

Abstract
The ventrobasal (VB) thalamus is innervated by GABAergic afferents from the thalamic reticular nucleus (TRN) and participates in nociception. But how the TRN-VB pathway regulates pain is not fully understood. In the present study, we reported decreased extracellular GABA levels in the VB of rats with CFA-induced chronic inflammatory pain, measured by microdialysis with HPLC analysis. In vitro whole-cell patch-clamp recording showed decreased amplitudes of tonic currents, increased frequencies of mIPSCs, and increased paired-pulse ratios in thalamic slices from chronic inflammatory rats (7 days). Microinjection of the GABAAR agonist muscimol and optogenetic activation of the TRN-VB pathway relieved thermal hyperalgesia in chronic inflammatory pain. By contrast, microinjecting the extrasynaptic GABAAR agonist THIP or selective knockout of synaptic GABAAR γ2 subunits aggravated thermal hyperalgesia in the chronic stage of inflammatory pain. Our findings indicate that reduced GABAergic transmission in the VB contributes to thermal hyperalgesia in chronic inflammatory pain, which could be a synaptic target for pharmacotherapy.
The ventrobasal thalamus (VB), including the ventral posterolateral (VPL) and the ventral posteromedial (VPM) nuclei, gates nociceptive information to the cerebral cortex. Both VPM and VPL are subject to GABAergic modulation from the thalamic reticular nucleus (TRN)1,2,3,4,5. GABAergic modulation in the VB is preferentially mediated by GABA type A receptors (GABAARs), despite the involvement of GABA type B receptors4,6,7. GABAARs are pentametric ligand-gated ion channels composed of two α (1–6), two β (1–3) subunits and one special subunit δ, γ (1–3), θ, ε, π, ρ (1–3)8,9. In the thalamus, GABAARs are mainly composed of α1, α4, β2, δ and γ2 subunits10. GABA released from presynaptic vesicles as a neurotransmitter binds to synaptic GABAARs (α1β2γ2) and mediates the transient or phasic inhibition; whereas ambient GABA binds to extra-synaptic GABAARs (α4β2δ) to mediate the persistent or tonic inhibition6,11. Functionally, the diversity of GABAergic inhibition in the thalamus participates in a variety of physiological and pathological conditions, such as attention, sleep, absence epilepsy and tinnitus5.
Anatomical, biochemical and electrophysiological changes occur in the VB under inflammatory and other chronic pain conditions12,13,14,15,16. Patients with neuropathic pain display significant volume loss of the somatosensory thalamus associated with decreased TRN activities13. However, the exact role of thalamic GABAergic transmission in chronic pain has not been sufficiently investigated. Based on previous findings, we hypothesize that chronic pain is accompanied by altered thalamic GABAergic transmission, which contributes to the thermal hyperalgesia commonly seen in this disorder. In the present study, we examined this hypothesis with a combinative application of microdialysis, electrophysiological recording, behavioral testing, and pharmacological and optogenetic manipulations in rats or mice with complete Freund’s adjuvant (CFA)-induced chronic inflammatory pain.
Results
Decreased extracellular GABA levels in the VB of rats with CFA-induced chronic inflammatory pain
We first examined the level of extracellular GABA in vivo in the VB of rats with CFA-induced inflammatory pain using microdialysis at the perfusion rate 0.1 μl/min (Fig. 1A). Figure 1B showed the chromatograms of O-phthalaldehyde (OPA) derivatives of GABA from the CFA and control groups at different time points (baseline, and days 1 and 7 after CFA/NS injection). GABA levels in the VB showed no obvious changes in rats in the control group, but were significantly lower in the CFA group on day 7 (CFA-7D) [Fig. 1C, F(2, 24) = 4.765, p = 0.018]. These results indicate decreased extracellular GABA levels in the VB of rats with chronic inflammatory pain.
Figure 1. Decreased extracellular GABA levels in the VB of rats with chronic inflammatory pain.

(A) Histological confirmation of microdialysis probe tips. Brain slices at the level of 2.6 mm/3.2 mm caudal to the Bregma showed the probe tip in the right VB in the control and CFA groups. (B) Brain perfusate samples (black) and internal standards (green) were detected with high-performance liquid chromatography (HPLC). GABA was eluted at ~14.5 min. Chromatograms of OPA derivatives of GABA from a control rat (top) and an inflammatory rat (bottom) were shown at different time points: Baseline (black line); day 1 (blue line) and day 7 (red line). (C) Saline injection did not affect GABA levels in the VB, whereas the level of GABA decreased 7 days after CFA injection (CFA-7D). Error bars indicated SEMs, *P < 0.05, two-way ANOVA with Bonferroni post-test. BL: baseline.
Altered thalamic GABAergic transmission in rats with CFA-induced chronic inflammatory pain
To explore the electrophysiological dynamics of thalamic GABAergic transmission in chronic inflammatory pain, we performed whole-cell patch-clamping in the thalamic slice contralateral to the CFA injection side. With a −70 mV holding potential, we first observed tonic currents of extra-synaptic GABAARs under the blockage of the GABAAR antagonist gabazine (25 μM) (Fig. 2A). As shown in Fig. 2B, amplitudes of tonic currents in the CFA-1D group were similar to those in the control group [100 ± 19.50 pA vs. 78.75 ± 14.20 pA, F(2, 25) = 1.22, p = 0.281], but decreased in the CFA-7D group [50.38 ± 7.13 pA, F(2, 25) = 5.63, p = 0.026]. No significant differences were observed between control-1D and control-7D rats and the data were pooled into the control group (control-1D, 111.1 ± 31.19 pA; control-7D, 90.25 ± 27.02 pA, p = 0.63). These results, consistent with decreased extracellular levels of GABA, indicate decreased tonic currents of extra-synaptic GABAARs in the VB in chronic inflammatory pain rats.
Figure 2. Decreased tonic inhibition of extra-synaptic GABAARs in the VB in CFA-induced chronic inflammatory pain rats (CFA-7D).

Representative traces of tonic inhibition recordings from thalamic relay neurons in the control (n = 8 cells), CFA-1D (n = 10 cells) and CFA-7D (n = 10 cells) groups after inhibiting ionotropic glutamatergic components (AP5 50 μM, DNQX 20 μM) and sodium currents (TTX 1 μM). Gabazine (25 μM), a competitive GABAARs antagonist, produced a positive shift and blocked both phasic and tonic currents. (B) Maximal current changes after gabazine (25 μM) application measured as the shift of GABAergic tonic inhibition (∆IPSCtonic = IPSCafter − IPSCbefore). The histogram of tonic inhibitory currents showed a reduction of tonic inhibitory currents in the CFA-7D, but not the CFA-1D groups. Error bars indicated SEMs, *P < 0.05 compared with the control group, one-way ANOVA with Tukey post-test.
We further examined other electrophysiological properties of thalamic GABAergic transmission. Miniature inhibitory postsynaptic currents (mIPSCs), resulting from spontaneous GABA vesicle release, were recorded for 3–5 min at a −70 mV holding potential, with NMDA and AMPA receptors blocked by AP5 (50 μM) and CNQX (25 μM), respectively (Fig. 2A). No significant differences were observed between control-1D and control-7D rats (amplitudes: 23.01 ± 3.62 pA vs. 25.30 ± 3.56 pA; frequencies: 0.89 ± 0.27 Hz vs. 0.73 ± 0.19 Hz; p > 0.05) and the data were pooled into the control group. Compared with the control or CFA-1D groups, mIPSCs in the CFA-7D group showed similar amplitudes (Fig. 3B, control: 24.23 ± 2.48 pA; CFA-1D: 21.57 ± 2.83 pA; CFA-7D: 22.04 ± 1.96 pA; F2,50 = 0.333, p = 0.718), but significantly higher frequencies [Fig. 3C, control: 0.69 ± 0.12 Hz; CFA-1D: 0.91 ± 0.16 Hz; CFA-7D: 1.18 ± 0.12 Hz, F(2, 50) = 3.586, p = 0.035]. These changes indicate an increased probability of spontaneous GABA release in chronic inflammatory pain rats17.
Figure 3. GABAergic transmission in the VB in chronic inflammatory pain revealed by whole-cell patch clamp recording.

(A) Representative synaptic GABAARs-mediated mIPSC traces in thalamic relay neurons in the control (normal saline, n = 16 cells), CFA-1D (n = 14 cells) and CFA-7D (n = 20 cells) groups. (B) Similar amplitudes of mIPSCs in three groups. (C) Compared with the control group, the frequency of mIPSCs remained unchanged in the CFA-1D group, but significantly increased in the CFA-7D group. (D) Representative IPSC pairs evoked by paired-pulse stimulations with a 50-ms interval in the control (n = 7 cells) and CFA-7D (chronic inflammatory pain, n = 9 cells) groups. (E) Increased paired-pulse ratios (PPRs = IPSC2nd/IPSC1st) induced by paired-pulse stimulation with 25-, 50-, 100- or 200-ms intervals in the CFA-7D group. Error bars indicated SEMs. *P < 0.05, **P < 0.01, one-way ANOVA with Tukey post-test for mIPSCs and two-way ANOVA with Bonferroni post-test for PPR analysis.
We next recorded the paired-pulse ratios (PPRs) of IPSCs, which inversely correlated with activity-driven presynaptic GABA release18,19,20. Figure 3D showed representative IPSCs evoked by paired-pulse stimulations with intervals of 50 ms. PPRs significantly increased on day 7 after CFA injection [Fig. 3E, F(1, 49) = 24.46, p < 0.001, at 25-ms interval: 0.69 ± 0.21 vs. 1.90 ± 0.62, p < 0.05; at 50-ms interval: 0.69 ± 0.22 vs. 1.65 ± 0.40, p < 0.05; at 100-ms interval: 0.56 ± 0.15 vs. CFA-7D 1.67 ± 0.28, p < 0.01. at 200-ms interval: 0.67 ± 0.20 vs. 1.18 ± 0.47, p > 0.05]. These results indicate a deceased activity-driven GABA release capacity derived from TRN neurons in chronic inflammatory pain rats.
Changes in both mIPSC frequencies and PPRs indicate a presynaptic origin of the altered GABAergic transmission21,22. Consistently, Western blotting did not reveal significant changes of protein expression of gamma2 or delta subunits of GABAARs (Fig. 4 and Supplementary Fig. S1), or GAD65, a GABA synthesizing enzyme, in chronic inflammatory pain rats (Fig. 5 and Supplementary Fig. S2).
Figure 4. Lack of significant changes in the expression of GABAAR gamma2 and delta subunits in the VB of inflammatory pain rats.

We did no detect significant changes in protein expression of the GABAAR gamma2 subunit ((A) ~45 KD) or delta subunit ((B) ~55 KD) in CFA-induced inflammatory pain rats. Na+, K+-ATPase was used as the reference in the Western blotting. Control: normal saline injection into hindpaws of mice. CFA-1D or CFA-7D: day 1 or day 7 after CFA injection to hindpaws of mice. Error bars indicated SEMs, unpaired t test.
Figure 5. Lack of significant changes in the expression of GAD65 in the VB of inflammatory pain rats.
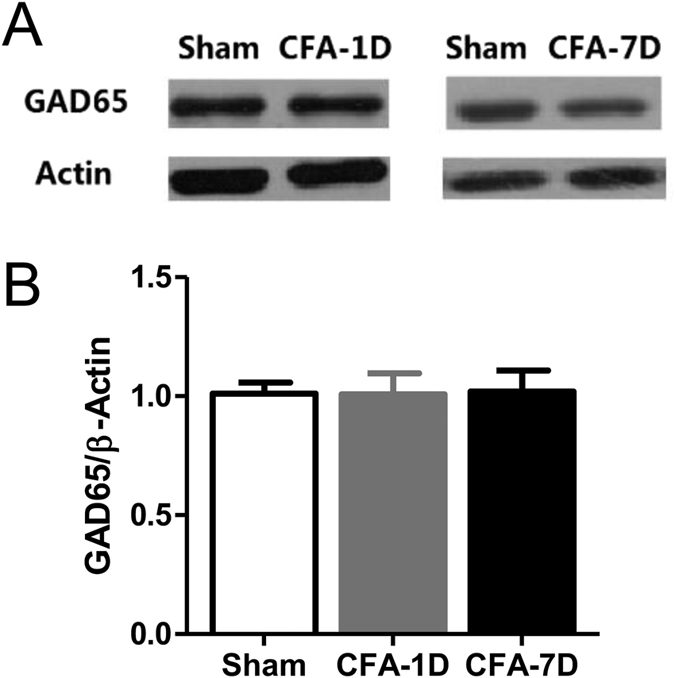
(A) Representative Western blotting bands of GAD65 (~65 KD). β-actin was used as the reference. (B) Statistics of relative quantification of GAD65 in the control, CFA-1D (day 1 after CFA injection) and CFA-7D (day 7 after CFA injection) groups. Error bars indicated SEMs, unpaired t test.
Overall, the above results indicate altered GABAergic transmission in the VB in rats with chronic inflammatory pain. However, it is not known whether these changes exacerbate or alleviate chronic inflammatory pain. We next performed a series of pharmacological, transgenic and optogenetic manipulations to rescue or simulate the changes in thalamic GABAergic transmission, and examined their effects on nociceptive behaviors.
Activation of GABAARs with muscimol in the VB: facilitation of physiological and acute inflammatory pain but attenuation of chronic inflammatory pain
We first examined the effects of intra-VB injection of the GABAAR agonist muscimol on thermal nociceptive thresholds in normal and inflammatory rats (Fig. 6A). As a control, intra-VB injection of vehicle (normal saline) did not affect the paw withdrawal latency (PWL) in naïve (Fig. 6B, 13.47 ± 0.57 s vs. 13.61 ± 0.54 s, p = 0.889) or inflammatory rats (CFA-7D: 6.31 ± 0.78 s vs. 6.10 ± 0.38 s, p = 0.449; CFA-14D: 9.42 ± 0.99 s vs. 8.14 ± 0.54 s, p = 0.667). Intra-VB injection of muscimol significantly reduced PWLs in naïve (13.71 ± 0.35 s vs. 10.29 ± 0.48 s, p < 0.001) and acute inflammatory pain rats (CFA-1D: 3.2 ± 0.21 s vs. 2.57 ± 0.08 s, p = 0.028), indicating an algesic effect of thalamic GABAergic transmission under physiological and acute inflammatory pain conditions. By contrast, intra-VB injection of muscimol produced significant relief of the thermal hyperalgesia in CFA-induced chronic inflammatory pain rats (CFA-7D: 6.35 ± 0.59 s vs. 8.35 ± 0.53 s, p = 0.043; CFA-14D: 8.74 ± 0.70 s vs. 11.56 ± 0.66 s, p = 0.003).
Figure 6. Effects of VB injection of muscimol and THIP on paw withdrawal latencies (PWLs).

Histological illustration of microinjection sites at Bregma −2.8 mm. (B) Vehicle (normal saline) injection did not affect paw withdrawal latencies (PWLs). (C) Muscimol decreased PWLs in naïve and acute inflammatory pain rats (CFA-1D), but increased PWLs in chronic inflammatory pain rats (CFA-7D and CFA-14D). (D) Activation of extra-synaptic GABAARs with THIP had limited effects on PWLs in naïve or acute inflammatory pain rats (CFA-1D), but significantly attenuated thermal hyperalgesia in chronic inflammatory pain rats (CFA-7D and 14D). Error bars indicated SEMs, n = 10–12, *P < 0.05, **P < 0.01, ***P < 0.001, paired t tests.
These results indicate dual effects of thalamic GABAAR activation on nociceptive behaviors: it facilitates physiological and acute inflammatory pain, but attenuates thermal hyperalgesia in chronic inflammatory pain.
Activation of extra-synaptic GABAARs with THIP: facilitation on chronic inflammatory pain
Muscimol activates both synaptic and extra-synaptic GABAARs, which have distinct electrophysiological dynamics in controlling neuronal activities23. THIP is a preferential agonist for delta subunits, the most abundantly expressed extra-synaptic subunit of GABAARs in the VB24,25,26. Intra-VB injection of THIP did not affect PWLs in naïve (14.11 ± 0.83 s vs. 13.35 ± 0.76 s, p = 0.504) or acute inflammatory pain rats (CFA-1D: 3.19 ± 0.26 s vs. 3.53 ± 022 s, p = 0.132), but significantly reduced PWLs in chronic inflammatory pain rats (CFA-7D: 9.32 ± 0.30 s vs. 6.98 ± 0.43 s, p = 0.0003; CFA-14D: 10.01 ± 0.70 s vs. 7.69 ± 0.61 s, p = 0.0037, Fig. 6D). These results suggest that selective activation of extra-synaptic GABAARs with THIP facilitates thermal hyperalgesia in CFA-induced chronic inflammatory pain.
Overall, the different effects of muscimol and THIP injections indicate distinct roles of synaptic and ambient thalamic GABA in thermal hyperalgesia in chronic inflammatory pain, with the former exerting analgesic and the latter algesic effects.
Optogenetic activation of GABAergic transmission in the VB: attenuation of CFA-induced chronic inflammatory pain
The currents of synaptic GABAARs are mainly mediated by presynaptic GABA release from vesicles. We next applied optogenetics to examine the hypothesized protective role of synaptic GABAergic transmission in chronic inflammatory pain. The majority of GABAergic innervations to the VB come from the TRN in rodents5. To mimic the phasic inhibition mediated by synaptic GABAARs, optical fibers were implanted in the VB to activate ChR2-expressing TRN terminals in VGAT-ChR2-EYFP transgenic mice (Fig. 7A). Effective illumination parameters were first determined using in vitro whole-cell patch-clamp recordings. 20 ms pulses of a 465 nm blue light with an intensity of 0.5 or 1.0 mW induced IPSCs (Fig. 7B).
Figure 7. Optogenetic activation of GABAergic transmission in the VB attenuated CFA-induced thermal hyperalgesia.

(A) Experimental configuration showing the location of optical fibers. Light was delivered to ChR2-expressing TRN terminals in the VB of VGAT-ChR2 mice with evoked IPSCs recorded from a thalamic relay neuron. (B) In vitro whole-cell patch clamp recordings of evoked IPSCs with 10 Hz, 465 nm blue light. IPSCs were successfully evoked with 0.5 mW and 1.0 mW stimulation and inhibited by 25 μm gabazine. (C) Activation of contralateral thalamic GABAergic neurons with 465 nm blue light stimulation decreased paw withdrawal latencies (PWLs) in naïve mice but attenuated CFA-induced thermal hyperalgesia in VGAT-ChR2 mice (ChR+). Photo-stimuli did not affect mice without ChR2 protein (ChR−). Error bars indicated SEMs, *P < 0.05, **P < 0.01, paired t test. ChR: Channelrhodopsin-2; Off: light off; On: light on.
Previous studies have shown a 10–20 Hz spontaneous discharge rate of TRN neurons in freely-moving rodents27,28. IPSCs were effectively evoked by 10 Hz trains (Fig. 7B). At 20 Hz and 50 Hz, however, IPSCs could only be evoked by the first couple of photo-stimuli, but not the following stimuli (data not shown), possibly due to the desensitization of ChR2 currents29. An activation power of 3.0 mW could activate a much broader VB response30, prompting us to observe nociceptive behaviors of VGAT-ChR2 mice under 10 Hz and 3.0 mW blue light (465 nm) stimulations in the following experiments.
Activation of contralateral thalamic TRN terminals in the VB facilitated nociceptive behaviors in naïve mice (Fig. 7C, baseline, ChR2(−): 12.81 ± 0.77 s vs. 12.81 ± 0.52 s, p = 0.970; ChR2(+): 11.62 ± 0.62 s vs. 9.21 ± 0.61 s, p = 0.028, light off vs. light on), similar to the data of muscimol injection. In inflammatory mice, by contrast, activation of contralateral thalamic TRN terminals attenuated the thermal hyperalgesia (CFA-7D, ChR2(−): 6.99 ± 1.24 s vs. 7.99 ± 3.06 s, p = 0.354; ChR2(+): 6.24 ± 1.37 s vs. 10.77 ± 1.80 s, p = 0.003). These data confirm the analgesic effects of synaptic GABAergic transmission in the VB in chronic inflammatory pain.
Selective knockout of synaptic GABAAR gamma2 subunits in the VB: delay in the recovery of CFA-induced chronic inflammatory pain
The above findings led to the assumption that inhibiting synaptic GABAergic transmission would delay the recovery of inflammatory pain. To test this hypothesis, we examined CFA-induced inflammatory pain in mice with selective knockout of VB GABAAR gamma2 subunits, the most abundant synaptic subunits in the thalamus7,9. Immunofluorescence staining and Western blotting confirmed the knockout of GABAAR gamma2 subunits in the VB by injection of AAV-Cre virus into Gabrg2tm2Lusc/J mice (Fig. 8A–F).
Figure 8. Selective knockout of synaptic GABAAR gamma2 subunits in the VB delayed the recovery of CFA-induced chronic inflammatory pain.

(A–C) GABAAR gamma2 subunit knockout using the Cre-LoxP system. Immunofluorescent double staining of GABAAR gamma2 subunits (red) with AAV-Cre vector (green) into the VB of Gabrg2tm2Lusc/J mice. Gamma2 subunits were knocked-out in AAV-Cre-transfected thalamic relay cells. (D) and (E) Localization and expression of AAV-Cre/AAV-eGFP in the right VB at Bregma −1.7 mm. (F) GABAAR gamma2 subunit expression in AAV-Cre/AAV-eGFP-injected Gabrg2tm2Lusc/J mice with Western blot. The protein level of gamma2 subunits significantly decreased in AAV-Cre-injected, but not AAV-eGFP-injected mice. (G) Thalamic GABAAR gamma2 subunit knockout delayed the recovery from CFA-induced inflammatory pain (Left paw, CFA-14D and CFA-21D) without affecting contralateral PWLs (H) or acute inflammatory pain (CFA-1D). Error bars indicated SEMs, *P < 0.05, **P < 0.01, two-way ANOVA with Bonferroni post-test. PWL: paw withdrawal latency.
Gamma2 subunit knockout had limited effects on the nociceptive thermal threshold in naïve or acute inflammatory mice (Fig. 8G). In contrast, gamma2 subunit knockout significantly delayed the recovery from chronic inflammatory pain [Fig. 8G, 14 day: 9.34 ± 0.96 s vs. 6.15 ± 0.64 s, F(4, 72) = 2.89, p = 0.028; 21 day: 11.14 ± 0.43 s vs. 8.01 ± 0.68 s, F(4, 72) = 3.65, p < 0.01] during the whole process. These results indicate a protective role of synaptic GABAergic transmission on thermal hyperalgesia in chronic inflammatory pain.
Discussion
Pain pathways share a common projection in the somatosensory thalamic nuclei (VPL/VPM). While the VPM is the area representing the body, both VPL and VPM are subject to GABAergic modulation by conditioning stimulation of hindlimb afferents4. The majority of GABAergic afferents of the VB come from thalamic TRN in rodents, with a small part form the zona incerta and local interneurons5. The TRN is a reservoir of GABAergic interneurons and concerns with most, if not all, functional modalities resulted from its complexity31. The reduction in extracellular GABA levels (Fig. 1) and concomitant decreased tonic inhibition (Fig. 2) thus most possibly result from decreased GABA release from the presynaptic TRN.
We also detected increased frequencies of mIPSCs and increased PPRs (Fig. 3). These two measurements both reflect presynaptic functions but are distinct in their mechanisms: mIPSCs result from spontaneous GABA release from presynaptic vesicles, whereas PPRs reflect release capacity of GABA evoked by activities (action potentials). Spontaneous and activity-dependent releases originate from distinct vesicle pools with limited cross-talk between each other32,33. In addition, the highly regulated nature of evoked neurotransmitter release is in sharp contrast to spontaneous vesicle fusion, which is only loosely regulated by extracellular or intracellular calcium34. Knockout of synaptotagmin 1 or complexins attenuates evoked synchronous neurotransmitter release without affecting spontaneous release35, whereas deletion of synaptobrevin-2/VAMP2 reduces spontaneous fusion rate36. Finally, vesicles in the spontaneously release pool are more likely to refuse spontaneously compared to vesicles released with activity, enabling an increased frequency32. Increased PPRs indicate decreased GABA release capacity driven by activities, consistent with decreased extracellular GABA levels and the fact that repeated noxious stimuli depress neuronal firing in the TRN37. By contrast, spontaneously release events, as measured with mIPSCs, are closely associated with synaptic development and plasticity, including stabilization of synaptic networks38,39. Thus, neuroplastic changes of spontaneous releases may occur in chronic inflammatory pain, which give rise to structural and/or functional alterations of the TRN-VB circuit.
These results support several previous reports. Partial peripheral nerve injury promotes a loss of GABAergic inhibition in the dorsal horn21, whereas neuroimaging studies show a significant reduction of gray matter volumes and blood flow of the TRN in patients with chronic pain13,40. Another recent study in chronic neuropathic pain reports increased oscillatory activities in the thalamus were associated with altered neuron-astrocyte couplings41. Altered GABA signaling by astrocytes leads to local circuit imbalance in some pathological conditions, such as Alzheimer’s disease42 and epilepsy43. Similar mechanisms may also contribute to chronic pain. This hypothesis is also consistent with stronger nociceptive perception in females than males: synaptic GABAARs subunits in the VB are lower in the female44. In addition, GABAergic transmission in the VB could be increased by the high levels of plasma estradiol during the preestrous and estrous phases, which are associated with attenuated pain in the estrous cycle45. With various manipulations, we show that manipulating thalamic GABAergic transmission significantly affects thermal hyperalgesia in chronic inflammatory pain.
In addition to these reflex behaviors, the VB has also been implicated in spontaneous pain behaviors, such as conditioned place preference46. However, we did not include this paradigm in the present study. In addition, although evoked activities of thalamic neurons are preferentially mediated by GABAARs47, some studies have also implicated a significant role of GABABRs48,49. GABAARs and GABABRs could interact with each other though different mechanisms47. Decreased mRNA expression of GABABR1/2 subunits has been reported in the thalamus of chronic monoarthritic animals48,50. But the exact mechanisms of GABABRs’ involvement in chronic pain require further investigation.
Thalamic GABAergic transmission plays different roles in thermal hyperalgesia under acute and chronic inflammatory conditions. One interesting finding in the present study is the distinct effects of various manipulations on physiological or acute thermal pain versus chronic thermal hyperalgesia22. VB injection of muscimol or photo-stimulation of the TRN-VB GABAergic pathway significantly facilitated physiological thermal pain or acute thermal hyperalgesia but inhibited chronic thermal hyperalgesia (Figs 6C and 7C). Similar biphasic effects, which have also been reported in the primary somatosensory cortex51, most likely result from distinct neuronal effects of GABAA receptor activation52, depending on the basal tone of GABAergic transmission and the sensitivity of GABAARs under different conditions53. The polarity of GABAergic currents critically depends on the intracellular chloride concentration and the expression of Na+-K+-2Cl− cotransporters (NKCC1) and K+-Cl− cotransporters (KCC2)54. High expression of NKCC1 and low (or no) expression of KCC2 keep a high intracellular Cl− concentration, which induces depolarization or excitatory responses to GABA. Recent studies indicate that peripheral nerve injury-induced neuropathic pain is associated with decreased KCC2 levels in the thalamus, especially in the chronic phase15. Similar changes might happen in chronic inflammatory pain, which would give a possible interpretation of the opposite effects of optogenetics in acute and chronic pain. In addition to intracellular Cl− concentrations, the excitatory or inhibitory effect of GABA is also affected by the biphasic change in GABAA conductance and shunting inhibition in TRN interneurons52.
One direct way to elucidate the distinct effects of thalamic GABAergic transmission on acute and chronic pain would be in vivo recording of thalamic neurons. Thalamic reticular cells demonstrate electrophysiological diversities: burst firing mode in slow-wave sleep, single spike mode or tonic activities during the waking state28, and more complex firing mode under anaesthesia55. Our previous studies and many others have found that chronic pain is accompanied by altered EEG56,57 or thalamocortical dysrhythmia, where the TRN plays a leading role in the generation of pathological oscillations58. In the present study, optogenetic activation of contralateral thalamic TRN GABAergic interneurons or pharmacological activation of GABAARs with muscimol attenuated CFA-induced chronic inflammatory pain, confirming this hypothesis.
For thalamic relay cells, two main firing modes have been reported: tonic and burst firing. Activation of extra-synaptic GABAARs hyperpolarizes neurons and promotes burst firing, whereas blockage of these receptors promotes tonic-over-burst firing1. However, conflicting evidence is present regarding the relationship between these firing modes and pain behaviors. Several studies correlate the tonic firing of thalamic relay cells to nociceptive pain, and the burst firing to inhibition on nociceptive information processing12,59. Mice with T-type calcium channel knockout, for example, show depleted burst firing in the thalamus and stronger visceral pain responses59. By contrast, positive or lack of correlations between burst firing in the sensory thalamus with pain symptoms have been reported by other studies1,60,61,62. Finally, some researchers suggest that only specific bursting firing in the VB has the anti-nociceptive effect63,64.
Another complexity of findings from the present study lies in the distinct effects of synaptic and ambient GABA, which binds to synaptic and extra-synaptic GABAARs different in their molecular heterogeneity and effects on neuronal excitability. Unlike the dual effects of phasic GABAergic transmission between low and high GABA levels, tonic GABAergic inhibition positively and dose-dependently correlates with the levels of GABA65. In the present study, a decreased tonic inhibition in the VB was observed in chronic inflammatory pain rats (Fig. 2B). Optogenetic activation of the TRN-VB GABAergic pathway produced similar analgesic effects to muscimol injection, which was more sensitive to synaptic than extra-synaptic GABAARs. This conclusion was further confirmed with knockout of γ2 subunits, the most abundant synaptic GABAAR subunit in the thalamus. Injection of THIP, a preferential agonist of the extra-synaptic δ subunit, yielded an opposite, algesic effect in chronic inflammatory pain. These findings suggest that reduced synaptic GABAergic transmission in the VB contributes to thermal hyperalgesia in chronic inflammatory pain, whereas the reduction of tonic inhibition of extra-synaptic GABAARs in the VB might play a protective role. However, it needs to be noted that the distinction between synaptic and extra-synaptic GABAergic transmission could not be dissociated unequivocally. In vivo recording would be required to more directly correlate synaptic and ambient GABAergic transmission with neuronal firing modes and pain behaviors.
Conclusions
In conclusion, the present study supports a hypothesis that chronic inflammatory pain is associated with reduced GABAergic transmission in the VB, which contributes to persistent thermal hyperalgesia. Therapeutic interventions targeting GABAergic transmission in the VB might benefit the relief from chronic inflammatory pain.
Materials and Methods
Animals
Male Sprague-Dawley rats were provided by the Department of Experimental Animal Sciences, Peking University Health Science Center. VGAT-ChR2-EYFP line 8 BAC transgenic mice (https://www.jax.org/strain/014548) and Gabrg2tm2Lusc/J mice (https://www.jax.org/strain/016830) were purchased from Jackson Laboratories. Animals were housed under a 12-hour alternating light/dark cycle with food and water available ad libitum, and handled before behavioral experiments. All experimental procedures were conducted in accordance with the guidelines of the International Association for the Study of Pain and were approved by the Animal Care and Use Committee of Peking University.
CFA model of chronic inflammatory pain and behavioral testing
CFA (100 μl for rats of ~300 g or 50 μl for mice of ~8 weeks, Sigma-Aldrich) was intraplantarly injected into the left hind paw to induce inflammatory pain66. The same volume of normal saline (NS) was injected into the left hind paw as the control group. Paw withdrawal latencies (PWLs) were measured with 40 W (for rats) or 15 W (for mice) radiant light focused on the left plantar surface. A 30 s (for rats) or 20 s (for mice) cut-off time was applied to avoid unnecessary tissue damage. Animals were adapted to the test environment for at least 20 min before tests. PWLs were measured before (as the baseline), and 1, 7, and 14 days after CFA injection. The tests were repeated 3 times with 3–5 min intervals.
Microdialysis analysis of GABA in the VB
Microdialysis of extracellular levels of GABA was performed in rat VB, and GABA in the perfusates was analyzed with high performance liquid chromatography (HPLC) coupled with electrochemical detector (ECD), as described previously67. In brief, the guiding cannula (CMA/11, Harvard Apparatus, Holliston, MA) was implanted into the right VB according the following coordinates: anterior–posterior (AP) 3.6 mm; medial–lateral (ML) 3.6 mm; and dorsal–ventral (DV) 6.5 mm relative to the Bregma. Four or five skull screws were used for securing the guiding cannula to the skull surface with dental acrylic. Before sampling, probes (CMA/11, Harvard Apparatus, Holliston, MA) with 2-mm cuprophane membranes were inserted carefully into contralateral VB overnight for stabilization with a very slow perfusion rate (0.1 μl/min). Previous studies had shown good efficiency (>90%) for detection of neurotransmitters at the 0.1 μl/min perfusion rate29,68,69. O-phthalaldehyde (OPA, Sigma-Aldrich) with sodium sulfite (Sigma-Aldrich) was chosen as the derivate reagent70. Standards were obtained from Sinopharm Chemical Reagent.
Thalamocortical slice preparation and whole-cell patch-clamp recordings
Male Sprague-Dawley rats, weight 200 ± 20 g, were anesthetized with isoflurane (Shenzhen RWD Life Science Co., Ltd.) and perfused transcardially with chilled sucrose-based cutting solution (in mM): 234 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgSO4 and 10 glucose (pH 7.4; 335–340 mOsm). The brain was removed quickly, and somatosensory thalamocortical slices71 were cut at 350 μm with a vibratome (VT1000S, Leica Geosystems, Heidelburg, Germany). These slices were incubated in 37 °C artificial cerebral spinal fluid (aCSF) solution (in mM) consisting of 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2 and 10 glucose (305–310 mOsm), aerated with 95% O2 and 5% CO2 to a final pH of 7.40. After 1 h incubation, slices were transferred carefully to the recording chamber by super-fusing aCSF at the room temperature.
A Multiclamp 700B amplifier and Clampex software (Molecular Device, Sunnyvale, CA) were used for electrophysiological recording. Thalamic relay cells were viewed using a 60× water-immersion lens with infrared differential interference contrast optics. Miniature inhibitory postsynaptic currents (mIPSCs) and tonic currents were recorded at a −70 mV holding potential. Pipettes (about 3–5 MΩ) were filled with an internal solution containing (in mM): 140 CsCl, 10 Hepes, 10 EGTA, 2 MgSO4, 1 CaCl2 and 2 Mg-ATP (pH 7.20–7.30, 290–300 mOsm). Data were filtered at 2 kHz and digitized at 10 kHz. D(−) 2-amino-5-phosphonovaleric acid (AP5, 50 μM), 6-cyano-7-nitroquinoxaline-2.3-dione sodium (CNQX, 20 μM), and tetrodotoxin (TTX, 0.5 μM) were used to block glutamatergic and sodium currents, respectively. All drugs used were purchased from Sigma-Aldrich except TTX (Institute for Aquatic Science and Technology, Qinhuangdao, China). The mIPSCs were analyzed with the Mini-Analysis software (version 6.0.7, Synaptosoft). Amplitudes of mIPSC thresholds were defined at 10 pA. Populations of mIPSCs were verified visually and the average amplitude and frequency were measured. Paired-pulse stimulations were given with 25, 50, 100 and 200 ms intervals using a monoelectrode. Tonic inhibition and paired-pulse ratios (PPRs) were analyzed using Clamp 10.1.
Expression of GABAAR gamma2/delta subunits and GAD65
Contralateral VB from mice with the adeno-associated virus (AAV) vector expression (see below) or from rats 1 or 7 days after CFA injection were collected and immediately stored at −80 °C. Membrane proteins were extracted using the Membrane and Cytosol Protein Extraction Kit (Applygen Technologies Inc., Beijing). Equal amounts (15 μg) of membrane proteins of gamma2/delta subunits and total proteins of GAD65 were separated by SDS–PAGE on 10% gels and transferred to PVDF membranes (Merck Millipore). Membranes were blocked with 5% milk for 30 min at the room temperature and incubated at 4 °C overnight with rabbit anti-GABAAR primary antibodies (for the gamma2 subunit, 1:1000, Novus; for the delta subunit, 1:1000, Merck Millipore) and anti-GAD65 primary antibodies (1:5000, Abcam). The blots were then incubated with HRP-conjugated anti-rabbit secondary antibody (1:2000, Jackson, Bar Harbor, Maine). The bands were detected using an enhanced chemiluminescence detection kit (ECL, Santa Cruz Biotechnology, Inc.). Band intensity was quantified using the software Quantity One 4.6.2 (Bio-Rad Laboratories).
Cannula implantation and microinjection of GABAAR agonists
Rats were anaesthetized deeply with 1% sodium pentobarbital (0.5 ml/100 g, i.p.) and placed in a stereotaxic frame. Guide cannulas (O.D. 0.56 mm/I.D. 0.38 mm, Shenzhen RWD Life Science Co., Ltd., China) were implanted bilaterally into the VB according to the following coordinates relative to the Bregma: AP −3.6 mm; ML −3.6 mm; and DV −6.5 mm. Four or five skull screws were used for securing the guide cannula to the skull surface with dental acrylic. Matching caps (Shenzhen RWD Life Science Co., Ltd., China) were inserted into guide cannulas and checked every day to prevent possible clogging. All animals were given at least one week for recovery before further experiments. The injection needle (Shenzhen RWD Life Science Co., Ltd., China), 0.5 mm lower than the guide cannula, was used for microinjection with a polyethylene catheter connecting a micro-syringe. GABAAR agonists muscimol (0.15 μg/μl, 0.5 μl, Tocris Bioscience) or THIP (4,5,6,7-tetrahydroisoxazolo(5, 4-c) pyridin-3-ol, 50 μM, 0.5 μl, Sigma-Aldrich)51,72 or vehicle (normal saline, 0.5 μl) were injected into the VB on either side over 2 min. The injection needle was held on for at least 1 min to allow for drug diffusion. Behavioral tests were performed 30 min after drug/vehicle injection. Rats with incorrect sites of the guide cannula were excluded from analysis and the final numbers of rats in each group were more than 10.
Adeno-associated virus (AAV) vectors and knockout of the GABAAR gamma2
AAV-mCaMKII-Cre-eGFP (AAV-Cre) and AAV-mCaMKII-eGFP (AAV-eGFP) viruses were packaged and purchased from Sunbio Medical Biotechnology, Shanghai, China. Male Gabrg2tm2Lusc/J mice were microinjected in the VB (AP −1.6 mm; ML ±1.7 mm; and DV −3.5 mm) with 0.3 μl AAV-Cre or AAV-eGFP (as a control) at a speed of 0.1 μl/min after being anesthetized with 1% sodium pentobarbital (0.5 ml/100 g body weight, i.p.). The needle was kept on the site for 2 min to allow for virus diffusion and gradually withdrawn over 2 min to prevent possible leakage from the needle track. Two baseline measurements were performed: one before the injection of AAV virus and the other before CFA injection 3 weeks later. On days 1, 7, 14 and 21, PWLs were measured following the protocol described above. Only mice with correct and effective delivery of AAV virus into the VB were analyzed in behavioral tests.
Immunohistochemistry for the GABAAR gamma2 subunit
Immunostaining and Western blotting (performed according to the protocol described above) were used to verify the knockout of AAV-Cre for GABAAR gamma2 subunits with Gabrg2tm2Lusc/J mice. For immunohistochemistry, thalamic sections (30-μm in thickness) were incubated with the primary antibody (a rabbit anti-GABAAR gamma2, 1:1000, Novus) in 0.5% BSA followed by Alexa Fluor 594-conjugated goat anti-rabbit IgG (1:200, Vitrogen Life Technologies) at 4 °C overnight. Sections were observed with a Leica DMI4000 microscope (Leica, Wetzlar).
Optogenetics
Male VGAT-ChR2-EYFP line 8 (http://jaxmice.jax.org/strain/014548.html) mice (~8 weeks) were used in the optogenetic experiment. Following the same coordinates of the virus injection, the optical fiber (NA 0.37, O.D. 200 μm, Fiblaser Technology Co., Ltd, Shanghai, China) was implanted into the contralateral VB to drive the TRN terminals. Light-evoked IPSCs were recorded following the protocol of mIPSCs. A 465 nm blue light (PlexBright LED, Hong Kong Plexon) was delivered for evoked IPSCs at 0.1–3.0 mW with 10 Hz, 20 Hz or 50 Hz. In animal behavioral tests, in order to stimulate a larger area of the VB, 3.0 mW instead of 1.0 mW, 10 Hz, 465 nm blue light, was chosen to activate axonal terminals from the TRN. Based on predicted irradiance values measured in mammalian brain tissues, the light applied in the present study mainly activated neurons within an extent of no more than 1 mm (http://web.stanford.edu/group/dlab/cgi-bin/graph/chart.php), ensuring the specificity of the stimulation.
Histology
After all experiments, animals were deeply anesthetized and perfused with 4% paraformaldehyde in phosphate buffer. Rats in the microdialysis and drug injection experiments received blue ink injection before perfusion to mark the site of sampling or injection. Thalamic sections (30-μm in thickness) were sliced coronally using a cryostat microtome to identify the tips of microdialysis probes, microinjection needles or optical fibers, according to The Rat/Mouse Brain Stereotaxic Coordinates73,74. Data from animals with incorrect localization excluded from analysis.
Statistical analysis
All data were presented as means ± SEMs. Unpaired or paired student’s t tests were used for the comparisons of two groups. Data analysis of three groups was performed with one-way ANOVA with Tukey post-test. Comparisons of two groups with different time points were preformed using two-way ANOVA with Bonferroni post-test. The differences were calculated by the GraphPad Prism 5.0 and statistical significance was defined as p < 0.05.
Additional Information
How to cite this article: Zhang, C. et al. Reduced GABAergic transmission in the ventrobasal thalamus contributes to thermal hyperalgesia in chronic inflammatory pain. Sci. Rep. 7, 41439; doi: 10.1038/srep41439 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The authors thank Prof. Min-Min Luo and Dr. Zheng-Wei Yuan (National Institute of Biological Sciences, Beijing, China) for the VGAT-ChR2-EYFP mouse line as a gift and in vivo verification of the optogenetic system. This research was supported by: National Basic Research Program of the Ministry of Science and Technology of China (2013CB531905, 2014CB548200 and 2015CB554503). National Natural Science Foundation of China (81230023, 81221002, and 81571067). Key Project of Chinese Ministry of Education (109003). “111” Project of the Ministry of Education of China (B07001).
Footnotes
The authors declare no competing financial interests.
Author Contributions C.Z., M.Y. and Y.W. conceived the experiment, C.Z., R.X.C., J.W., and F.Q.X. conducted the experiment and analyzed the results. Y.Z., F.F.L. and F.Y.L. participated to animal behavioral test. J.C. participated to the whole-cell path-clamp recording. C.Z., M.Y. and Y.W. wrote the main manuscript text, C.Z. prepared all the figures. All authors reviewed the manuscript.
References
- Cope D. W., Hughes S. W. & Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosc. 25, 11553–63 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery R. W., Feig S. L. & Lozsadi D. A. Paying attention to the thalamic reticular nucleus. Trends Neurosci 21, 28–32 (1998). [DOI] [PubMed] [Google Scholar]
- Halassa M. M. et al. State-dependent architecture of thalamic reticular subnetworks. Cell 158, 808–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts W. A., Eaton S. A. & Salt T. E. Widely distributed GABA-mediated afferent inhibition processes within the ventrobasal thalamus of rat and their possible relevance to pathological pain states and somatotopic plasticity. Exp Brain Res 89, 363–72 (1992). [DOI] [PubMed] [Google Scholar]
- Steriade M. The GABAergic reticular nucleus: a preferential target of corticothalamic projections. Proc Natl Acad Sci 98, 3625–7 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M. & Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci 6, 215–29 (2005). [DOI] [PubMed] [Google Scholar]
- Krosigk M., Bal T. & McCormick D. A. Cellular mechanisms of a synchronized oscillation in the thalamus. Science 261, 361–4 (1993). [DOI] [PubMed] [Google Scholar]
- Braat S. & Kooy R. F. The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86, 1119–30 (2015). [DOI] [PubMed] [Google Scholar]
- Pirker S., Schwarzer C., Wieselthaler A., Sieghart W. & Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815–50 (2000). [DOI] [PubMed] [Google Scholar]
- Hortnagl H. et al. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 236, 345–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley S. G., Revilla V., Cull-Candy S. G., Wisden W. & Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature 409, 88–92 (2001). [DOI] [PubMed] [Google Scholar]
- Cheong E. et al. Tuning thalamic firing modes via simultaneous modulation of T- and L-type Ca2+ channels controls pain sensory gating in the thalamus. J Neurosci 28, 13331–40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson L. A. et al. Chronic pain: lost inhibition? J Neurosci 33, 7574–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc B. W., Lii T. R., Silverman A. E., Alleyne R. T. & Saab C. Y. Cortical theta is increased while thalamocortical coherence is decreased in rat models of acute and chronic pain. Pain 155, 773–82 (2014). [DOI] [PubMed] [Google Scholar]
- Modol L., Cobianchi S. & Navarro X. Prevention of NKCC1 phosphorylation avoids downregulation of KCC2 in central sensory pathways and reduces neuropathic pain after peripheral nerve injury. Pain 155, 1577–90 (2014). [DOI] [PubMed] [Google Scholar]
- Walton K. D., Dubois M. & Llinas R. R. Abnormal thalamocortical activity in patients with Complex Regional Pain Syndrome (CRPS) type I. Pain 150, 41–51 (2010). [DOI] [PubMed] [Google Scholar]
- Borgkvist A. et al. Loss of striatonigral GABAergic presynaptic inhibition enables motor sensitization in Parkinsonian mice. Neuron 87, 976–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P., Hestrin S. & Nicoll R. A. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246, 815–8 (1989). [DOI] [PubMed] [Google Scholar]
- Zhang Z. & Pan Z. Z. Synaptic mechanism for functional synergism between δ- and μ-opioid receptors. J Neurosci 30, 4735–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Cai Y. Q., Zou F., Bie B. & Pan Z. Z. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med 17, 1448–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K. A. et al. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22, 6724–31 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M., Wu G. & Wu L. J. Neuronal and microglial mechanisms of neuropathic pain. Mol Brain 4, 31–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M., Ebert B., Wafford K. & Smart T. G. Distinct activities of GABA agonists at synaptic- and extrasynaptic-type GABAA receptors. J Physiol 588, 1251–68 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N., Kerby J., Bonnert T. P., Whiting P. J. & Wafford K. A. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol 136, 965–74 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd M. B. et al. Inhibition of thalamic excitability by 4,5,6,7-tetrahydroisoxazolo (4,5-c)pyridine-3-ol: a selective role for δ-GABAA receptors. Eur J Neurosci 29, 1177–87 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storustovu S. I. & Ebert B. Pharmacological characterization of agonists at δ-containing GABAA receptors: Functional selectivity for extrasynaptic receptors is dependent on the absence of γ2. J Pharmacol Exp Ther 316, 1351–9 (2006). [DOI] [PubMed] [Google Scholar]
- Gardner R. J., Hughes S. W. & Jones M. W. Differential spike timing and phase dynamics of reticular thalamic and prefrontal cortical neuronal populations during sleep spindles. J Neurosci 33, 18469–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks G. A. & Roffwarg H. P. Spontaneous activity in the thalamic reticular nucleus during the sleep/wake cycle of the freely-moving rat. Brain Res 623, 241–8 (1993). [DOI] [PubMed] [Google Scholar]
- Lin J. Y., Lin M. Z., Steinbach P. & Tsien R. Y. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J 96, 1803–14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay A., Thomas S., Lesage F. & Casanova C. Repetitive and retinotopically restricted activation of the dorsal lateral geniculate nucleus with optogenetics. PLoS One 9, 94633–41 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Rev 46, 1–31 (2004). [DOI] [PubMed] [Google Scholar]
- Sara Y. et al. An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45, 563–73 (2005). [DOI] [PubMed] [Google Scholar]
- Zucker R. S. Minis: whence and wherefore? Neuron 45, 482–4 (2005). [DOI] [PubMed] [Google Scholar]
- Angleson J. K. & Betz W. J. Intraterminal Ca2+ and spontaneous transmitter release at the frog neuromuscular junction. J Neurophysiol 85, 287–294 (2001). [DOI] [PubMed] [Google Scholar]
- Geppert M. et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727 (1994). [DOI] [PubMed] [Google Scholar]
- Schoch S. et al. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294, 1117–1122 (2001). [DOI] [PubMed] [Google Scholar]
- Peschanski M., Guilbaud G. & Gautron M. Neuronal responses to cutaneous electrical and noxious mechanical stimuli in the nucleus reticularis thalami of the rat. Neurosci Lett 20, 65–70 (1980). [DOI] [PubMed] [Google Scholar]
- Verhage M. et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287, 864–869 (2000). [DOI] [PubMed] [Google Scholar]
- McKinney et al. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci 2, 44–49 (1999). [DOI] [PubMed] [Google Scholar]
- Gustin S. M. et al. Thalamic activity and biochemical changes in individuals with neuropathic pain after spinal injury. Pain 155, 1027–1036 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshelh Z. et al. Chronic Neuropathic pain: it’s about the rhythm. J Neurosci 36, 1008–18 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn L. M., Kamphuis W., Wadman W. J. & Hol E. M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol 144, 121–41 (2016). [DOI] [PubMed] [Google Scholar]
- Robel S. & Sontheimer H. Glia as drivers of abnormal neuronal activity. Nat Neurosci 19, 28–33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Huguenard J. R. & Fisher R. S. Gender and age differences in expression of GABAA receptor subunits in rat somatosensory thalamus and cortex in an absence epilepsy model. Neurobiol Dis 25, 623–30 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umorin M., Stinson C., Bellinger L. L. & Kramer P. R. Genes in the GABA pathway increase in the lateral thalamus of Sprague-Dawley rats during the proestrus/estrus phase. J Cell Physiol 231, 1057–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T. et al. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci 12, 1364–1366 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A., Hoffman K. & Keller A. Roles of GABAA and GABAB receptors in regulating thalamic activity by the zona incerta: a computational study. J Neurophysiol 112, 2580–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-Gomes J., Neto F. L. & Castro-Lopes J. M. GABAB2 receptor subunit mRNA decreases in the thalamus of monoarthritic animals. Brain Res Bull 71, 252–8 (2006). [DOI] [PubMed] [Google Scholar]
- Martins I. et al. GABA acting on GABAB receptors located in a medullary pain facilitatory area enhances nociceptive behaviors evoked by intraplantar formalin injection. Pain 156, 1555–65 (2015). [DOI] [PubMed] [Google Scholar]
- Ferreira-Gomes J., Neto F. L. & Castro-Lopes J. M. Differential expression of GABAB(1b) receptor mRNA in the thalamus of normal and monoarthritic animals. Biochem Pharmacol 68, 1603–11 (2004). [DOI] [PubMed] [Google Scholar]
- Wang N., Wang J. Y. & Luo F. Corticofugal outputs facilitate acute, but inhibit chronic pain in rats. Pain 142, 108–15 (2009). [DOI] [PubMed] [Google Scholar]
- Song I., Savtchenko L. & Semyanov A. Tonic excitation or inhibition is set by GABAA conductance in hippocampal interneurons. Nat Commun 2, 376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarson K. E. & Enna S. J. GABA pharmacology: the search for analgesics. Neurochem Res 39, 1948–63 (2014). [DOI] [PubMed] [Google Scholar]
- Payne J. A., Rivera C., Voipio J. & Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci 26, 199–206 (2003). [DOI] [PubMed] [Google Scholar]
- Pinault D. & Deschenes M. Voltage-dependent 40-Hz oscillations in rat reticular thalamic neurons in vivo. Neuroscience 51, 245–58 (1992). [DOI] [PubMed] [Google Scholar]
- Liao F. et al. Characterizing heat-sensitization responses in suspended moxibustion with high-density EEG. Pain Med 15, 1272–81 (2014). [DOI] [PubMed] [Google Scholar]
- Wang J. et al. Cortical activities of heat-sensitization responses in suspended moxibustion: an EEG source analysis with sLORETA. Cogn Neurodyn 9, 581–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning P. J., Jeanmonod D. & Verschure P. F. A computational model of thalamocortical dysrhythmia. Eur J Neurosci 33, 1281–1290 (2011). [DOI] [PubMed] [Google Scholar]
- Kim D. et al. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science 302, 117–9 (2003). [DOI] [PubMed] [Google Scholar]
- Hains B. C. Saab C. Y. & Waxman S. G. Alterations in burst firing of thalamic VPL neurons and reversal by Na(v)1.3 antisense after spinal cord injury. J Neurophysiol 95, 3343–52 (2006). [DOI] [PubMed] [Google Scholar]
- Lenz F. A., Kwan H. C., Dostrovsky J. O. & Tasker R. R. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res 496, 357–60 (1989). [DOI] [PubMed] [Google Scholar]
- Wang G. & Thompson S. M. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. J Neurosci 28, 11959–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerina M. et al. Thalamic Kv 7 channels: pharmacological properties and activity control during noxious signal processing. Br J Pharmacol 172, 3126–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh Y. & Cho J. Discrete pattern of burst stimulation in the ventrobasal thalamus for anti-nociception. PLoS One 8, 67655–65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sametsky E. A., Turner J. G., Larsen D., Ling L. & Caspary D. M. Enhanced GABAA-mediated tonic inhibition in auditory thalamus of rats with behavioral evidence of tinnitus. J Neurosci 35, 9369–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X. et al. Shp-1 dephosphorylates TRPV1 in dorsal root ganglion neurons and alleviates CFA-induced inflammatory pain in rats. Pain 156, 597–608 (2015). [DOI] [PubMed] [Google Scholar]
- Rea K., Cremers T. I. & Westerink B. H. HPLC conditions are critical for the detection of GABA by microdialysis. J Neurochem 94, 672–9 (2005). [DOI] [PubMed] [Google Scholar]
- Menacherry S., Hubert W. & Justice J. J. In vivo calibration of microdialysis probes for exogenous compounds. Anal Chem 64, 577–83 (1992). [DOI] [PubMed] [Google Scholar]
- Wang M., Hershey N. D., Mabrouk O. S. & Kennedy R. T. Collection, storage, and electrophoretic analysis of nanoliter microdialysis samples collected from awake animals in vivo. Anal Bioanal Chem 400, 2013–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. L. et al. Changes of some amino acid concentrations in the medial vestibular nucleus of conscious rats following acute hypotension. Neurosci Lett 477, 11–4 (2010). [DOI] [PubMed] [Google Scholar]
- Agmon A. & Connors B. W. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–79 (1991). [DOI] [PubMed] [Google Scholar]
- Monconduit L., Lopez-Avila A., Molat J. L., Chalus M. & Villanueva L. Corticofugal output from the primary somatosensory cortex selectively modulates innocuous and noxious inputs in the rat spinothalamic system. J Neurosci 26, 8441–50 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G. & Watson C. The rat brian in stereotaxic coordinates (ed. Paxinos G. & Watson C.) Figure 27–32 (Elsevier, 1997). [Google Scholar]
- Paxinos G. & Franklin K. B. J. The mouse brian in stereotaxic coordinates (ed. Paxinos G. & Franklin K. B. J.) Figure 42–46 (Acdemic, 2001). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.