as a PKR substrate as previously described (57). We observed similar autophosphorylation of PKR and PKR-mediated phosphorylation of GST-eIF2α in the presence of the substrate (Fig. 8H). Based on current understanding of the mechanism of activation of PKR, it is possible that interaction of other proteins with the dsRBMs or the region of the kinase domain that binds the dsRBMs would induce a conformational change that prevents said binding, thereby allowing PKR to adopt the active conformation. The data suggest that activation of PKR during hyperosmotic stress is independent of its RNA-binding function. Further support of this conclusion was obtained by a kinase assay performed on immunoprecipitated PKR from poly(I·C)-treated WT hPKR and Mut hPKR cells. Autophosphorylation of PKR was significantly higher in poly(I·C)-treated WT hPKR cells (Fig. 8I), supporting the idea that RNA binding is the major mechanism of PKR activation by poly(I·C).
Taken together, all of these data suggest a signaling pathway during the hyperosmotic stress response and DSS treatment, connecting the kinase ability of PKR to the phosphorylation and nuclear localization of NF-κB subunit p65, which amplifies the induction of proinflammatory genes such as the iNOS gene, ultimately contributing to cell death. The mechanism via which PKR influences induction of a subgroup of NF-κB p65 target genes is not known.
DISCUSSION
In this report, we have found that PKR is activated independently of RNA binding in response to extracellular hyperosmolarity. This activation corresponds to and is necessary for nuclear localization of NF-κB subunit p65 phosphorylated at serine-536, amplification of the induction of iNOS mRNA, and subsequent cell death in MEFs. In addition, PKR amplifies induction of a subset of NF-κB p65 target proinflammatory genes in a DSS-induced colitis mouse model, as well as in a cell culture model of DSS-induced hyperosmotic stress. Together, these data indicate that PKR plays a proapoptotic role in the hyperosmotic and inflammatory cellular responses and may act as a link to connect these responses in disease conditions such as IBD.
Of the known mechanisms by which PKR can initiate an inflammatory response, several are not supported by our data. PKR has been shown to directly phosphorylate IκBα (58), which is then ubiquitinated and degraded by the proteasome, freeing NF-κB to induce expression of inflammatory genes. However, we show no difference in the phosphorylation or levels of IκBα in response to PKR suppression (Fig. 1C). It is therefore expected that the IKKα/β complex is activated and phosphorylates IκBα independently of PKR. PKR may also initiate an inflammatory response by phosphorylation of eIF2α, which inhibits global protein synthesis, including synthesis of de novo IκBα, which removes NF-κB from the nucleus and limits downstream gene transcription. However, protein synthesis rates are unchanged in wild-type and shPKR cell lines (Fig. 4F). The NLRP3 inflammasome has also been suggested to play a role in sensing extracellular hyperosmolarity in macrophages (59), and it is possible that PKR is involved in the activation of this complex (60). Although the inflammasome has been shown to be assembled and activated in macrophages and in vivo, NLRP3 and Pycard subunits are not expressed in MEFs (61); caspase-1 is also not cleaved, and IL-1β is not induced (59).
It remains a possibility that PKR acts in complex with other kinases to refine the phosphorylation profile of individual NF-κB subunits, affecting the ability of these subunits to localize to the nucleus. Various phosphorylation sites have been shown to enhance transactivation potential upon treatment by different biological stimuli; serine-536 of NF-κB can be phosphorylated by IKKα in human T-cell leukemia virus type 1 (HTLV-1) infection or lymphotoxin β stimulation (62, 63), by IKKβ in TNF-α or CD3/CD28 costimulation (64), and by Akt, TBK1, or IKKε upon IL-1β stimulation (65–68). Phosphorylation can affect the nuclear translocation of the NF-κB dimer, and modification at individual sites can affect the profile of target genes induced by each individual species (31). Phosphorylation of p65 at serine-468 was shown to be necessary for induction of Saa3 and Mmp3 upon TNF-α stimulation in macrophages, while induction of Cxcl2 was insensitive to phosphorylation status at this site or at serine-536 (31). The expression of IL-2, Csf2, and gamma interferon (IFN-γ) was shown to be mediated by O-GlcNAcylation of serine-350 of NF-κB c-Rel in response to T cell receptor (TCR) activation, while other genes were insensitive to this modification (69). We were unable to pull down PKR protein with NF-κB antibodies, or vice versa, which does not eliminate the possibility of modification via complex formation.
iNOS induction by NF-κB p65 has been previously shown although cell type, stress condition, and other factors may contribute to expression levels. Other situations in which iNOS could potentially be induced by PKR activation include lipopolysaccharide (LPS) treatment or TLR-mediated inflammatory signaling (70), and models of aggregate-mediated neuronal cell death such as in Alzheimer's disease (71). One mechanism by which iNOS is induced by NF-κB involves the juxtaposition of AP-1 and κB sites in the promoter of target genes and subsequent release of corepressor factors when c-Jun becomes phosphorylated by nuclearly localized IKKε (43). We were unable to find differences in nuclear localization or phosphorylation of IKKε in MEF cells by Western blotting but cannot disprove this hypothesis.
The mouse studies used here represent a more physiological model of the role of PKR in DSS-induced colitis than either of the previous investigations. Previous studies used a whole-mouse C-terminal mutant (kinase deficient) of PKR (72) or a whole-mouse knockout of PKR with bone marrow reconstitution of wild-type or N-terminal mutant (dimerization-impaired) PKR (73). In this study, full-length wild-type PKR is present, allowing any complex formation or interaction to occur as normal; and no artificial system is used to create mutant mice, so naturally occurring signaling pathways are used, as opposed to secondary or alternative pathways adapted to carry out the function of PKR. Rath et al. (72) found that PKR is necessary for induction of the mitochondrial unfolded protein response (mtUPR) in DSS-induced colitis, that PKR-deficient mice were protected from deleterious effects of DSS, and that human patients diagnosed with IBD had upregulated PKR and the mtUPR marker CPN60. Cao et al. (73) found that reconstitution of hematopoietic cells of PKR-deficient mice with wild-type PKR upon DSS treatment led to activation of the ER chaperone response and increased ER-associated degradation and protection from more severe histological disease phenotypes. Both of these studies found induction of the unfolded protein response, which was not observed in our mice (Fig. 2F), perhaps indicating that the involvement of the UPR is an artifact of the PKR-deficient mice in general. Although our results do not support the UPR as being involved in the response to DSS, they do support the proapoptotic role of PKR, agreeing with Rath et al. (72). The molecular inhibitor of PKR, C16, has been used previously in mice to protect neurons against amyloid-β aggregation-mediated cell death (74), but it has not been published in any model of inflammation in the bowel. The inhibitor has not been observed to have any adverse off-target effects, but no study has been published in which treated mice were monitored in any great depth.
The finding that distal colon also displays iNOS mRNA induction in DSS-induced colitis is novel and may indicate that iNOS amplification is a signaling precursor to DSS- or inflammation-induced tissue damage and not the cause of damage itself. However, while insufficient to cause colitis, iNOS may be required for disease development. As distal colon tissue is not inflamed in DSS-induced colitis, the presence of iNOS mRNA may represent upstream signaling, a protective mechanism in the absence of further proinflammatory cytokines, or merely a localized inflammation profile that is insufficient to induce tissue damage. Although iNOS is expressed in IECs, this does not mean that iNOS or NO produced by macrophages or other innate immune cells does not contribute to the inflammatory phenotype; we hypothesize that iNOS in IECs is one, but not the sole, factor contributing to inflammation in localized microenvironments that also express interleukins and chemokines necessary for initiation of inflammation. Future studies will be required to elucidate the role of regional and cell type differences in gene induction in contributing to the inflammatory response. It remains to be seen whether PKR activation is seen at a higher level in rectal colon than in distal colon and whether PKR can target differential genes based on a colonic microenvironment. Although the iNOS, IL-6, and IL-1β genes were affected by PKR inhibition (Fig. 6D), it is unclear how many or which other proinflammatory genes follow this expression pattern. Exploration of the specific PKR-mediated amplification transcription program will require further genome-wide studies.
It was discovered long ago that colonic fluids in patients diagnosed with IBD have higher osmolarity and ionic content (12, 13). It has been previously hypothesized that DSS contributes to induction of colitis via a hyperosmotic stress response (50, 53). Here, we provide direct evidence that DSS increases the osmolarity of extracellular fluid and that osmoadaptive genes are upregulated in response to DSS-induced extracellular osmolarities of 395 mosM. However, a switch from osmoadaptation to proinflammatory gene expression occurs before DSS-induced extracellular osmolarity reaches 470 mosM. PKR is shown to be part of the switch between induction of expression of osmoadaptive genes and proinflammatory genes, thus contributing to cell fate. It remains to be explored if and by what mechanism(s) PKR may inhibit osmoadaptation at greater stress intensities and by what mechanisms it exacerbates the colitis phenotype in vivo. As low levels of DSS treatment in MEFs induced adaptive genes (SNAT2 and AR) and yet these genes were not induced by DSS treatment in mice, it may be that DSS accumulation in the mouse colon leads to a hyperosmotic environment beyond the point at which adaptation is a viable option. That TonEBP was also not expressed at the same level in the mouse colonic tissue as in the cultured fibroblasts (Fig. 7B) may also play a role in the switch from osmoadaptation to inflammation. It is likely that other factors may be involved, such as activation of transcription factor 3 (ATF3), which is known to participate in osmoadaptation and repression of iNOS in renal medullary cells (75, 76) and macrophages (77). Although GAPDH was determined to be modified and demonstrated altered functionality in our MEF culture model, we hypothesize that it is likely that nitrosylation of lower-abundance proteins will have a more profound impact on cell fate in vivo.
The exact mechanism by which PKR becomes activated under hyperosmotic stress is unknown. However, our data show that the binding of dsRNA to PKR is not required for activation as mutation-generated impairment of this binding does not affect phosphorylation or downstream signaling events (Fig. 8). Other methods of PKR activation have been identified, such as the binding of PACT to PKR to disrupt the closed conformation and promote the open active conformation (36). It is also possible that PKR becomes activated due to increased proximity as a result of the molecular crowding occurring during cell shrinkage in response to a hyperosmotic environment. Our data showed that PKR immunopurified from stressed cells was inactive and was acutely activated with the addition of the substrate (Fig. 8H). This finding suggests that PKR may be phosphorylated during hyperosmotic stress upon interaction with proteins that cause its activation. Additional experiments are necessary to demonstrate the validity of this hypothesis.
Our data define a PKR/NF-κB/iNOS signaling axis that amplifies induction of a subset of proinflammatory genes that mediate cell fate in response to hyperosmotic stress and DSS treatment. This suggests that the response to hyperosmotic stress may be a mechanism that contributes to the development of inflammatory diseases such as IBD. In addition, the use of the PKR inhibitor to alleviate the expression of proinflammatory iNOS and epithelial barrier disruption may represent a novel therapeutic use for this molecule in IBD or other inflammatory diseases. The identification of this PKR/NF-κB/iNOS signaling pathway may lead to new insights or novel therapeutic strategies for the treatment of patients suffering from IBD or other inflammatory diseases.
MATERIALS AND METHODS
Cells and reagents.
Mouse embryonic fibroblasts (MEFs) were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were cultured at 37°C and 5% CO2. Hyperosmotic medium was made by addition of 300 mM sucrose to culture medium for a final osmolarity of 600 mosM. DSS culture medium was made by adding DSS powder to DMEM (5% or 10%, wt/vol). DSS was purchased from MP Biomedicals, LLC (catalog no. 160110). Small molecules used in culture include the following: C16/PKRi (1.25 μM) (catalog no. 527450; Calbiochem), IKKε-kinase inhibitor II (100 nM) (1811-1; BioVision), and cPTIO (100 μM) (81540; Cayman Chemicals). Antibodies were used against the following proteins: tubulin (catalog no. T9026; Sigma), cleaved caspase-3 (9664; Cell Signaling), p65/RelA (301-824A; Bethyl), phospho-p65 Ser-536 (3033; Cell Signaling), phospho-p65 Ser-468 (3039; Cell Signaling), PKR (sc-6282; Santa Cruz), phospho-PKR Ser-451 (sc-101784; Santa Cruz), IκBα (14D4; Cell Signaling), phospho-IκBα Ser-32 (44D4; Cell Signaling), IKKε (3146; Cell Signaling), phospho-IKKε Ser-172 (D1B7; Cell Signaling), TonEBP (a generous gift of H. M. Kwon, University of Maryland—Baltimore), histone H3 (9715; Cell Signaling), FLAG (200472; Stratagene), and GAPDH (ab9485; Abcam).
Western blotting.
Whole-cell extracts were prepared by lysing the cells in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-Cl, pH 8, 150 mM NaCl, 1% NP-40, 0.1% SDS, 2 mM EDTA, protease and phosphatase inhibitor tablets [Roche]) on ice. Whole-cell extracts were collected after centrifugation at 12,000 rpm for 15 min at 4°C. Proteins were quantified by Bradford assay, and immunoblot analysis was performed as described previously (2). Protein bands were quantified using ImageJ software.
Nuclear fractionation.
Nuclear extracts were prepared by a two-step lysis process. First, cells were lysed in a hypotonic lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol [DTT], 0.3% NP-40, and phosphatase and protease inhibitor tablets) on ice for 10 min. Supernatants containing the cytoplasmic fractions were collected by centrifugation for 4 min at 2,500 × g at 4°C. Nuclear pellets were washed in lysis buffer lacking NP-40 detergent and then resuspended in extraction buffer (20 mM HEPES, pH 7.9, 0.45 M NaCl, 1 mM EDTA, 0.5 mM EDTA, and protease and phosphatase inhibitor tablets). Nuclei were lysed by sonication and slowly rotated at 4°C for 30 min, and supernatant was collected after centrifugation at 12,000 rpm for 15 min at 4°C.
shRNA.
shRNA directed against PKR was obtained from Sigma-Aldrich (TRCN0000026987). HEK cells were transfected with 10 μg of shRNA, 5 μg of Rev-responsive element (RRE), 5 μg of vesicular stomatitis virus (VSV), and 5 μg of REV. Control plasmid consisted of the shRNA-plasmid backbone with a puromycin resistance cassette lacking any shRNA target sequence. Medium was collected at 12 h, 36 h, and 60 h posttransfection. Collected medium was filtered through a 0.45-μm-pore-size filter syringe to obtain viral transduction particles. MEF cells were then infected with medium containing viral particles for 3 days. Cells were selected for infection by puromycin treatment (30 μg/ml) and verified by Western blotting prior to use.
Caspase-3 activity assay.
MEFs were treated with hyperosmotic medium as described above. Cells were lysed in lysis buffer [1% CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate), 150 mM NaCl, 10 mM HEPES, 10 mM EDTA, pH 7.4]. Protein (100 μg) was mixed with ICE buffer (50 mM Tris-Cl, pH 7.2, 100 mM KCl, 10% sucrose, 0.1% CHAPS, 5 mM DTT) to 100 μl. Ac-DEVD-AFC (N-acetyl–Asp–Glu–Val–Asp–amido-4-trifluoromethylcoumarin) caspase substrate (1 μM) (50-230-4108; Calbiochem) was added, and lysates were incubated at 37°C for 60 min. Caspase-3 activity was measured using a SpectraMax M3 Molecular Device fluorimeter at 360-nm excitation and 460-nm emission wavelengths.
RT-qPCR.
Total RNA was prepared using TRIzol reagent (Ambion) according to the manufacturer's instructions. RNA was subsequently treated with DNase (AM2238, Turbo DNase; Ambion) for 1 h at 37°C, precipitated for 2 h at 4°C in 0.8 M LiCl twice, precipitated in 100% ethanol at −20°C for 30 min, and washed in 75% ethanol according to Viennois et al. (78). cDNA was synthesized using Superscript III First-Strand Synthesis SuperMix (Invitrogen), and mRNA levels were determined by RT-qPCR using FastStart Universal SYBR green Master (Roche) according to the manufacturer's protocol. Data were normalized to GAPDH, β-actin, or villin mRNA signal and analyzed using Student's t test.
Luciferase assays.
Transient transfections were performed using the iNOS-firefly luciferase reporter and the cytomegalovirus (CMV)-Renilla luciferase reporter at a 9:1 ratio. Cells were used for subsequent experiments 24 h after transfection. Luciferase activity was determined using a dual-luciferase reporter system (Promega) as described by the manufacturer.
Nitric oxide detection.
For detection of NO, cells were incubated with 10 μM difluorofluorescein (DAF-FM) diacetate (Molecular Probes) for 1 h at 37°C. Cells were washed to remove excess probes, and fluorescence was detected at 495-nm excitation and 515-nm emission using a SpectraMax M3 Molecular Device fluorimeter.
Arginine transport assay.
Arginine uptake by system y+ was measured as described previously (79). Briefly, cells were seeded in 24-well plates at 5 × 104 cells/well, grown for 24 h, and subjected to treatment as described above. Cells were washed twice, and assays were performed in Earle's balanced salt solution (EBSS) with 100 μM Arg supplemented with [3H]Arg (8 μCi/ml; PerkinElmer) for 30 s in the presence of 2 mM Leu to prevent Arg uptake through system y+L. Amino acids were extracted in ethanol, and radioactivity was measured in a liquid scintillation counter. Protein concentration was determined by the Lowry method using bovine serum albumin (BSA) as a standard.
Cell survival assay.
MEFs were seeded in 96-well clear, flat-bottom plates. Cells were treated with hyperosmotic medium as described above in the presence or absence of PKR inhibitor (1.25 μM; 1 h prior to addition of hyperosmotic medium). Cell viability was determined by following the Cell Titer-Blue cell viability assay protocol (Promega). Cell viability was measured using a GENios Pro fluorimeter at 535-nm excitation and 590-nm emission wavelengths.
Detection of protein nitrosylation.
Untreated and treated MEF cells were collected and subjected for the biotin switch technique (BST) assay as described previously (47). Briefly, all free cysteine thiols from cell extracts were alkylated by methyl methanethiosulfonate (MMTS) (20 mM; Thermo Scientific) at 45°C for 1 h following protein precipitation by cold acetone. The pellets were suspended and incubated with sodium ascorbate (30 mM) in the presence of hexyl pyridyldithiopropionamide (HPDP)-biotin (0.5 mM) for 1 h at 25°C. Excess HPDP-biotin was removed by acetone precipitation. The biotinylated samples were mixed with streptavidin beads and rotated gently overnight at 4°C. The beads were collected and washed, and proteins were eluted by addition of DTT (20 mM), followed by SDS-PAGE Western blot analysis.
GAPDH activity assay.
MEF cells were lysed using RIPA buffer without SDS. Five micrograms of the lysate was added to the reaction mixture (100 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 3 mM 3-phosphoglycerate, 5 units/ml of Saccharomyces cerevisiae 3-phosphoglycerate kinase, 2 mM ATP, 0.25 mM NAD) in a 96-well plate. Activity was monitored via spectrophotometric absorbance at 340 nm for 3 min.
Immunofluorescence.
MEFs transfected with GAPDH-HA plasmid were grown on glass coverslips. Cells were fixed and stained as described previously (2) and visualized using a Leica SP2 confocal microscope with an argon laser (488 nm) and two-photon excitation (760 nm). Total HA fluorescence signal area and nuclear 4′,6′-diamidino-2-phenylindole (DAPI) signal area were calculated, and HA signal overlapping DAPI was quantified using ImageJ (n = 10 per condition).
Mouse studies.
Mouse experiments were approved by Case Western Reserve University's Institutional Animal Care and Use Committee. Eight-week-old C57BL/6 mice (Jackson) were obtained and fed and watered ad libitum. At the start of experiment, water was replaced with water containing 2.5% dextran sodium sulfate (DSS; MP Biomedicals) solution. After 5 days, mice were sacrificed, and colons were removed and fixed in UW cold storage solution (Belzer).
Colon sectioning.
Distal and rectal colonic sections were separated at the juncture of the transverse and descending colon for RNA isolation in TRIzol. Intestinal epithelial cells were isolated from whole colonic tissue sections by incubation and rotation in Hanks balanced salt solution (HBSS) supplemented with 1 mM EDTA, as described by Grossmann et al. (80). Colon tissue fragments were washed in prewarmed (37°C) HBSS three times for 10 min. Cells were washed in phosphate-buffered saline (PBS) and pelleted for RNA isolation.
PKRi injection.
Mice were intraperitoneally injected with PKR inhibitor (PKRi) C16 (500 μg/kg) or equivalent dimethyl sulfoxide (DMSO) each day, beginning 1 day prior to DSS treatment, until day 5 of DSS treatment. Intestinal epithelial cells were isolated as described above.
Histology.
Whole sections of colon were used for histology. Following dissection of colon, a 1-cm portion of the colon most proximal to the rectum was cut and fixed in HistoChoice (Sigma). A Leica Peloris automated processor was used to dehydrate and infiltrate tissues with paraffin, followed by embedding on a Sakura embedding station. Sectioning was performed on a Leica microtome. Sections were stained with hematoxylin and eosin, and color images were collected at a magnification of ×20 (0.5 μm/pixel) using an SCN400FL slide scanner (Leica Microsystems, GmbH, Wetzlar, Germany).
Measuring osmolarity.
Osmolarity of DSS medium was measured using a vapor pressure osmometer (Wescor 5500).
Sequencing.
Total RNA was prepared from PKR knockout MEFs reconstituted with wild-type or mutant human PKR, and cDNA was synthesized as described above. Transcripts corresponding to PKR were amplified by PCR using GoTaq Green master mix (Promega) as described by the manufacturer. Sequencing was performed on an ABI Prism 3730 DNA analyzer.
In vitro kinase assay.
MEFs were lysed in RIPA buffer as described above. Forty microliters of protein G Dynabeads (Fisher) was washed with 0.1 M Na-phosphate buffer (pH 8.1) three times, followed by incubation with Flag antibody (4 μg) for 45 min at room temperature (RT). Beads were then washed once in 0.1 M Na-phosphate buffer (pH 8.1), followed by three washes in RIPA buffer. Five hundred micrograms of cell lysates was incubated with beads for 2 h at 4°C. Beads were then washed three times in RIPA buffer, followed by three washes in kinase buffer (20 mM Tris-HCl [pH 8.0], 50 mM KCl, 25 mM MgCl2, 1 protease tablet [Roche] per 10 ml). Five microliters of beads was incubated with 5 μl of purified eIF2α substrate and 0.1 μl of [γ-32P]ATP in a 15-μl reaction volume for 20 min at 30°C. Beads were then washed three times in kinase buffer, boiled in Laemmli sample buffer, and run on precast SDS-PAGE gels (Invitrogen). Blots were analyzed via autoradiograph at −80°C overnight.
ACKNOWLEDGMENTS
We thank Paul Fox for the donation of GAPDH-HA plasmids and Josephine Kam, Tai Dermawan, and George Stark for critical discussion of the manuscript. We also graciously thank Gokhan Hotamisligil for the generous gift of PKR-deficient and -reconstituted cell lines.
Author contributions are as follows. K.T.F., M.M., D.K., X.-H.G., A.E.K., and M.H. conceived and designed the experiments. K.T.F., M.M., B.-J.G., R.J., J.W., D.K., X.-H.G., A.S., P.R., and M.B. performed the experiments. K.T.F., M.M., B.-J.G., R.J., D.K., X.-H.G., M.L., P.R., and M.H. analyzed the data. M.L., E.D.C., M.D., P.R., and M.H. provided reagents/materials/analysis tools. K.T.F. and M.H. wrote the paper, with edits by B.-J.G., D.K., and P.R.
We declare that we have no competing financial interests.
REFERENCES
- 1.Alfieri R, Petronini PG. 2007. Hyperosmotic stress response: comparison with other cellular stresses. Pflugers Arch 454:173–185. doi: 10.1007/s00424-006-0195-x. [DOI] [PubMed] [Google Scholar]
- 2.Bevilacqua E, Wang X, Majumder M, Gaccioli F, Yuan CL, Wang C, Zhu X, Jordan LE, Scheuner D, Kaufman RJ, Koromilas AE, Snider MD, Holcik M, Hatzoglou M. 2010. eIF2α phosphorylation tips the balance to apoptosis during osmotic stress. J Biol Chem 285:17098–17111. doi: 10.1074/jbc.M110.109439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritsch RM, Schneider G, Saur D, Scheibel M, Schmid RM. 2007. Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem 282:22551–22562. doi: 10.1074/jbc.M702673200. [DOI] [PubMed] [Google Scholar]
- 5.Scheuner D, Patel R, Wang F, Lee K, Kumar K, Wu J, Nilsson A, Karin M, Kaufman RJ. 2006. Double-stranded RNA-dependent protein kinase phosphorylation of the alpha-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J Biol Chem 281:21458–21468. doi: 10.1074/jbc.M603784200. [DOI] [PubMed] [Google Scholar]
- 6.Saikia M, Jobava R, Parisien M, Putnam A, Krokowski D, Gao XH, Guan BJ, Yuan Y, Jankowsky E, Feng Z, Hu GF, Pusztai-Carey M, Gorla M, Sepuri NB, Pan T, Hatzoglou M. 2014. Angiogenin-cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress. Mol Cell Biol 34:2450–2463. doi: 10.1128/MCB.00136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura K, Hayashi H, Kubokawa M. 2015. Proinflammatory cytokines and potassium channels in the kidney. Mediators Inflamm 2015:362768. doi: 10.1155/2015/362768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aramburu J, López-Rodríguez C. 2009. Brx shines a light on the route from hyperosmolarity to NFAT5. Sci Signal 2:pe20. doi: 10.1126/scisignal.265pe20. [DOI] [PubMed] [Google Scholar]
- 9.Neuhofer W. 2010. Role of NFAT5 in inflammatory disorders associated with osmotic stress. Curr Genomics 11:584–590. doi: 10.2174/138920210793360961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baudouin C, Aragona P, Messmer EM, Tomlinson A, Calonge M, Boboridis KG, Akova YA, Geerling G, Labetoulle M, Rolando M. 2013. Role of hyperosmolarity in the pathogenesis and management of dry eye disease: proceedings of the OCEAN group meeting. Ocul Surf 11:246–258. doi: 10.1016/j.jtos.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Tibrewal S, Ivanir Y, Sarkar J, Nayeb-Hashemi N, Bouchard CS, Kim E, Jain S. 2014. Hyperosmolar stress induces neutrophil extracellular trap formation: implications for dry eye disease. Invest Ophthalmol Vis Sci 55:7961–7969. doi: 10.1167/iovs.14-15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schilli R, Breuer RI, Klein F, Dunn K, Gnaedinger A, Bernstein J, Paige M, Kaufman M. 1982. Comparison of the composition of faecal fluid in Crohn's disease and ulcerative colitis. Gut 23:326–332. doi: 10.1136/gut.23.4.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vernia P, Gnaedinger A, Hauck W, Breuer RI. 1988. Organic anions and the diarrhea of inflammatory bowel disease. Dig Dis Sci 33:1353–1358. doi: 10.1007/BF01536987. [DOI] [PubMed] [Google Scholar]
- 14.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. 2011. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 15.Bae J, Leo CP, Hsu SY, Hsueh AJ. 2000. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem 275:25255–25261. doi: 10.1074/jbc.M909826199. [DOI] [PubMed] [Google Scholar]
- 16.Sharma JN, Al-Omran A, Parvathy SS. 2007. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 15:252–259. doi: 10.1007/s10787-007-0013-x. [DOI] [PubMed] [Google Scholar]
- 17.Alican I, Kubes P. 1996. A critical role for nitric oxide in intestinal barrier function and dysfunction. Am J Physiol 270:G225–G237. [DOI] [PubMed] [Google Scholar]
- 18.Kolios G, Valatas V, Ward SG. 2004. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology 113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzik TJ, Korbut R, Adamek-Guzik T. 2003. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol 54:469–487. [PubMed] [Google Scholar]
- 20.Shiratora Y, Aoki S, Takada H, Kiriyama H, Ohto K, Hai K, Teraoka H, Matano S, Matsumoto K, Kamii K. 1989. Oxygen-derived free radical generating capacity of polymorphonuclear cells in patients with ulcerative colitis. Digestion 44:163–171. [DOI] [PubMed] [Google Scholar]
- 21.Avdagić N, Zaćiragić A, Babić N, Hukić M, Seremet M, Lepara O, Nakaš-Ićindić E. 2013. Nitric oxide as a potential biomarker in inflammatory bowel disease. Bosn J Basic Med Sci 13:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guihot G, Guimbaud R, Bertrand V, Narcy-Lambare B, Couturier D, Duée PH, Chaussade S, Blachier F. 2000. Inducible nitric oxide synthase activity in colon biopsies from inflammatory areas: correlation with inflammation intensity in patients with ulcerative colitis but not with Crohn's disease. Amino Acids 18:229–237. doi: 10.1007/s007260050020. [DOI] [PubMed] [Google Scholar]
- 23.Dhillon SS, Mastropaolo LA, Murchie R, Griffiths C, Thöni C, Elkadri A, Xu W, Mack A, Walters T, Guo C, Mack D, Huynh H, Baksh S, Silverberg MS, Brumell JH, Snapper SB, Muise AM. 2014. Higher activity of the inducible nitric oxide synthase contributes to very early onset inflammatory bowel disease. Clin Transl Gastroenterol 5:e46. doi: 10.1038/ctg.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleinert H, Schwarz PM, Förstermann U. 2003. Regulation of the expression of inducible nitric oxide synthase. Biol Chem 384:1343–1364. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Wang S, Qin J, Lv Y, Ma X, Liu C. 2010. Ethanol upregulates iNOS expression in colon through activation of nuclear factor-kappa B in rats. Alcohol Clin Exp Res 34:57–63. doi: 10.1111/j.1530-0277.2009.01066.x. [DOI] [PubMed] [Google Scholar]
- 26.Kawai T, Akira S. 2007. Signaling to NF-κB by Toll-like receptors. Trends Mol Med 13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Pingle SC, Sanchez JF, Hallam DM, Williamson AL, Maggirwar SB, Ramkumar V. 2003. Hypertonicity inhibits lipopolysaccharide-induced nitric oxide synthase expression in smooth muscle cells by inhibiting nuclear factor κB. Mol Pharmacol 63:1238–1247. doi: 10.1124/mol.63.6.1238. [DOI] [PubMed] [Google Scholar]
- 28.Gilmore TD. 2006. Introduction to NF-κB: players, pathways, perspectives. Oncogene 25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 29.Sen R, Smale ST. 2010. Selectivity of the NF-κB response. Cold Spring Harb Perspect Biol 2:a000257. doi: 10.1101/cshperspect.a000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hochrainer K, Racchumi G, Anrather J. 2013. Site-specific phosphorylation of the p65 protein subunit mediates selective gene expression by differential NF-κB and RNA polymerase II promoter recruitment. J Biol Chem 288:285–293. doi: 10.1074/jbc.M112.385625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno R, Sobotzik JM, Schultz C, Schmitz ML. 2010. Specification of the NF-κB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucleic Acids Res 38:6029–6044. doi: 10.1093/nar/gkq439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence T. 2009. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol 1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoesel B, Schmid JA. 2013. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer 12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aramburu J, Drews-Elger K, Estrada-Gelonch A, Minguillón J, Morancho B, Santiago V, López-Rodríguez C. 2006. Regulation of the hypertonic stress response and other cellular functions by the Rel-like transcription factor NFAT5. Biochem Pharmacol 72:1597–1604. doi: 10.1016/j.bcp.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Németh ZH, Deitch EA, Szabó C, Haskó G. 2002. Hyperosmotic stress induces nuclear factor-kappaB activation and interleukin-8 production in human intestinal epithelial cells. Am J Pathol 161:987–996. doi: 10.1016/S0002-9440(10)64259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S, Peters GA, Ding K, Zhang X, Qin J, Sen GC. 2006. Molecular basis for PKR activation by PACT or dsRNA. Proc Natl Acad Sci U S A 103:10005–10010. doi: 10.1073/pnas.0602317103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemaire PA, Anderson E, Lary J, Cole JL. 2008. Mechanism of PKR Activation by dsRNA. J Mol Biol 381:351–360. doi: 10.1016/j.jmb.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kapil P, Stohlman SA, Hinton DR, Bergmann CC. 2014. PKR mediated regulation of inflammation and IL-10 during viral encephalomyelitis. J Neuroimmunol 270:1–12. doi: 10.1016/j.jneuroim.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viatour P, Merville MP, Bours V, Chariot A. 2005. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci 30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Zandi E, Chen Y, Karin M. 1998. Direct phosphorylation of IκB by IKKα and IKKβ: discrimination between free and NF-κB-bound substrate. Science 281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 42.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, Glass CK. 2009. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell 35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Kang F, Li J, Zhang J, Shan B. 2013. Overexpression of p65 attenuates celecoxib-induced cell death in MDA-MB-231 human breast cancer cell line. Cancer Cell Int 13:14. doi: 10.1186/1475-2867-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collett GP, Campbell FC. 2006. Overexpression of p65/RelA potentiates curcumin-induced apoptosis in HCT116 human colon cancer cells. Carcinogenesis 27:1285–1291. doi: 10.1093/carcin/bgi368. [DOI] [PubMed] [Google Scholar]
- 45.Masters BS, McMillan K, Sheta EA, Nishimura JS, Roman LJ, Martasek P. 1996. Neuronal nitric oxide synthase, a modular enzyme formed by convergent evolution: structure studies of a cysteine thiolate-liganded heme protein that hydroxylates l-arginine to produce NO as a cellular signal. FASEB J 10:552–558. [DOI] [PubMed] [Google Scholar]
- 46.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. 2005. S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 47.Morisse S, Zaffagnini M, Gao XH, Lemaire SD, Marchand CH. 2014. Insight into protein S-nitrosylation in Chlamydomonas reinhardtii. Antioxid Redox Signal 21:1271–1284. doi: 10.1089/ars.2013.5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiesler P, Fuss IJ, Strober W. 2015. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol 1:154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. 1994. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz L, Abolhassani M, Pooya M, Steyaert JM, Wertz X, Israël M, Guais A, Chaumet-Riffaud P. 2008. Hyperosmotic stress contributes to mouse colonic inflammation through the methylation of protein phosphatase 2A. Am J Physiol Gastrointest Liver Physiol 295:G934–G941. doi: 10.1152/ajpgi.90296.2008. [DOI] [PubMed] [Google Scholar]
- 51.Colgan SP, Curtis VF, Campbell EL. 2013. The inflammatory tissue microenvironment in IBD. Inflamm Bowel Dis 19:2238–2244. doi: 10.1097/MIB.0b013e31828dcaaf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zenewicz LA, Flavell RA. 2011. Recent advances in IL-22 biology. Int Immunol 23:159–163. doi: 10.1093/intimm/dxr001. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz L, Guais A, Pooya M, Abolhassani M. 2009. Is inflammation a consequence of extracellular hyperosmolarity? J Inflamm (Lond.) 6:21. doi: 10.1186/1476-9255-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roth I, Leroy V, Kwon HM, Martin PY, Féraille E, Hasler U. 2010. Osmoprotective transcription factor NFAT5/TonEBP modulates nuclear factor-κB activity. Mol Biol Cell 21:3459–3474. doi: 10.1091/mbc.E10-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee HH, Sanada S, An SM, Ye BJ, Lee JH, Seo YK, Lee C, Lee-Kwon W, Küper C, Neuhofer W, Choi SY, Kwon HM. 2016. LPS-induced NFκB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci Rep 6:24921. doi: 10.1038/srep24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Youssef OA, Safran SA, Nakamura T, Nix DA, Hotamisligil GS, Bass BL. 2015. Potential role for snoRNAs in PKR activation during metabolic stress. Proc Nat Acad Sci 112:5023–5028. doi: 10.1073/pnas.1424044112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dey M, Velyvis A, Li JJ, Chiu E, Chiovitti D, Kay LE, Sicheri F, Dever TE. 2011. Requirement for kinase-induced conformational change in eukaryotic initiation factor 2α (eIF2α) restricts phosphorylation of Ser51. Proc Natl Acad Sci U S A 108:4316–4321. doi: 10.1073/pnas.1014872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. 1994. Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proc Natl Acad Sci U S A 91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ip WK, Medzhitov R. 2015. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun 6:6931. doi: 10.1038/ncomms7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. 2012. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. 2009. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O'Mahony AM, Montano M, Van Beneden K, Chen LF, Greene WC. 2004. Human T-cell lymphotropic virus type 1 tax induction of biologically active NF-κB requires IκB kinase-1-mediated phosphorylation of RelA/p65. J Biol Chem 279:18137–18145. doi: 10.1074/jbc.M401397200. [DOI] [PubMed] [Google Scholar]
- 63.Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. 2003. The NF-κB activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem 278:919–926. doi: 10.1074/jbc.M208696200. [DOI] [PubMed] [Google Scholar]
- 64.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. 1999. IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 65.Madrid LV, Mayo MW, Reuther JY, Baldwin AS Jr. 2001. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-κB through utilization of the IκB kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem 276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 66.Sizemore N, Leung S, Stark GR. 1999. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-κB p65/RelA subunit. Mol Cell Biol 19:4798–4805. doi: 10.1128/MCB.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M. 2003. Identification of NAP1, a regulatory subunit of IκB kinase-related kinases that potentiates NF-κB signaling. Mol Cell Biol 23:7780–7793. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. 2004. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem 279:55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 69.Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. 2013. Activation of the transcriptional function of the NF-κB protein c-Rel by O-GlcNAc glycosylation. Sci Signal 6:ra75. doi: 10.1126/scisignal.2004097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He Y, Franchi L, Núñez G. 2013. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur J Immunol 43:1147–1152. doi: 10.1002/eji.201243187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peel AL, Bredesen DE. 2003. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiol Dis 14:52–62. doi: 10.1016/S0969-9961(03)00086-X. [DOI] [PubMed] [Google Scholar]
- 72.Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D. 2012. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61:1269–1278. doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao SS, Song B, Kaufman RJ. 2012. PKR protects colonic epithelium against colitis through the unfolded protein response and prosurvival signaling. Inflamm Bowel Dis 18:1735–1742. doi: 10.1002/ibd.22878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tronel C, Page G, Bodard S, Chalon S, Antier D. 2014. The specific PKR inhibitor C16 prevents apoptosis and IL-1β production in an acute excitotoxic rat model with a neuroinflammatory component. Neurochem Int 64:73–83. doi: 10.1016/j.neuint.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 75.Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. 2004. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cai Q, Brooks HL. 2011. Phosphorylation of eIF2α via the general control kinase, GCN2, modulates the ability of renal medullary cells to survive high urea stress. Am J Physiol Renal Physiol 301:F1202–F1207. doi: 10.1152/ajprenal.00272.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jungda H, Kim KH, Byeon HE, Park HJ, Park B, Rhee DK, Um SH, Pyo S. 2015. Involvement of ATF3 in the negative regulation of iNOS expression and NO production in activated macrophages. Immunol Res 62:35–45. doi: 10.1007/s12026-015-8633-5. [DOI] [PubMed] [Google Scholar]
- 78.Viennois E, Chen F, Laroui H, Baker MT, Merlin D. Dextran sodium sulfate inhibits the activities of both polymerase and reverse transcriptase: lithium chloride purification, a rapid and efficient technique to purify RNA. BMC Res Notes 6:360. doi: 10.1186/1756-0500-6-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krokowski D, Jobava R, Guan BJ, Farabaugh K, Wu J, Majumder M, Bianchi MG, Snider MD, Bussolati O, Hatzoglou M. 2015. Coordinated regulation of the neutral amino acid transporter SNAT2 and the protein phosphatase subunit GADD34 promotes adaptation to increased extracellular osmolarity. J Biol Chem 290:17822–17837. doi: 10.1074/jbc.M114.636217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grossmann J, Maxson JM, Whitacre CM, Orosz DE, Berger NA, Fiocchi C, Levine AD. 1998. New isolation technique to study apoptosis in human intestinal epithelial cells. Am J Pathol 153:53–62. doi: 10.1016/S0002-9440(10)65545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]