

Abstract
Multiple factors help shape the infant intestinal microbiota early in life. Environmental conditions such as the presence of bioactive molecules from breast milk dictate gut microbial growth and survival. Infants also receive distinct, personalized, bacterial exposures leading to differential colonization. Microbial exposures and gut environmental conditions differ between infants in different locations, as does the typical microbial community structure in an infant’s gut. Here we evaluate potential influences on the infant gut microbiota through a longitudinal study on cohorts of breast-fed infants from the neighboring countries of Armenia and Georgia, an area of the world for which the infant microbiome has not been previously investigated. Marker gene sequencing of 16S ribosomal genes revealed that the gut microbial communities of infants from these countries were dominated by bifidobacteria, were different from each other, and were marginally influenced by their mother’s secretor status. Species-level differences in the bifidobacterial communities of each country and birth method were also observed. These community differences suggest that environmental variation between individuals in different locations may influence the gut microbiota of infants.
The time period directly following birth is a critical developmental window where there is a need to both protect the vulnerable infant from disease, and to educate the neonatal immune system1,2,3. Dysbiosis, an imbalanced pattern of microbiota composition or colonization during early development, is associated with a number of disease states including obesity, metabolic syndrome, chronic inflammatory bowel diseases, nonalcoholic steatohepatitis, atherosclerosis, type 1 diabetes, allergy, asthma, celiac disease, kwashiorkor, autism, atopy, and other autoimmune diseases (reviewed in refs 4 and 5). The structure of an infant’s intestinal microbiome is influenced both by the selective environmental conditions in the gut such as carbohydrate availability, pH, activity of the immune system and the arrival of bacteria into the ecosystem due to the sources of microbes to which an infant is exposed. A mechanism by which a mother could beneficially influence the microbiota of her infant via manipulation of the environmental conditions in the gut or the control of microbial exposures may help prevent dysbiosis, and thus be evolutionarily advantageous5.
Breast milk has been shaped over mammalian evolution to promote the growth and development of infants. Non-digestible sugars in breast milk, known as human milk oligosaccharides (HMOs), are beneficial to the infant in a number of ways, such as providing protection from pathogens6,7,8. HMOs can be bound to other compounds in milk as glycoconjugates, which are known as human milk glycans (HMGs)7. HMGs may play a similar role to free HMOs9. These maternally-provided glycans are hypothesized to guide the assembly of the microbial community in the gut due to the ability of only select microbial species to metabolize them7,10. Bifidobacteria are often the major HMG-consuming constituent of the gut microbiota in breast-fed infants11,12,13,14,15,16, and may provide unique benefits to the newborn, including reducing inflammation, improving gut permeability, and improving responses to both oral and parenteral vaccines17,18,19,20,21,22. Specific analysis of infant-borne bifidobacteria clearly demonstrate that certain species and strains possess a unique genetic capacity to consume HMOs and HMGs and B. longum subsp. infantis (B. infantis) in particular is the only species shown to consume the complete constellation of HMO/Gs present in milk9,23. We have recently shown how the genetic capacity of the mother, specifically the FUT2 gene status (also known as secretor status), can influence the bifidobacterial colonization phenotype24. Thus, colonization of the infant gut is driven by a mother’s genetics (i.e. expressed milk glycome), infant environmental exposure, and the genetic makeup of the bifidobacteria transferred to the infant—components of which appear to have co-evolved over mammalian history25.
However, due to the observed differences in the microbiota of breast fed infants (especially in bifidobacterial abundances) from different countries (Norway26, Sweden27, Canada28, Italy29, Switzerland30, Bangladesh22, the USA31, Malawi and Finland32) we sought to examine the influences on the microbiome of infants from an area of the world that has not previously been studied. The present study investigates country of origin and birth method (differential sources of microbial exposure) and mother’s secretor-status (an influence on environmental conditions) as possible factors influencing the infant gut microbiome in two cohorts, one from Yerevan, Armenia and one from Tbilisi, Georgia. These countries, like any area on earth, have unique exposure patterns and environmental conditions, however their proximity to one another allows us to test the effect of national boundaries while maintaining relatively low separation by geographic distance.
Methods
Sample collection
Fecal samples were collected from exclusively breastfed healthy infants at three time points, one week of life, one month of life, and 3 months of life. Exclusion criteria included formula or other supplemental feeding, mothers who received antibiotic treatments in the year before enrollment, and infants receiving antibiotic treatment. Parents were provided with 50 ml tubes and wooden tongue depressors to collect infant feces. Samples were delivered to the laboratory no more than 2 hours after collection. Metadata questionnaires were filled out at the subject’s home before obtaining samples. Informed consent was obtained from all mothers while they were in the maternity ward. The study protocol was approved by an institutional review board at the Georgian Maternal and Child Care Union (IRB approval #2011-009), and by the Ethics Committee at the Ministry of Education and Science of Armenia (ISTC Project #A-1957) and the Armenian Ministry of Health (M2/938-14 06.02.2014) and all methods were carried out in accordance with the approved guidelines.
DNA Extraction
DNA was extracted using the ZR Fecal DNA Miniprep Kit (Zymo Research Irvine, CA) with a bead-beating step as per manufacturer’s instructions33.
Sequencing and Analysis
Illumina sequencing—V4 region
Extracted DNA was prepared for marker gene sequencing as previously described34 with the following modifications. The following universal barcoded primers were used to PCR amplify the V4 region of the 16S rRNA gene, and Illumina sequencing adapters were subsequently ligated to the amplicons (adapters are italicized and an example barcode is highlighted in bold): V4F (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT ACTGCTGAGTGTGCCAGCMGCCGCGGTAA-3′) and V4Rev (5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCT GCTGAACCGCTCTTCCGATCTCCGGACTACHVGGGTWTCTAAT-3′)34. PCRs were heated to 94 °C for 3 min to denature the DNA, amplified for 25 cycles of 94 °C for 45 s, 50 °C for 60 s, and 72 °C for 90 s, after which a final extension of 10 min at 72 °C was added to ensure complete amplification. PCR reactions contained 7.5 μl 2x GoTaq Green Master Mix (Promega, Madison, WI), 0.6 μl 25 mM MgCl2, 3.6 μl water, 1.5 μl forward and 0.3 μl reverse primers (0.2 μM final concentration), and 1.5 μl DNA. A negative control was also included into which water was added in the place of DNA. A portion of each reaction was electrophoresed in a 0.8% agarose gel and stained with GelGreen (Phenix, Candler, NC). The DNA band for each sample was visually categorized by brightness and size for quality control. Samples were pooled (5 μl of each reaction for samples with bright bands, 10 μl for faint samples with bands, and 12 μl for samples with non-visible bands) and purified with the QIAquick PCR Purification Kit (QIAGEN, Valencia, CA) according to the manufacturer’s instructions. Pooled, purified amplicons were sequenced at the University of California-Davis DNA Technologies Core Facility on an Illumina MiSeq sequencing platform.
Sequence Analysis
The QIIME software package (version 1.8) was used to analyze the results of the Illumina sequencing run. llumina V4 16S rRNA gene sequences were demultiplexed and quality filtered with default settings unless otherwise specified35. Reads were truncated after a maximum number of 3 consecutive low quality scores. The minimum number of consecutive high quality base calls to include a read (per single end read) as a fraction of the input read length was 0.75. The minimum acceptable Phred quality score was set at 20. Similar sequences were clustered into operational taxonomic units (OTUs) using open reference OTU picking with UCLUST software36. Taxonomy was assigned to each OTU with the Ribosomal Database Project (RDP) classifier37 and the RDP taxonomic nomenclature38. OTU representatives were aligned against the Greengenes core set39 with PyNAST software40. The alignment was filtered with the filter_alignment.py script to remove positions which are gaps in every sequence and a phylogeny was constructed with FastTree41. Beta diversity analysis was performed using a weighted UniFrac distance matrix42. The OTU table was normalized by cumulative sum scaling and the adonis function with 999 permutations was used to detect differences in the community structure (as a whole) by sample category43,44. OTUs were then merged at the genus level, Bifidobacterium reads apportioned into (sub) species as measured by Bif-TRFLP/BLIR (see below) and metagenomeSeq used to detect differential taxa abundance between sample classes, with FDR correction44.
Community State Analysis
After taxa were aggregated at the species level, microbiomes were clustered using the Bray-Curtis distance metric and a Non-metric multidimensional scaling (NMDS) ordination method. The Bray-Curtis metric was chosen because it does not account for phylogenetic relatedness and therefore provides a better separation of communities that are dominated by closely related species, as occurs with infant gut microbiomes. Based on a screeplot and eigenvalues, the first 6 dimensions were retained for the NMDS ordination. The number of clusters (K = 5) was determined using the gap statistic. The members of the five clusters were determined using the partition around medoids algorithm, pam, in R.
Bif-TRFLP (terminal restriction fragment length polymorphism)
Because short amplicon-based sequencing often lacks the ability to resolve species-level taxonomic distinctions45, the method of Lewis et al. was used to perform the Bifidobacterium-specific terminal restriction fragment length polymorphism (Bif-TRFLP) assay to clarify the composition of bifidobacteria in infants46. Briefly, DNA from feces was amplified in triplicate by PCR using primers NBIF389 (5′-[HEX]-GCCTTCGGGTTGTAAAC) and NBIF1018 REV (5′-GACCATGCACCACCTGTG) (Table S1). DNA was purified using the Qiagen Qiaquick PCR purification kit (QIAGEN, Valencia, CA) and then cut with restriction enzymes AluI and HaeIII. The resulting fragments were analyzed on an ABI 3100 Capillary Electrophoresis Genetic Analyzer at the UC Davis College of Biological Sciences Sequencing Facility. The data was analyzed with PeakScanner 2.0 software (Applied Biosystems, Carlsbad, CA) and sizes were compared against the published database for species identification.
Bifidobacterium Longum-Infantis Ratio (BLIR)
A PCR-based assay, BLIR, was used to determine which subspecies of B. longum were present in each sample and to gain an estimate of their abundance relative to each other47. Three primers (FWD_BL_BI (5′-[HEX]-AAAACGTCCATCCATCACA), REV_BL (5′-ACGACCAGGTTCCACTTGAT), and REV_BI (5′-CGCCTCAGTTCTTTAATGT)) were used to amplify the targeted genomic regions. FWD_BL_BI is complementary to a sequence in both subspecies while REV_BL and REV_BI are complementary to nearby sequences in only B. longum subsp. longum and B. longum subsp. infantis, respectively. FWD_BL_BI and REV_BL amplify a fragment of the B. longum spp. longum genome 145 bp in length, while FWD_BL_BI amplify a fragment of the B. longum subsp. infantis genome 114 bp in length.
DNA from each sample was amplified by PCR using 0.5 μl of 10 μM stock of each of the above primers, 12.5 μl GoTaq Green Master Mix (Promega), 1 μl of 25 mM MgCl2, 1 μl of template DNA, and 9 μl of nuclease free water. Cycling conditions were 95 °C for 2 minutes, 30 cycles of 95 °C for 1 minute, 54 °C for 1 minute, and 72 °C for 30 seconds, followed by a 72 °C extension for 5 minutes. PCR products were purified from the mixture using the QIAquick PCR purification kit (Qiagen) and diluted 1:10. 1.5 μl of the dilutions were analyzed by capillary electrophoresis on an ABI 3100 Capillary Electrophoresis Genetic Analyzer at the UC Davis College of Biological Sciences Sequencing Facility. The HEX fluorophore on the common primer allowed detection and differentiation of amplicon sizes and a rough quantitation of the abundance of each amplicon based on peak area when the samples were analyzed with PeakScanner 2.0 software (Applied Biosystems, Carlsbad, CA). A positive control was included with each PCR run to ensure potential amplification of both B. longum subsp. longum and B. longum subsp. infantis products.
Determination of Secretor Genotype
DNA from saliva was extracted using the Qiagen Blood & Tissue Kit and protocol (QIAGEN, Valencia, CA). Secretor genotype was determined as in Lewis et al.31. Briefly, genomic DNA was amplified with primers FUT2-F (5′-CCTGGCAGAACTACCACCTG) and FUT2-R (5′-GGCTGCCTCTGGCTTAAAGA) (Georgia- TECHNE TC-5000 thermocycler, Armenia- BOECO THERMAL CYCLER TC-PRO), which produces a 608 bp amplicon. Samples with low amplicon concentrations were reamplified with 35 cycles of PCR for all samples, and 50 for some difficult samples. Negative controls were run with each set of PCRs, and results were not used unless the controls were free of amplification in the expected size range. The individual performing the PCRs’ genotype was non-secretor, which should limit false secretor designations due to lab worker contamination.
The amplicons were then digested with BfaI, which cuts the DNA of individuals containing the mutated non-secretor rs601338 SNP (G → A, Trp → Ter) allele of the FUT2 gene, the predominant non-secretor mutation in Caucasians. The resulting digests were electrophoresed on a 2% agarose gel for ~2 hours at 80 V and the resulting bands were visualized using GelGreen dye under UV light. Individuals possessing a secretor allele produce a 608 bp band on the gel after digestion, while a non-secretor allele produces two bands with sizes of 516 bp and 92 bp. In this way it is possible to easily distinguish both homozygote genotypes from each other and from heterozygotes. Heterozygotes were counted as secretors for statistical analysis, therefore contamination by non-secretor DNA would not change the designation of individuals for this purpose.
Results
Cohort Metadata
In total, 205 samples were collected, 133 from Armenia, and 72 from Georgia. Mother’s saliva (for secretor genotyping) was unavailable for 31 of the 53 Armenian infants, and 11 of the 28 Georgian infants. Four samples (38b, 48.a, 48.b, 48.c) were found to be duplicates and excluded from analysis. A full description of the metadata of the two cohorts is found in Table 1, including classification by country, mother’s secretor status, and day of sample collection.
Table 1. Cohort metadata.
| Country | ||||||||
|---|---|---|---|---|---|---|---|---|
| Armenia | Georgia | |||||||
| Day | Day | |||||||
| Secretor Status | 7 | 30 | 90 | All | 7 | 30 | 90 | All |
| H (Heterozygote) | 11 | 12 | 10 | 33 | 9 | 8 | 7 | 24 |
| N (Homozygote non-secretor) | 7 | 6 | 6 | 19 | 3 | 3 | 2 | 8 |
| S (Homozygote secretor) | 2 | 2 | 1 | 5 | 5 | 6 | 3 | 14 |
| U (Unknown) | 30 | 25 | 21 | 76 | 11 | 11 | 4 | 26 |
| All | 50 | 45 | 38 | 133 | 28 | 28 | 16 | 72 |
Breakdown of samples by time point, secretor status, and country.
H = heterozygote, S = homozygote secretor, N = non-secretor, U = unknown.
General Microbiome Trends
Four hundred and twenty six genus-level taxa were identified in the sequencing data. Table 2 lists all taxa with a minimum average relative abundance of 0.5%, and breaks down the bifidobacteria by species according to the Bif-TRFLP/BLIR data. Bifidobacteria were the dominant taxa in the infant gut microbiota in these infants, comprising 44% of all reads. The most abundant bifidobacterial species was B. breve, which accounted for 16.3% of the total microbiota.
Table 2. Summary of all taxa with a minimum average relative abundance of 0.5%, and breakdown of the bifidobacteria by species according to the Bif-TRFLP/BLIR data.
| Taxa | Average Abundance (%) |
|---|---|
| g_Bifidobacterium; s_All | 44.0 |
| s_breve | 16.3 |
| s_longum subsp. longum | 8.3 |
| s_pseudocatenulatum | 8.1 |
| s_bifidum | 4.2 |
| s_Unknown | 3.5 |
| s_adolescentis | 1.7 |
| s_bifidum/pseudocatenulatum | 1.0 |
| s_longum subsp. infantis | 0.8 |
| s_animalis | 0.1 |
| f_Enterobacteriaceae; g_ | 13.5 |
| g_Streptococcus | 11.6 |
| g_Enterococcus | 7.6 |
| g_Lactobacillus | 5.6 |
| g_Bacteroides | 3.1 |
| f_Planococcaceae; g_ | 1.8 |
| g_Staphylococcus | 1.8 |
| g_Rothia | 0.8 |
| g_Clostridium | 0.7 |
| g_Blautia | 0.7 |
| f_Enterobacteriaceae; Other | 0.5 |
| Total | 91.9 |
A community state analysis was conducted on the sequencing-based data to explore microbial community structure (see Methods). This unsupervised exploration of the data yielded a set of 5 community state types (Fig. 1): a community dominated by an OTU assigned to the B. longum group (Figures S1, S6), a community dominated by an OTU assigned to B. adolescentis (Figures S2, S7), a community dominated by Bifidobacterium of unresolved species (Figures S3, S8), a mixed community low in Bifidobacterium (Figures S4, S9), and an Enterococcus-dominant community (Figures S5, S10). The biggest driver of community state seems to be whether or not the community is dominated by bifidobacteria. The samples associated with the Enterococcus-dominant group were mostly from Day 7. Also, none of the samples in this group came from babies with homozygous secretor mothers. To avoid overemphasizing the importance of minor community members, the analysis of differentially abundant taxa was confined to only bifidobacteria (at a species level determined by the Bif-TRFLP and BLIR methods) and other major genera. Figure 2 shows a breakdown of the levels of these taxa over time by various metadata categories and Fig. 3 shows the breakdown of bifidobacterial species in the same way.
Figure 1. Infant gut microbiome clusters using a Bray-Curtis distance metric and a Non-metric multidimensional scaling (NMDS) ordination.
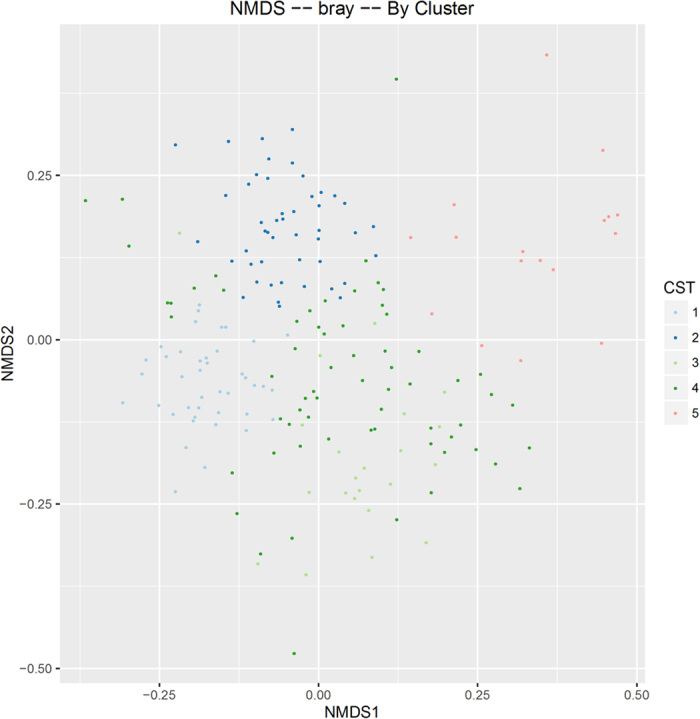
CST = community state type.
Figure 2. Members of the infant gut microbiome with greater than 0.05% average relative abundance over time based on proportion of sequencing reads.

Error bars are standard error. (A) Average proportion of major gut microbes by country. (B) Average proportion of major gut microbes by birth method. (C) Average proportion of major gut microbes by mother’s secretor status. H = heterozygote, S = homozygote secretor, N = non-secretor, U = unknown.
Figure 3. Average relative abundance of each member within the genus Bifidobacterium over time, based on the Bif-TRFLP/BLIR data.

Error bars are standard error. (A) Average proportion of bifidobacteria by country. (B) Average proportion of bifidobacteria by birth method. (C) Average proportion of bifidobacteria by mother’s secretor status. H = heterozygote, S = homozygote secretor, N = non-secretor, U = unknown.
Individual infants were found to be similar to themselves over time, with an Adonis R2 value of 0.027 for subject number, indicating that individuality was responsible for 2.7% of the variation in the microbiota (Pr(>F) = 0.001). To investigate the pattern of bacterial colonization of the gastrointestinal tract over time, the composition of the infant microbiota was compared at each time point. Time was found to be a significant influence on the infant microbiota (Pr(>F) = 0.001) and was responsible for 2.3% of the variation in the microbiota (Adonis R2 of 0.023). Between days 7 and 90, B. breve, B. longum subsp. infantis, and lactobacilli increased in abundance (p = 0.004, 0.039, and 9.29 × 10−5 respectively), while staphylococci, and the Planococcaceae family decreased in abundance (p = 3.18 × 10−8 and 4.86 × 10−5 respectively). In total, 30 of the 426 genera were differentially abundant between the earliest time point and the latest. Table 3 contains a list of the major taxa enriched in each time point and metadata group, while Supplementary Table S1 contains a complete list of taxa differentially abundant between each pair of time points and other metadata categories.
Table 3. List of the major taxa and bifidobacterial species enriched (p < 0.05) in each time point and metadata group.
| Major Taxon | Country Comparison |
Birth Method Comparison |
Day 7 vs. Day 30 |
Day 7 vs. Day 90 |
Day 30 vs. Day 90 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Country | Adjusted p value | β coefficient | Birth Method | Adjusted p value | β coefficient | Day | Adjusted p value | β coefficient | Day | Adjusted p value | β coefficient | Day | Adjusted p value | β coefficient | |
| g_Bifidobacterium | |||||||||||||||
| s_breve | Day 7 | 0.046 | −2.60 | Day 90 | 0.004 | 3.27 | |||||||||
| s_longum subsp. longum | Georgia | 0.012 | −2.27 | ||||||||||||
| s_pseudocatenulatum | Georgia | 0.0004 | −3.33 | Vaginal | 0.031 | 3.38 | |||||||||
| s_bifidum | |||||||||||||||
| s_other | Day 7 | 0.046 | 2.12 | ||||||||||||
| s_adolescentis | |||||||||||||||
| s_bifidum/ pseudocatenulatum | |||||||||||||||
| s_longum subsp. infantis | Day 90 | 0.039 | 1.60 | ||||||||||||
| s_animalis | |||||||||||||||
| f_Enterobacteriaceae;g_ | Georgia | 1.95E-06 | −2.44 | Vaginal | 0.012 | 2.10 | |||||||||
| g_Streptococcus | Georgia | 0.0001 | −1.59 | Vaginal | 0.008 | 1.73 | |||||||||
| g_Enterococcus | Georgia | 0.022 | −1.37 | Day 7 | 0.021 | 2.02 | |||||||||
| g_Lactobacillus | Day 7 | 5.31E-05 | −2.78 | Day 90 | 9.29E-05 | 2.50 | |||||||||
| g_Bacteroides | Georgia | 0.042 | −1.21 | ||||||||||||
| f_Planococcaceae;g_ | Georgia | 7.04E-11 | −2.74 | Vaginal | 0.0007 | 2.29 | Day 90 | 4.86E-05 | −2.43 | Day 90 | 0.002 | −1.78 | |||
| g_Staphylococcus | Georgia | 2.86E-08 | −2.86 | Vaginal | 0.005 | 2.40 | Day 90 | 3.18E-08 | −3.72 | Day 90 | 4.43E-08 | −2.78 | |||
| g_Rothia | |||||||||||||||
| g_Clostridium | Georgia | 0.024 | −1.04 | ||||||||||||
| g_Blautia | |||||||||||||||
| f_Enterobacteriaceae; Other | Georgia | 8.65E-05 | −2.10 | ||||||||||||
The “Country”, “Birth Method”, and “Day” columns indicate the base group for comparison, and the sign of the corresponding β coefficient indicates whether the taxa is enriched (+) or depleted (−) in the group specified in the column to its left. Non-significant results are left blank.
Comparison Between Countries
The overall composition of the microbiota of the infant gastrointestinal tract was tested for differences between infants in the two countries. Figure 2A shows a summary of the major constituents of the infant gut bacterial community of the two countries (average abundance cutoff of 0.5%), and how they shift over the first 90 days of life. The overall patterns of colonization were similar between the two countries, as bifidobacteria dominated the infant fecal microbiome of both countries, with significant amounts of enterobacteria, streptococci, enterococci, lactobacilli, Bacteroides, and staphylococci also present. Country of origin accounted for 8.4% of total variation between samples (Adonis R2 value of 0.084, (Pr(>F) = 0.001)). Of the 426 total genera, 196 were found to be differentially abundant between the two countries. There were statistically significant differences (p < 0.05) found between the major community members of the two countries. In general infants in Armenia had more Enterobacteriaceae, Planococcaceae, Streptococcus, Enterococcus, Bacteroides, Clostridium, and Staphylococcus. B. breve was the most abundant bifidobacterial species in both countries. Of the bifidobacteria, Armenian infants had more B. pseudocatenulatum and B. longum subsp. longum. There were no bifidobacterial taxa more abundant in Georgian infants that reached statistical significance. Figure 3A shows the results of the Bif-TRFLP/BLIR analysis by country.
C-section vs. Vaginal Birth
In addition to the other metadata collected, birth method was tracked and a subset of 11 of the 28 infants from the Georgian cohort were born by Caesarian section. We explored the impact of this first exposure to microbes on the gut microbiota of the infants. Figure 2B shows a summary of the major constituents of the infant gut bacterial community of infants born by both methods, and Fig. 3B shows the bifidobacterial species-level data. Bifidobacteria, for example, dominated the communities of both types of infants. About 3.1% of the variation in the overall community structure was attributable to birth method (Pr(>F) = 0.001). Several major community members (average abundance cutoff of 0.5%) were generally different between infants born by C-section and vaginally. Vaginally born infants were higher in B. pseudocatenulatum (p = 0.03), Enterobacteriacaea; g_(p = 0.01), Streptococcus (p = 0.008), Staphylococcus (p = 0.005), and Planococcaceae; g_(p = 0.0007). Due to the lack of C-section born infants in Armenia, however, it is difficult to fully disentangle the effect of country from that of birth method.
Secretor Status Differences
We also wished to investigate the effect of the mother’s secretor status on the infant microbiota. We were able to determine the secretor genotype for 22/53 (41.5%) Armenian mothers and 17/28 (60.7%) of Georgian mothers. A breakdown of the overall infant gut bacterial community data by mother’s genotype in found in Fig. 2C. We grouped the infants samples into 4 subsets depending on the mother’s secretor genotype, homozygote secretors (S), homozygote non-secretors (N), heterozygotes (H), and “unknown” mothers for which salivary DNA was not available (U). Bifidobacteria were the dominant member of the microbiota in all four groups, and their abundance generally increased over time (except for the H group from day 30 to 90, where there was a small decrease) (Fig. 2C). Approximately 1.6% of the variation in the microbial community was attributable to mother’s secretor status, though the effect was only marginally significant (Pr(>F) = 0.066, mothers of known secretor status classes only with heterozygotes are counted as secretors).
However, a trend was observed where the average amount of bifidobacteria increased more quickly in secretor-fed infants than non-secretor-fed infants while heterozygote-fed and unknown-fed infants contained intermediate levels of bifidobacteria by day 90. This trend appears to be driven in part by fewer infants having near-zero levels of bifidobacteria in the secretor-fed infants (Fig. 4). There were no differentially abundant major taxa between infants fed by mothers of known secretor status (heterozygotes included as secretors) and infants fed by know non-secretor mothers. At the level of bifidobacterial species, B. breve was the present at the highest level across all infants (Fig. 3C).
Figure 4. Bifidobacterial relative abundance over time by secretor genotype.
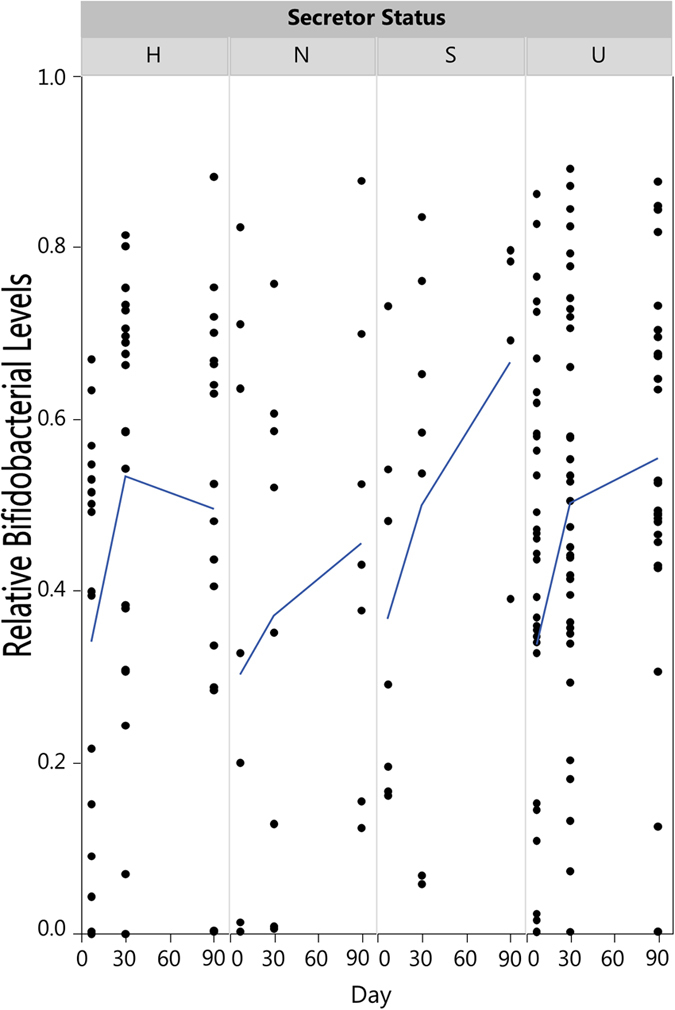
The lines show the average bifidobacterial abundance of each group over time. H = heterozygote, S = homozygote secretor, N = non-secretor, U = unknown.
Discussion
The composition of the gut microbiota is thought to impact infant health in numerous ways. The establishment of the gut microbiota can influence an individual’s lifelong health48,49. In the first years of life the infant gut microbiome progresses through age-associated compositional changes50. The infant gut microbiota is sensitive to disruption early in life, as it has unfilled niches and has not developed mature levels of colonization resistance5,51,52,53. From an evolutionary perspective, alterations in gene content or expression that assist an infant in fostering select, beneficial, microbial communities would be advantageous. For example, infants appear to suppress their immune system early in life to aid in shaping their microbiota54.
Maternal genes influencing the infant gut microbiota may be under selection as well, as long as the maternal investment benefits the infant sufficiently to overcome the inherent parent-offspring conflict over resources55. For example, mother’s milk has been shown to affect the infant gut microbiota in numerous ways7. Moreover, exposure to microbes via birth method (C-section vs. Vaginal birth) and other behavioral or circumstantial factors may also play a role in the development of the microbiota independent of the selective environmental conditions within the gut. Due to the observed differences in the microbiota of infants from different countries (Norway26, Sweden27, Canada28, Italy29, Switzerland30, Bangladesh22, the USA31, Malawi and Finland32) we sought to investigate and disentangle community differences caused by selection (i.e. maternal secretor status) from differences caused by environmental exposure (regionality and birth method).
Armenia and Georgia are neighboring countries, but possess distinct languages and cultural differences56. Country of birth was found to have the highest Adonis R2 value of any factor tested, indicating that regionality has a relatively large impact on the microbial community. Lacking both a thorough ethnography focused on behavioral differences between the two groups that may influence the exposure to colonizing microbes and relevant data on the environmental pressures experienced by the microbes in the gut, the mechanisms which drive the differences in the infant microbiota of the two countries are not identifiable. It is however striking to observe the differences in infants from cities which, while in two different countries, are only approximately 175 km in distance from each other. Despite these differences, bifidobacteria dominated the community of infants in both countries, and B. breve, a species of bifidobacteria common to infants, was the found in the highest levels in both cohorts. Similar to other reports on the infant microbiome, the major non-bifidobacterial taxa present in the infants in our study were enterobacteria, streptococci, enterococci, lactobacilli, Bacteroides, Clostridium, and staphylococci22,27,28,29,30,31,57. While this study is not focused on the minor taxa in these infants, there were many differences in the lower abundance taxa between the two countries, perhaps indicating differential exposure to allochthonous environmental microbes.
C-section birth has been associated with altered community assembly in infants, often thought to be the result of differential exposure to microbes from vaginally-born infants. Currently, over 30% of babies are born by Cesarean section in the United States, and approximately 82% of cases of methicillin-resistant Staphylococcus aureus infections occur in this subset of infants58. In other countries, C-sections are known to reduce the rate of bifidobacterial colonization59, but that trend was not observed in this and other studies22. We also failed to observe early enrichment of lactobacilli in the vaginally-born infants, a commonly observed phenomenon ascribed to the transfer of vaginal lactobacilli to the infant60.
Apart from the effect of microbial exposure, many studies suggest that the consumption of glycans found in milk influences the initial microbial colonization of the neonatal gastrointestinal tract10,31,61,62. Specifically, the glycan content in milk may shape the microbiome through its selective fermentability by members of the microbial community25,31,61,63,64. In this study the major bifidobacterial species present was B. breve, a common infant commensal known to consume human some milk oligosaccharides65,66,67. Indeed, B. breve and B. longum subsp. infantis, two species known to consume HMOs65,68, were both found to be enriched over the course of extended breastfeeding, supporting the hypothesis that the ability to consume these carbon sources is an adaptive trait for infant microbes. Early establishment of bifidobacteria is thought to be beneficial in numerous ways. High levels of bifidobacteria were shown to be predictive of better immune response to vaccines22. Other benefits associated with higher bifidobacteria include better development of an infant’s immature immune system and protection from colonization by pathogens17,22,69,70.
Secretor status in general is known to affect the gut microbiota of both adults and infants31,71,72. Secretor status also alters both the bifidobacterial species profiles and absolute levels of bifidobacteria in the gastrointestinal tract of adults and infants, where secretors have higher bifidobacterial abundance31,73,74. Maternal secretor status has been shown to impact infant resistance to infection by enteric pathogens, including members of the Proteobacteria phylum75,76. Maternal secretor status has also been shown to impact the types and amounts of bifidobacteria in the infant31. The infant’s own glycosylation system is also a potential influence on the development of the gut microbiota early in life, however the temporal variation of an infant’s glcosyltransferase expression, including the FUT2 gene, remains to be elucidated. While they do not reach statistical significance, the trends reported here support previous data that suggest that secretor mothers promote bifidobacteria establishment more strongly than non-secretor mothers. The failure to reach statistical significance, may possibly be due in part to the low number of subjects for which we had secretor status data. We were also unable to confirm our previous results correlating the abundance of B. longum subsp. infantis with the mother’s secretor status in this cohort, likely due to the low amount of B. longum subsp. infantis observed in these infants (n = 6 for infants with B. longum subsp. infantis levels of over 1%). We also did not observe co-enrichment of B. breve and B. bifidum abundances that would be indicative of previously hypothesized in vivo cross-feeding between the two species, even in infants fed by mothers of known secretor status77,78. The low percentages of variation explained by the results of the Adonis tests indicate that the sample metadata recorded this study may not fully capture the suite of factors that explain the majority of the differences between sample classes. Indeed, the unsupervised clustering of the data demonstrated that there were few relationships between the sample metadata and the resulting community state types, suggesting that microbial community structure in breastfed babies transcends geography, mode of delivery, and secretor status. The Enterococcus-rich communities were mostly from the earliest time point, implying that nearly all breastfed babies later reach the Bifidobacteria-dominant state.
In summary, this study adds valuable information about the gut microbiome of previously unstudied populations. Breast-fed infants in Georgia and Armenia appear to follow well-established patterns of microbial colonization5. Country of birth was found be the largest influence on microbial community structure of the factors tested, signaling differences in the infant microbiota between the two closely-situated countries and raising the question of the degree to which the developing intestinal microbiota might be influenced by national boundaries and the cultural and genetic differences they represent. Previous findings regarding the influence of maternal secretor status of infant gastrointestinal microbiota colonization patterns were supported by our results here, including a non-significant trend of enriched bifidobacteria in secretor-fed infants. These findings may be useful as guidance in the future application of pre- and probiotic treatments for vulnerable infants in the new era of easier genetic testing and personalized medicine.
Additional Information
How to cite this article: Lewis, Z. T. et al. The Fecal Microbial Community of Breast-fed Infants from Armenia and Georgia. Sci. Rep. 7, 40932; doi: 10.1038/srep40932 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors thank Khatuna Varsimashvili, Lana Tolordava, Lela Tinikashvili, Susanna Mirzabekyan, and Marika Gamkrelidze for technical support, and Astghik Harutyunyan and Evrik Afrikyan for guidance and advice on this work. This work was supported, in part, by the U.S. Department of Energy Global Initiatives for Proliferation Prevention (GIPP) program (LBNL- 291 0225-GE and LBNL-0231-AM). The project in Georgia (P509) was funded through the Science and Technology Center in Ukraine (STCU). The International Science and Technology Center (ISTC) in Moscow, Russia provided the financial support to Armenia (A-1957). This work was also supported in part by funding from National Institutes of Health awards R01AT007079 and R01AT008759 and the Peter J. Shields Endowed Chair in Dairy Food Science (DAM). ZTL is supported by an Alfred P. Sloan Foundation Microbiology of the Built Environment postdoctoral fellowship.
Footnotes
DAM is a co-founder of Evolve Biosystems, a company focused on diet-based manipulation of the gut microbiota. Evolve Biosystems played no role in the design, execution, interpretation, or publication of this study.
Author Contributions Z.L., T.T. and D.M. planned and coordinated the research. Z.L., D.L. and D.M. wrote the manuscript. Z.L. performed the Bif-TRFLP, BLIR, secretor status, and sequencing analyses. K.S., E.Z., N.K., L.T., V.T., A.M. and M.B. conducted laboratory work for the experiments. D.T. collected sample metadata. V.T., P.I. and A.P. participated in the design and interpretation of the studies. Z.L. and D.L. analyzed data. All authors read and approved the manuscript.
References
- Rook G. a W., Raison C. L. & Lowry C. a. Microbial ‘Old Friends’, immunoregulation and socio-economic status. Clin. Exp. Immunol. 1–24, doi: 10.1111/cei.12269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson L. A. et al. Protective factors in milk and the development of the immune system. Pediatrics 75, 172–176 (1985). [PubMed] [Google Scholar]
- Brown E. M., Arrieta M.-C. & Finlay B. B. A fresh look at the hygiene hypothesis: how intestinal microbial exposure drives immune effector responses in atopic disease. Semin. Immunol. 25, 378–87 (2013). [DOI] [PubMed] [Google Scholar]
- Bäckhed F. et al. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12, 611–22 (2012). [DOI] [PubMed] [Google Scholar]
- Scholtens P. A. M. J., Oozeer R., Martin R., Amor K. Ben & Knol J. The Early Settlers: Intestinal Microbiology in Early Life. Annu. Rev. Food Sci. Technol. 3, 425–447 (2012). [DOI] [PubMed] [Google Scholar]
- Hickey R. M. The role of oligosaccharides from human milk and other sources in prevention of pathogen adhesion. Int. Dairy J. 22, 141–146 (2012). [Google Scholar]
- Zivkovic A. M., Lewis Z. T., German J. B. & Mills D. A. Establishment of a Milk-Oriented Microbiota (MOM) in early life : How Babies Meet Their MOMs. Funct. Food Rev. 5, 3–12 (2013). [Google Scholar]
- Kunz C. & Rudloff S. Health promoting aspects of milk oligosaccharides. Int. Dairy J. 16, 1341–1346 (2006). [Google Scholar]
- Garrido D., Dallas D. C. & Mills D. a. Consumption of human milk glycoconjugates by infant-associated bifidobacteria: mechanisms and implications. Microbiology 159, 649–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela D. A. & Mills D. A. Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol. 18, 298–307 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivkovic A. M., German J. B., Lebrilla C. B. & Mills D. a. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl. Acad. Sci. USA 108 Suppl, 4653–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela D. A. et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. USA 105, 18964–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido D., Kim J. H., German J. B., Raybould H. E. & Mills D. A. Oligosaccharide binding proteins from Bifidobacterium longum subsp. infantis reveal a preference for host glycans. PLoS One 6, e17315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela D. A. et al. Bifidobacterium longum subsp. infantis ATCC 15697 α-fucosidases are active on fucosylated human milk oligosaccharides. Appl. Environ. Microbiol. 78, 795–803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakuma S. et al. Physiology of consumption of human milk oligosaccharides by infant gut-associated bifidobacteria. J. Biol. Chem. 286, 34583–92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S. et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469, 543–7 (2011). [DOI] [PubMed] [Google Scholar]
- Sheil B. et al. Role of interleukin (IL-10) in probiotic-mediated immune modulation: an assessment in wild-type and IL-10 knock-out mice. Clin. Exp. Immunol. 144, 273–80 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe S., Kinuta Y. & Saito Y. Bifidobacterium infantis suppresses proinflammatory interleukin-17 production in murine splenocytes and dextran sodium sulfate-induced intestinal inflammation. Int. J. Mol. Med. 22, 181–185 (2008). [PubMed] [Google Scholar]
- Preising J. et al. Selection of bifidobacteria based on adhesion and anti-inflammatory capacity in vitro for amelioration of murine colitis. Appl. Environ. Microbiol. 76, 3048–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichlowski M., De Lartigue G., German J. B., Raybould H. E. & Mills D. a. Bifidobacteria isolated from infants and cultured on human milk oligosaccharides affect intestinal epithelial function. J. Pediatr. Gastroenterol. Nutr. 55, 321–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huda M. N. et al. Stool Microbiota and Vaccine Responses of Infants. Pediatrics 134, e362–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T. et al. A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nat. Commun. 7, 11939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Z. T. et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichlowski M., German J. B., Lebrilla C. B. & Mills D. A. The influence of milk oligosaccharides on microbiota of infants: opportunities for formulas. Annu. Rev. Food Sci. Technol. 2, 331–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avershina E. et al. Major fecal microbiota shifts in composition and diversity with age in a geographically restricted cohort of mothers and their children. FEMS Microbiol. Ecol. 87, 280–90 (2014). [DOI] [PubMed] [Google Scholar]
- Abrahamsson T. R. et al. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin. Exp. Allergy 44, 842–50 (2014). [DOI] [PubMed] [Google Scholar]
- Azad M. B. et al. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ 185, 385–394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos S. et al. 454 pyrosequencing analysis on faecal samples from a randomized DBPC trial of colicky infants treated with Lactobacillus reuteri DSM 17938. PLoS One 8, e56710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost T., Lacroix C., Braegger C. P. & Chassard C. New insights in gut microbiota establishment in healthy breast fed neonates. PLoS One 7, e44595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Z. T. et al. Maternal Fucosyltransferase 2 Status Affects the Gut Bifidobacterial Communities of Breastfed Infants. Microbiome 3, 1–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grześkowiak Ł. et al. Distinct gut microbiota in southeastern African and northern European infants. J. Pediatr. Gastroenterol. Nutr. 54, 812–6 (2012). [DOI] [PubMed] [Google Scholar]
- Lewis Z. T. et al. The impact of freeze-drying infant fecal samples on measures of their bacterial community profiles and milk-derived oligosaccharide content. PeerJ 4, e1612 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108, 4516–4522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. QIIME allows analysis of high- throughput community sequencing data. Nat. Methods 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–1 (2010). [DOI] [PubMed] [Google Scholar]
- Wang Q., Garrity G. M., Tiedje J. M. & Cole J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–7 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J. R. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–72 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S. & Arkin A. P. Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C. A., Hamady M., Kelley S. T. & Knight R. Quantitative and qualitative B diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M. J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001). [Google Scholar]
- Paulson J. N., Stine O. C., Bravo H. C. & Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Desantis T. Z., Andersen G. L. & Knight R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 36, 1–11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Z. T. et al. Use of bifidobacterial specific terminal restriction fragment length polymorphisms to complement next generation sequence profiling of infant gut communities. Anaerobe 19, 62–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Z. T. et al. Validating bifidobacterial species and subspecies identity in commercial probiotic products. Pediatr. Res. 1–8, doi: 10.1038/pr.2015.244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasucci G., Benenati B., Morelli L., Bessi E. & Boehm G. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr 138, 1796S–1800S (2008). [DOI] [PubMed] [Google Scholar]
- Bäckhed F. et al. Defining a Healthy Human Gut Microbiome: Current Concepts, Future Directions, and Clinical Applications. Cell Host Microbe 12, 611–622 (2012). [DOI] [PubMed] [Google Scholar]
- Subramanian S. et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510, 417–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques T. M. et al. Programming infant gut microbiota: influence of dietary and environmental factors. Curr. Opin. Biotechnol. 21, 149–56 (2010). [DOI] [PubMed] [Google Scholar]
- Koenig J. E. et al. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 108, 4578–85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley T. D. & Walker A. W. Intestinal colonization resistance. Immunology 138, 1–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi S. et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature 504, 158–162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivers R. L. Parent-Offspring Conflict. Am. Zool. 14, 249–264 (1974). [Google Scholar]
- CIA World Factbook. At https://www.cia.gov/library/publications/the-world-factbook/geos/gg.html.
- Fouhy F. et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 56, 5811–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Community-associated methicillin-resistant Staphylococcus aureus infection among healthy newborns–Chicago and Los Angeles County, 2004. MMWR Morb Mortal Wkly Rep 55, 329–332 (2006). [PubMed]
- Huurre A. et al. Mode of Delivery – Effects on Gut Microbiota and Humoral Immunity. Neonatology 93, 236–240 (2008). [DOI] [PubMed] [Google Scholar]
- Hickey R. J., Zhou X., Pierson J. D., Ravel J. & Forney L. J. Understanding vaginal microbiome complexity from an ecological perspective. Transl. Res. 160, 267–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode L. Human Milk Oligosaccharides: Every Baby needs a Sugar Mama. Glycobiology 22, 1147–62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.-T. et al. The principal fucosylated oligosaccharides of human milk exhibit prebiotic properties on cultured infant microbiota. Glycobiology 23, 169–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leoz M. L. A. et al. Human Milk Glycomics and Gut Microbial Genomics in Infant Feces Shows Correlation between Human Milk Oligosaccharides and Gut Microbiota: A Proof-of-Concept Study. J. Proteome Res., doi: 10.1021/pr500759e (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. et al. Fecal microbiota composition of breast-fed infants is correlated with human milk oligosaccharides consumed. J. Pediatr. Gastroenterol. Nutr. 60, 825–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Moyano S. et al. Variation in Consumption of Human Milk Oligosaccharides by Infant Gut-Associated Strains of Bifidobacterium breve. Appl. Environ. Microbiol. 79, 6040–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avershina E. et al. Bifidobacterial succession and correlation networks in a large unselected cohort of mothers and their children. Appl. Environ. Microbiol. 79, 497–507 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottacini F. et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genomics 15, 170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoCascio R. G., Desai P., Sela D. A., Weimer B. & Mills D. A. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 76, 7373–81 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard C. L., Elson C. O., Hatton R. D. & Weaver C. T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 489, 231–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönlund M.-M. et al. Maternal breast-milk and intestinal bifidobacteria guide the compositional development of the Bifidobacterium microbiota in infants at risk of allergic disease. Clin. Exp. Allergy 37, 1764–72 (2007). [DOI] [PubMed] [Google Scholar]
- Wacklin P. et al. Faecal Microbiota Composition in Adults Is Associated with the FUT2 Gene Determining the Secretor Status. PLoS One 9, e94863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M. et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 1–14, doi: 10.1038/ismej.2014.64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch P. et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc. Natl. Acad. Sci. USA 108, 19030–19035 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacklin P. et al. Secretor genotype (FUT2 gene) is strongly associated with the composition of Bifidobacteria in the human intestine. PLoS One 6, e20113 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburg D. S., Ruiz-Palacios G. M. & Morrow A. L. Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr. 25, 37–58 (2005). [DOI] [PubMed] [Google Scholar]
- Newburg D. S. et al. Innate protection conferred by fucosylated oligosaccharides of human milk against diarrhea in breastfed infants. Glycobiology 14, 253–63 (2004). [DOI] [PubMed] [Google Scholar]
- Egan M. et al. Cross-feeding by Bifidobacterium breve UCC2003 during co-cultivation with Bifidobacterium bifidum PRL2010 in a mucin-based medium. BMC Microbiol. 14, 282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan M., O’Connell Motherway M., Ventura M. & van Sinderen D. Metabolism of sialic acid by Bifidobacterium breve UCC2003. Appl. Environ. Microbiol. 80, 4414–4426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.