

Abstract
Natural products are a major source of biological molecules. The 3‐methylfuran scaffold is found in a variety of plant secondary metabolite chemical elicitors that confer host‐plant resistance against insect pests. Herein, the diversity‐oriented synthesis of a natural‐product‐like library is reported, in which the 3‐methylfuran core is fused in an angular attachment to six common natural product scaffolds—coumarin, chalcone, flavone, flavonol, isoflavone and isoquinolinone. The structural diversity of this library is assessed computationally using cheminformatic analysis. Phenotypic high‐throughput screening of β‐glucuronidase activity uncovers several hits. Further in vivo screening confirms that these hits can induce resistance in rice to nymphs of the brown planthopper Nilaparvata lugens. This work validates the combination of diversity‐oriented synthesis and high‐throughput screening of β‐glucuronidase activity as a strategy for discovering new chemical elicitors.
Keywords: 3-methylbenzofurans, beta-glucuronidase activity, chemical elicitors, diversity-oriented synthesis, insect pests
1. Introduction
Plants possess innate immune responses to deter or overcome pests and attackers.1 These responses, their natural self‐defense mechanisms, are activated by the accumulation of chemical elicitors; these elicitors are generated in plants or are provided by the attackers.2 Because the chemical elicitors themselves are not toxic to pests or herbivores and decompose easily in the environment, a good way to develop safe plant protection agents is based on chemical elicitors.3
Natural products (NPs), which have been optimized over time in a long process of natural selection, are the result of the co‐evolution of plants with their biotic environments. NPs have been and continue to be a major source of the biological molecules from which many drugs and pesticides, as well as elicitors, have been developed.4 Because many NPs are defense molecules against other organisms, NPs are essential sources of new chemical elicitors.5 Therefore, the synthesis and screening of small‐molecule libraries based on NPs or NP‐like structures is a significant means of developing new chemical elicitors.6
Diversity‐oriented synthesis (DOS), which aims to synthesize libraries of diverse small molecules, has proven to be an essential tool for discovering bioactive small molecules.7 The incorporation of privileged molecular scaffolds has become an essential element in DOS pathways. In this work, we used DOS to create NP‐like libraries containing a 3‐methylfuran core.7b, 8
The 3‐methylfuran framework is found in a variety of NPs with diverse biological activities. Of particular interest are menthofuran (monoterpene),9 furanoeremophilane (sesquiterpene),10 cacalol,11 and tanshinone (quinone; Figure 1).12 Moreover, these compounds are plant secondary metabolites, conferring host resistance against plant invaders.9, 10, 11b Therefore, we predict that the 3‐methylfuran scaffold is an important factor in the activity of these molecules, and we expect to find chemical elicitors by synthesizing NP‐like libraries containing the 3‐methylfuran moiety. Furthermore, coumarin,13 chalcone,14 flavone,15 flavonol,16 isoflavone17 and isoquinolinone18 are six naturally occurring compounds that have exhibited a variety of biological activities. Inspired by these NPs and their inherent biological activities, we designed a divergent synthetic pathway to synthesize 88 NP‐like compounds based on a key 3‐methylfuran core obtained from available starting material cyclohexane‐1,3‐diketone (Scheme 1).13b, 19 These compounds have been applied in a phenotypic high‐throughput screening of β‐glucuronidase (GUS)20 and several new hits have been discovered. As chemical elicitors, these hits can induce resistance in rice to nymphs of the brown planthopper (BPH) Nilaparvata lugens, one of the most important rice insect pests in Asia.
Figure 1.
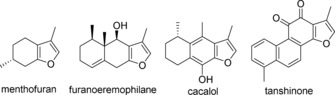
Bioactive natural products that contain a 3‐methylfuran core.
Scheme 1.
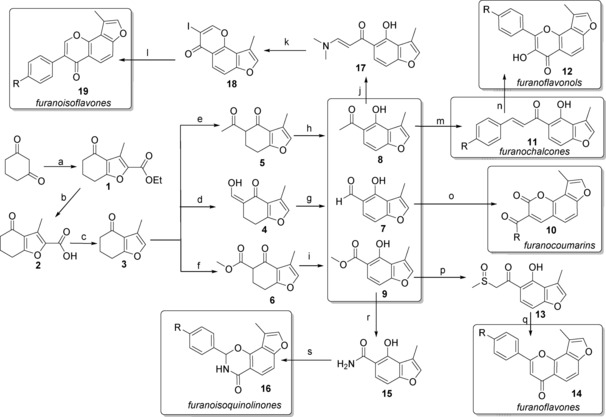
Diversity‐oriented synthesis of 88 compounds based on six natural product frameworks. Reagents and conditions: a) KOH, ethyl 2‐chloroacetoacetate, H2O/MeOH (6:1), RT, 5 days, ≈65 %; b) KOH, H2O/MeOH (1:2.5), RT, 6 h, >90 %; c) Cu, pyridine, DEG, 175 °C, 10 h, ≈85 %; d) NaH, ethyl formate, PhMe, 0 °C–RT, 10 h; e) NaH, ethyl acetate, DME, 0–90 °C, 4.5 h; f) NaH, dimethyl carbonate, DME, 0–90 °C, 4.5 h; g) DDQ, PhMe, 120 °C, 6 h, ≈75 % (d+g); h) DDQ, PhMe, 120 °C, 6 h, ≈70 % (e+h); i) DDQ, PhMe, 120 °C, 6 h, ≈75 % (f+i); j) DMF‐DMA, DMF, 75 °C, 4 h, 99 %; k) I2, CHCl3, RT, 15 h, 90 %; l) arylboronic acid, Na2CO3, Pd(OAc)2, PEG 10 000, MeOH, 50 °C, 4 h; m) benzaldehyde, NaH, THF, RT, 2 h; n) 25 % aq NaOH, 30 % aq H2O2, THF/MeOH (3:5), 0 °C–RT, 48 h; o) reactive methylene compound, piperidine, EtOH, 80 °C, 4 h; p) DMSO, NaH, PhMe, 80 °C, 2 h, 95 %; q) benzaldehyde, piperidine, PhMe, 120 °C, 3 h; r) MeOH (saturated with NH3), 65 °C, 24 h, 100 %; s) benzaldehyde, piperidine, PhMe, 120 °C, 12 h. DEG=diethylene glycol; DDQ=2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone; DMF‐DMA=N,N‐dimethylformamide dimethyl acetal; DME=glycol dimethyl ether; DMF=N,N‐dimethylformamide; PEG=polyethylene glycol; THF=tetrahydrofuran; DMSO=dimethylsulfoxide.
2. Results and Discussion
A divergent synthetic pathway to NP‐like libraries containing a 3‐methylfuran moiety is illustrated in Scheme 1. Our divergent synthetic pathway was achieved by identifying 3 as the versatile key substrate that can be transformed into the target compounds.
The compound 3 was prepared in three steps directly from commercially available cyclohexane‐1,3‐diketone and ethyl 2‐chloroacetoacetate. This process involved the cycloaddition of cyclohexane‐1,3‐diketones with ethyl 2‐chloroacetoacetate, followed by the base‐catalyzed dehydration to give the 3‐methyltetrahydrobenzofuran 1 in 65 % yield.21 This method was efficient and gave the product 1 on a 20 g scale. Ester 1 was hydrolyzed to afford 2 in 90 % yield by treatment with KOH in aqueous ethanol at RT. Treatment of 2 with Cu powder in diethylene glycol containing pyridine was heated at 170–175 °C for 10 h to give the product 3 in 85 %.21a After being acidified to pH<1, the excess of pyridine was removed, and the pure 3 could be easily obtained.
Three key intermediates 7, 8, 9 were prepared from cyclohexenone 3 using 2,3‐dichloro‐5,6‐dicyano‐l,4‐benzoquinone (DDQ)‐catalyzed dehydrogenation–aromatization reactions (Scheme 1).19c, 19e This transformation was a key step, because it is difficult to obtain the pure products 7, 8 and 9 by a classical electrophilic aromatic substitution reaction of phenol.19d, 22 Notably, the metal‐free oxidative aromatization of substituted cyclohexenones is an effective route to obtain ortho‐substituted phenols with high regioselectivity. For example, 3 was formylated using an excess of NaH in the presence of ethyl formate in toluene to give 4 followed by the dehydrogenation–aromatization reaction to 7 in an overall 75 % yield.19c The pure compound 7 was obtained by using column chromatography. Compounds 8 and 9 were also obtained under similar conditions.23 In a 1H NMR spectroscopic examination of 7, 8 and 9, the proton of OH in phenol displayed a typical chemical shift of 9.4 ppm due to the deshielding effect of the hydrogen bond with the neighboring C‐12 carbonyl oxygen atom.
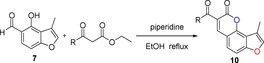
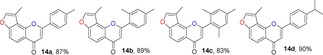
2.1. Synthesis of Angular 3‐Methylfuranocoumarins (10)
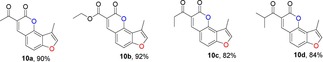
For the synthesis of 3‐methylfuranocoumarins 10 a, we first used l‐proline as the promoter to test the reaction between aldehyde 7 and ethyl acetoacetate, according the procedure reported by Kurt and co‐workers.24 Surprisingly, the reaction failed, probably due to the presence of an acidic hydroxy group in the phenol. After several trials, the use of piperidine13a, 25 as a promoter in ethanol under reflux conditions gave the products 10 a in 90 % yield (Table 1). Furthermore, the general scope of this reaction was tested with different active methylene compounds, and the desired products 10 b–10 h were directly precipitated from solution under identical reaction conditions in good yields (Table 1). This methodology is efficient, operationally simple and environmentally friendly.
Table 1.
Structures and yields[a, b] of angular 3‐methylfuranocoumarins derivatives.
|
|---|
|
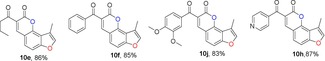
|
[a] All reactions were performed in ethanol at reflux. [b] Yield of isolated product.
2.2. Synthesis of Angular 3‐Methylfuranochalcones (11)
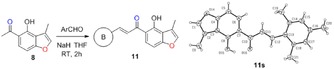
The synthesis of angular 3‐methylfuranochalcones 11 was accomplished from commercially available starting materials in the presence of bases through Claisen–Schmidt condensation (Table 2).14 First, we investigated the effect of bases on the reactions of 8 with benzaldehyde. It was found that 5 equivalents of NaOH can hardly catalyze the reaction (yield <5 %). KOH gave 57 % yield of the desired product 11 a. Further screening of other bases afforded a very good yield (82 %) of 11 a, and NaH was proved to be optimal for this transformation.26 Furthermore, a series of substituted aldehydes (1 equiv) were allowed to undergo Claisen–Schmidt condensation with 8 (1 equiv) in the presence of NaH (2 equiv) in anhydrous THF at room temperature (Table 2). The products 11 were readily obtained by recrystallization from ethanol after acidifying the solution to pH 1. Reactions involving electron‐withdrawing and electron‐donating substituents on benzene rings all proceeded well to afford good to very good yields of 11 (Table 2). Finally, we attempted to determine the E,Z configurations of 11. The trans (E) configuration of 11 was confirmed by the use of X‐ray structure analysis of a single crystal of 11 s obtained by recrystallization from ethanol.43
Table 2.
Structures and yields[a] of angular 3‐methylfuranochalcones derivatives. The X‐ray crystal structure of 11 s is also shown.
| ||||||
|---|---|---|---|---|---|---|
| Product | B | Yield [%] | Product | B | Yield [%] | |
| 11 a | C6H5 | 82 | 11 o | 2‐ClC6H4 | 83 | |
| 11 b | 4‐MeC6H4 | 85 | 11 p | 2‐BrC6H4 | 81 | |
| 11 c | 4‐iPrC6H4 | 87 | 11 q | 2‐MeOC6H4 | 86 | |
| 11 d | 4‐tBuC6H4 | 84 | 11 r | 2,4‐di‐MeC6H3 | 85 | |
| 11 e | 4‐MeOC6H4 | 88 | 11 s | 2,4,6‐tri‐MeC6H2 | 86 | |
| 11 f | 4‐EtOC6H4 | 85 | 11 t | 3,4,5‐tri‐MeOC6H2 | 87 | |
| 11 g | 4‐FC6H4 | 74 | 11 u | 2,4‐di‐ClC6H3 | 80 | |
| 11 h | 4‐ClC6H4 | 81 | 11 v | 2‐Br‐4‐FC6H3 | 82 | |
| 11 i | 4‐BrC6H4 | 76 | 11 w | 3‐ClC6H4 | 81 | |
| 11 j | 4‐(Me)2NC6H4 | 86 | 11 x | 3‐BrC6H4 | 80 | |
| 11 k | 4‐CNC6H4 | 73 | 11 y | 3‐MeC6H4 | 84 | |
| 11 l | 4‐(Et)2NC6H4 | 88 | 11 z | 3‐MeOC6H4 | 86 | |
| 11 m | 4‐NO2C6H4 | 81 | 11 aa | 2‐furaldehyde | 77 | |
| 11 n | 2‐FC6H4 | 73 | 11 ab | 2‐thenaldehyde | 79 | |
[a] Yield of isolated product.
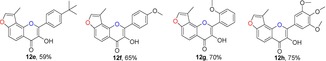
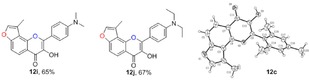
2.3. Synthesis of Angular 3‐Methylfuranoflavonols (12)
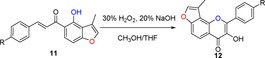
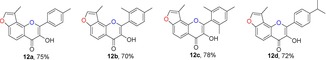
From a survey of the literature, we found that flavonol derivatives can be achieved by the epoxidation of corresponding flavone compounds27 or by the Baker–Venkataraman type of sequential cyclization‐dehydration reaction.28 Another Algar–Flynn–Oyamada reaction has also been widely used to make flavonols.16, 28b We chose this reaction for the transformation of 3‐methylfuranochalcones 11 to angular 3‐methylfuranoflavonols 12 in the presence of NaOH (2 equiv) and H2O2 (2.2 equiv). In this procedure, the use of THF and methanol as co‐solvents instead of methanol alone gave the best results, because of the poor solubility of these corresponding chalcones 11 in methanol (Table 3). The electron‐donating substituents on benzene rings were compatible.22a The product was purified by recrystallization from ethanol to afford the desired products 12. Single crystals of 12 c were obtained by recrystallization from petroleum ether/CH2Cl2, and the configurations of the products were unambiguously determined by X‐ray analysis.43
Table 3.
Structures and yields[a] of angular 3‐methylfuranoflavonol derivatives. The X‐ray crystal structure of 12 c is also shown.
|
|---|
|
|
|
[a] Yields of isolated products.
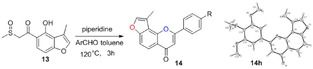
2.4. Synthesis of Angular 3‐Methylfuranoflavones (14)
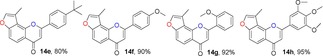
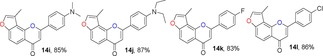
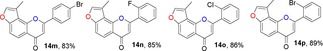
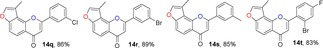
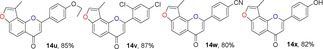
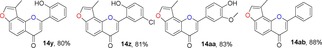
Many molecules of biological importance contain a 4‐pyrone moiety,19e and various methods are available for constructing this unit. These transformations have involved the Baker–Venkataraman rearrangement,29 Allan–Robinson reaction,30 and the intramolecular cyclization of 2‐hydroxychalcones.22a We tried to use the Von Strandtmann approach for constructing 4‐pyrone rings.22b, 31 The 3‐methylbenzofuran 9 was treated with the dimsyl anion in DMSO to form the β‐ketosulfoxide 13, which after treatment with benzaldehyde derivatives and piperidine in anhydrous toluene, first at 40 °C and then at 110 °C, produced the corresponding 3‐methylfuranoflavones 14 (Table 4). After removal of the solvent under reduced pressure, the residue was added to EtOH and stirred. The resulting precipitate was filtered, then recrystallized from EtOH to readily afford products 14. With the use of 13 as a highly activated methylene compound, both electron‐withdrawing and electron‐donating substituents were introduced into the aryl aldehyde to study the effects of the electronic and steric properties on reactivity. We found that all the reactions proceeded well to afford the 3‐methylfuranoflavones 14 in good yields ranging from 80 % to 95 % (Table 4). The identity of product 14 h was unambiguously determined by X‐ray analysis.43
Table 4.
Structures and yields[a] of angular 3‐methylfuranoflavone derivatives. The X‐ray crystal structure of 14 h is also shown.
|
|---|
|
|
|
|
|
|
|
[a] Yields of isolated products.
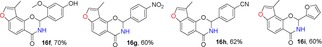
2.5. Synthesis of Angular 3‐Methylfuranoisoquinolinones (16)
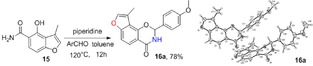
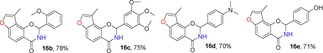
For the synthesis of 3‐methylfuranoisoquinolinones 16, we first synthesized amide intermediate 15 by treating ester 9 with a methanol solution saturated with ammonia in a Fisher–Porter bottle with 100 % conversion. Excess ammonia was removed under vacuum at 40 °C. The resulting intermediate 15 was allowed to undergo cyclization with a series of substituted aldehydes using piperidine as a catalyst at 110 °C to afford the products 16.18, 32 The crude products were readily purified by column chromatography in moderate to good yields (60–80 %) (Table 5). The structure of 3‐methylfuranoisoquinolinone 16 a was determined by X‐ray analysis.43
Table 5.
Structures and yields[a] of angular 3‐methylfuranoisoquinolinone derivatives. The X‐ray crystal structure of 16 a is also shown.
|
|---|
|
|
[a] Yields of isolated products.
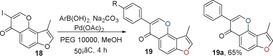
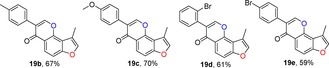
2.6. Synthesis of Angular 3‐Methylfuranoisoflavones (19)
Finally, the synthesis of 3‐methylfuranoisoflavones 19 was accomplished by the introduction of the key intermediate 18 in two steps. The first step was to treat intermediate 8 with N,N‐dimethylformamide dimethyl acetal (DMF‐DMA) to form the corresponding enamine 17. In the second step, this crude product 17 was filtered and directly cyclized to 3‐iodochromone 18 using iodine in CHCl3 17, 33 without further purification (Scheme 1). We were satisfied with the cross‐coupling of 3‐iodochromone 18 with 2 equivalents of substituted phenylboronic acids using polyethylene glycol 10 000 (PEG 10 0000) as a ligand in the presence of Pd(OAc)2 and Na2CO3 in MeOH to obtain 3‐methylfuranoisoflavones 19 in generally moderate to high yields (Table 6).16, 34
Table 6.
Structures and yields[a] of 3‐methylfuranoisoflavone derivatives.
|
|---|
|
[a] Yields of isolated products.
2.7. Chemoinformatic Analysis
To computationally assess the structural diversity of our 3‐methylfuran library, the structural features were analyzed in terms of their chemical properties by using principal component analysis (PCA).35 PCA computes the position of each compound in a two‐ or three‐dimensional coordinate system based on a set of molecular properties, such as physicochemical properties, to simplify the comparison with different sets of compounds.8a, 8b, 36 Using PCA, the chemical properties of our 3‐methylfuran library were compared with reference sets of 40 top‐selling brand name drugs used by Tan8b, 37 (Table S6) and 20 coumarin and flavonoid natural products (Table S7). Twenty physicochemical properties (Table S3) of these compounds were analyzed using a public, web‐based tool.38 These properties represent each compound as a vector in 20‐dimensional space. The 20‐dimensional vector can be reduced to two‐dimensional vectors by an orthogonal transformation and plotted as a scatter plot (Figure 2 A–C and Table S5). The first three principal components captured 80.7 % of the dataset variance (Table S4). As seen in Figure 3 a, our library accesses the chemical space occupied by top‐selling drugs considerably and overlapped with the space of coumarins and flavonoid natural products. This indicated the potential drug‐like properties of our library and possibly implied that these compounds overlapped with biologically relevant areas of the chemical space. Examination of the component loadings (Table S5) indicated that the major contributions of principal component one (PC1) are the number of all atoms, molecular surface areas and solvent‐accessible surface areas. The lipophilicity (log P), topological polarity surface area and relative hydrophobic surface area are the key factors associated with principal component two (PC2). The principal component three (PC3) is greatly influenced by relative negatively and positively charge surface areas. It is worth noting that in Figure 2 B and C, our library is even more distinct from the range of drug structures compared with the analysis in Figure 2 A. These suggest that the relative negatively and positively charged surface areas are two important properties that differentiate our libraries from the drug library in Figure 2 B and C. The understanding of such analyses might be advantageous for further structural optimization and provides some insight into the planning of future libraries.
Figure 2.
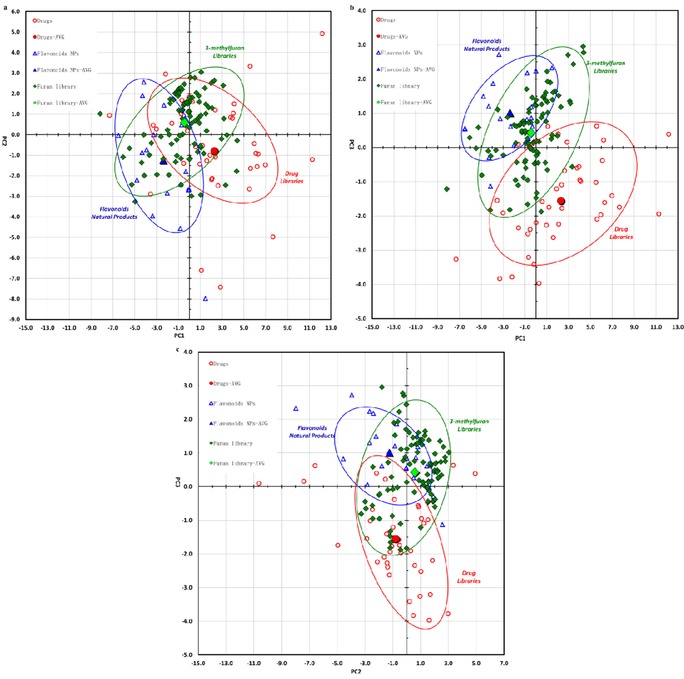
Comparative PCA plots. Ninety‐nine DOS library compounds (green), 40 top‐selling brand‐name drugs (red) and 20 coumarins and flavonoid natural products (blue) were used; a) PC1 versus PC2; b) PC1 versus PC3; c) PC2 versus PC3.
Figure 3.
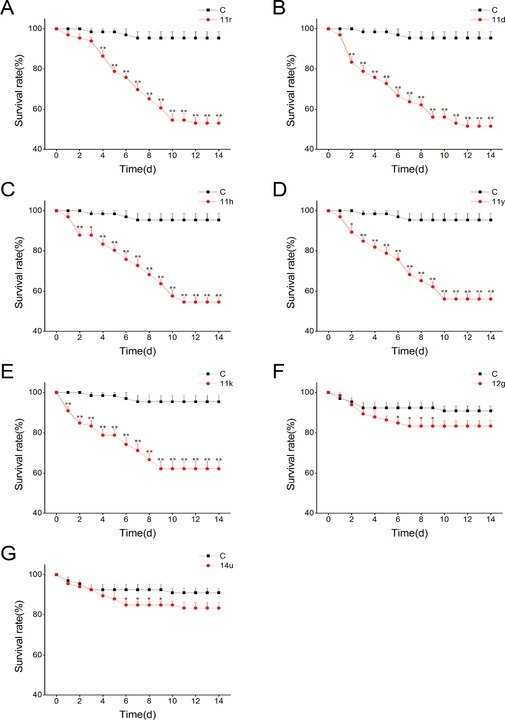
Mean survival rate of N. lugens nymphs (+SE, n=6) on plants, that had been treated with one of the compounds, at a concentration of 20 mg L−1 24 h before exposure versus control plants (C), 1–14 d after exposure. Asterisks indicate significant differences between treatments and controls at each time point (*P<0.05, **P<0.01, Student's t‐tests).
2.8. Screening of Candidate Elicitors from Synthesized Compounds
To discover new candidate elicitors,39 we used a high‐throughput screening system that we had previously established to evaluate these synthesized chemicals for their potential to induce GUS activity.20, 40 Table S2 summarizes the relative induction of GUS activity (comparing GUS activities in roots of plants that were grown in nutrient solution with that of compounds tested at a concentration of 5 mg L−1 versus GUS activities in roots of control plants) in rice plants after these were treated with each chemical. The higher the value, the higher the potential of the compound to induce GUS activity; also, the compound used might have high potential to elicit plant defenses. To our delight, 23 compounds were found to induce an increase in GUS activity (Table S2). Notably, compounds 3 (1.68**), 11 h (1.53**), 11 a (1.55**), 11 d (1.83**) 11 r (2.11**), 11 y (1.80**) and 11 k (1.51**) exhibited strong potential for the ability to induce GUS activity. Moreover, most of the 3‐methylfuranochalcones demonstrated moderate to good potential to induce GUS activity, and 3‐methylfuranochalcones derivatives showed the most potent activities of these compounds. Thus, 3‐methylfuranochalcones derivatives might be used as potential lead compounds to develop novel chemical elicitors.
2.9. Effects of Candidate Elicitors on the Survival Rates of the BPH
To investigate whether the candidate elicitors can provoke plant defenses and thus influence herbivore behavior, we chose the most promising compounds, including 3‐methylfuranochalcones (11 d, 11 h, 11 k, 11 r, and 11 y), a 3‐methylfuranoflavonol (12 g) and a 3‐methylfuranoflavone (14 u), to evaluate their effects on the survival rate of BPH nymphs. Results showed that compared to those feeding on control plants, the survival rates of BPH nymphs were significantly lower on plants whose roots had been treated with 3‐methylfuranochalcones compounds—such treatment caused 40–50 % mortality rate of BPH nymphs at 10–14 days after the release of the herbivore (Figure 3 A–E), whereas the survival rates on plants treated with compound 12 g or 14 u were only slightly lower than those on control plants (Figure 3 F and G). Moreover, the tested compounds themselves did not result in the death of the herbivore: the survival rates of BPH nymphs that fed on an artificial diet containing the test compounds did not differ from those of nymphs that fed on the artificial diet only (Figure S1); similarly, the survival rates of BPH nymphs that were sprayed with one of these chemicals had no difference with those of the control groups (Figure S2). In addition, the survival rates of BPH nymphs on plants treated with 3‐methylfuranochalcone compounds decreased continuously until 10–11 days after herbivore release, when the nymphs were in their third and fourth instars (Figure 3). These data suggest that the resistance of the candidate elicitor‐treated rice plant to the herbivore derives from the defense responses induced in the plant by these chemicals, not from the chemicals themselves, and that the elicited resistance is also effective against third–fourth instar BPH nymphs. Therefore, 3‐methylfuranochalcones can be used as possible lead compounds to develop chemical elicitors as safe plant protection agents.
3. Conclusions
We describe a divergent synthetic strategy for discovering chemical elicitors. The aim of DOS is to achieve high levels of skeletal diversity. A collection of 88 NP‐like small molecules with 3‐methylfuran cores was synthesized. Principal component analysis (PCA) was used to assess the structural diversity of our DOS library, and showed our molecules overlapping with drug and NP chemical spaces, indicating the drug‐ and NP‐like structures of our libraries. Phenotypic high‐throughput screening for GUS activity uncovered several hits. Further in vivo screenings confirmed that these elicitors can induce resistance to N. lugens in rice. The present work demonstrates that 3‐methylfuranochalcones can be used as potential lead compounds for developing chemical elicitors. Moreover, the discovery of new chemical elicitors serves to endorse DOS as an approach to discover new classes of biological small molecules.
Experimental Section
Chemical Synthesis
General Procedure for the Synthesis of Compounds 3
1,3‐Cyclohexanedione (20 g, 0.178 mol) was added to a solution of KOH (10 g, 0.178 mol) in water (240 mL). After 5 min, ethyl 2‐chloroacetoacetate (29.35 g, 0.178 mol) in MeOH (60 mL) was added. The mixture was stirred at room temperature for 5 days. After acidification with HCl (4 n, 300 mL), the solution was extracted with ethyl acetate (3×250 mL). The combined extracts were washed with brine (300 mL) and dried (Na2SO4), and the solvent was evaporated under vacuum to give the furan ester 1 (25.7 g, 65 %). Without further purification, compound 1 (20 g, 0.09 mol) was dissolved in MeOH (84 mL) and water (32 mL) and treated with KOH (35.4 g, 0.56 mol). After stirring overnight at RT, the reaction mixture was diluted with water (160 mL), acidified with HCl (6 n), and extracted with ethyl acetate (3×250 mL). The extract was washed with brine (250 mL), dried (Na2SO4), and the solvent was evaporated under vacuum to give the furan acid 2 (15.73 g, 90 %). Without further purification, the 2 (10 g, 0.052 mol) in diethylene glycol (80 mL) was treated with Cu powder (3.27 g, 0.052 mol) and anhydrous pyridine (8.23 g, 0.104 mol) and heated at 170–175 °C for 10 h. The mixture was cooled to room temperature, diluted with ice water (150 mL), acidified with HCl, (4 n, 100 mL), and extracted with ether (3×100 mL). The combined ether extracts were washed with water (100 mL) and saturated aqueous NaHCO3 solution (150 mL), dried (Na2SO4), and evaporated to give pale yellow crystals of 3 (6.6 g, 85 %).
General Procedure for the Synthesis of Compounds 7–9
NaH (2.47 g, 61.7 mmol, 60 % dispersion in mineral oil) was added to a solution of 3 (2.1 g, 14 mmol) in anhydrous toluene (150 mL) at 0 °C, and the mixture was stirred for 30 min. A solution of ethyl formate (3.42 g, 46.2 mmol) in anhydrous toluene (20 mL) was added to the reaction mixture over a period of 30 min at 0 °C. The mixture was stirred at 0 °C for 1 h, allowed to warm to RT then stirred for 8 h. Water (100 mL) was added to the mixture at 0 °C and acidified by HCl (2 n). The organic layer was separated, and the aqueous layer was extracted with diethyl ether (3×80 mL). The combined organic extracts were washed with brine and dried over anhydrous Na2SO4. Evaporation of the solvent gave crude product 4. Without further purification, 4 (2.0 g) in anhydrous toluene (20 mL) was treated with DDQ (3.06 g, 13.5 mmol), and the mixture was heated at reflux for 6 h. The reaction mixture was allowed to cool to RT and filtered. The solids were washed with toluene and the filtrate was evaporated in vacuo. The residue was purified by column chromatography to give the corresponding product 7. A solution of 3 (2.1 g, 14 mmol) in anhydrous DME (20 mL) at 0 °C was added to a stirred solution of NaH (2.8 g, 70 mmol, 60 % dispersion in mineral oil) in anhydrous DME (100 mL) under N2 was added and the mixture was stirred at 0 °C for 30 min. A solution of ethyl acetate (3.69 g, 42 mmol) in anhydrous DME (20 mL) was added to the reaction mixture over a period of 30 min at 0 °C. The mixture was heated slowly to reflux over 30 min and then heated at reflux for 3 h. After the mixture had cooled, water (40 mL) and saturated NH4Cl solution (120 mL) were added dropwise, and the aqueous layer was extracted with ethyl acetate (3×80 mL). The combined organic layers were washed with brine, dried over Na2SO4, and evaporation of the solvent gave crude product 5. Without further purification, 5 in anhydrous toluene (60 mL) was treated with DDQ (3.06 g, 13.5 mmol), and the mixture was heated at reflux for 6 h. The reaction mixture was allowed to cool to RT then filtered. The solids were washed with toluene and the filtrate was evaporated in vacuo. Purification by column chromatography gave the corresponding product 8 (1.87 g, 70 %, 2 steps). The target compound 9 was prepared by following the same procedure as for 8. Reaction of tetrahydrobenzofuran 3 (2.1 g, 14 mmol) with dimethyl carbonate (6.3 g, 70 mmol) in DME (150 mL) afforded 6 as a white solid. Product 6 (2.0 g) was treated with DDQ in anhydrous toluene (60 mL) to give compound 9 (2.16 g, 75 %, 2 steps).
General Procedure for the Synthesis of Angular 3‐Methylfuranocoumarins (10)
A solution of 7 (200 mg, 1.12 mmol), ethyl acetoacetate (146 mg, 1.12 mmol) and piperidine in anhydrous EtOH (10 mL) was heated at reflux for 4 h. The product precipitated quantitatively from the solution as it was formed. The precipitate was filtered and washed with ethanol to afford pure 10 a as a white solid in 90 % yield. The target compounds 10 b–10 h were prepared by following the same procedure as for 10 a.
General Procedure for the Synthesis of Angular 3‐Methylfuranochalcones (11)
NaH (2 mmol, 60 % dispersion in mineral oil) was added in portions to a solution of 8 (1 mmol) in anhydrous THF (5 mL), under N2 and with vigorous stirring. After stirring for 15 min, a solution of the corresponding benzaldehyde (1 mmol) in anhydrous THF (3 mL) was added dropwise over 5 min, and the reaction mixture was stirred at RT for 2 h. Water (10 mL) was added to quench the reaction. The pH of the mixture was adjusted to 1 by adding 2 n HCl. The product was precipitated as a colorful solid in aqueous solution. After filtration, the crude product was recrystallized from EtOH to afford the pure product as a colorful solid. Using this procedure, the target compounds 11 b–11 ab were obtained. An additional amount of product could be obtained from the mother liquors. To this end, the solvent was evaporated in vacuo, and the residue was purified by column chromatography.
General Procedure for the Synthesis of Angular 3‐Methylfuranoflavonols (12)
30 % aqueous H2O2 (2.2 mmol) was added dropwise to a stirred solution of the corresponding angular 3‐methylfuranochalcones (1 mmol) in THF (3 mL), methanol (5 mL), and 25 % aqueous NaOH solution (2 mmol) at 0 °C was added dropwise. The solution was sealed and stirred at 0 °C for 1 h, then stirred at RT for 48 h. The product precipitated from the solution as it was formed. After acidification with HCl (2 m), the resulting flavonols were filtered from aqueous solution. The crude product was recrystallized from EtOH to afford the pure product as a colorful solid. The target compounds 12 a–12 j were obtained using this procedure.
General Procedure for the Synthesis of Angular 3‐Methylfuranoflavones (14)
A mixture of anhydrous DMSO (5.2 mL) and NaH (36.4 mmol, 60 % dispersion in mineral oil) in anhydrous toluene (60 mL) was heated at 80 °C under N2 for 2 h. The solution was cooled to 40 °C, and a solution of 9 (7.2 mmol) in anhydrous toluene (8 mL) was added dropwise to the stirred solution. The reaction mixture was then stirred at 40 °C for 1 h, diluted with diethyl ether (75 mL), and quenched with a saturated solution of NH4Cl (60 mL). The organic layer was separated, and the aqueous layer was extracted with diethyl ether (30 mL×3). The combined organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The resulting residue was purified by column chromatography to afford 13 (95 %) as a yellow solid. A solution of substituted benzaldehyde (4.2 mmol) in anhydrous toluene was added dropwise to a warm solution (40 °C) of 13 (0.42 mmol) in anhydrous toluene (15 mL) containing a catalytic amount of piperidine (4 drops). The resulting mixture was heated at reflux for 3 h. After removal of the solvent under reduced pressure, EtOH (10 mL) was added to the residue and stirred to achieve precipitation. The resulting precipitate was filtered, then washed with EtOH to afford the pure product as a colorful solid. The products 14 a–14 ab were obtained in this way, which were of sufficient purity for most purposes.
General Procedure for the Synthesis of Angular 3‐Methylfuranoisoquinolinones (16)
A solution of 9 (3 mmol) in methanol (15 mL) was saturated with ammonia and heated to 65 °C for 24 h in a Fisher–Porter bottle. The excess of methanol and ammonia was removed under vacuum at 40 °C to provide pure intermediate 15. To a solution of 15 (0.42 mmol) and substituted benzaldehyde (1.26 mmol) in anhydrous toluene (20 mL), a catalytic amount of piperidine was added. The resulting mixture was heated at reflux for 12 h. After removal of the solvent under reduced pressure, the residue was purified by column chromatography to afford the product 16 as a white solid. The target compounds 16 a–16 i were obtained using this procedure.
General Procedure for the Synthesis of Angular 3‐Methylfuranoisoflavones (19)
A solution of 8 (1 mmol) and N,N‐dimethylformamide dimethyl acetal (3.5 mmol) in DMF (10 mmol) was heated to 70–75 °C for 4 h. Saturated brine (20 mL) was added to the cooled reaction mixture, and a green product precipitated from the solution. The reaction mixture was filtered, and a green precipitate of 17 (99 %) was obtained. I2 (3 mmol, 3 equiv) was added to a solution of 17 in CHCl3 (15 mL), and the mixture was stirred at room temperature for 12 h. The reaction was quenched by adding 5 % aqueous NaHSO3 (10 mL), and the aqueous layer was extracted with CHCl3 (3×10 mL). The combined organic layers were washed with 5 % aqueous NaHCO3, then dried over Na2SO4. Evaporation of the solvent gave a residue, which was purified by column chromatography to afford the product 18 (90 %). A mixture of Na2CO3 (0.212 g, 2 mmol), Pd(OAc)2 (2 mg, 1 mol %), PEG 10 000 (3.5 g), and methanol (3 mL) was heated to 50 °C with stirring. Then, 18 (1 mmol) and an arylboronic acid (1.5 mmol) were added to the solution and the mixture was stirred at 50 °C for 4 h. After being cooled to room temperature, the reaction mixture was diluted with H2O (30 mL) and extracted with diethyl ether (4×15 mL). The collected organic extracts were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography to afford the product 19. The target compounds 19 a–19 e were obtained using this procedure.
Principal Component Analysis
PCA was performed using the SPSS 20 software package. A total of 20 physicochemical properties (Table S3) were obtained for established reference sets of 40 top‐selling brand‐name drugs (Table S6) and 20 coumarins and flavonoid natural products (Table S7). The summary of the contribution of each principal component is shown in Table S4, and the component loadings are shown in Table S5. The first three principal components account for 80.7 % of the variance in the dataset and were used to generate Figure 2.
Quantitative β‐Glucuronidase Activity Assay
The rice genotypes used in this study were transgenic lines of L15–38.20 Pre‐germinated seeds of the different lines were cultured in plastic bottles (diameter 8 cm, height 10 cm) in a greenhouse (28 °C, 14 h light, 10 h dark). Ten‐day‐old seedlings were transferred to 20 L hydroponic boxes with a rice nutrient solution.41 After 25–30 days, plants were transferred to individual 500 mL hydroponic plastic pots. Four to five days later, individual rice plants were randomly divided into two groups: chemical treatment and control. For chemical treatment, individual plants were grown in the nutrient solution that contained one of the tested compounds at the concentration of 5 mg L−1. Non‐manipulated plants grown in the nutrient solution without tested compounds were used as controls. GUS activities in transgenic plants were analyzed according to the method described by He et al.40 Each treatment at each time interval was replicated six times. The relative GUS activity for one chemical is equal to the GUS activity induced by the tested compound (5 mg L−1) divided by that induced by the control 48 h after treatment.
Herbivore Bioassay
Plant Growth
The rice genotypes used in this study were cultivar Xiushui 110. The method for plant growth was the same as stated above.
Insects
Colonies of BPH were originally obtained from rice fields in Hangzhou, China, and maintained on TN1 seedlings in a controlled climate chamber at 26±2 °C, with a 12/12 h (light/dark) photoperiod and 80 % relative humidity.
Survival Rate of N. lugens Nymphs Feeding on Elicitor‐Treated Plants
Individual rice seedlings were grown in nutrient solution that contained one of the tested compounds (each compound was first dissolved in a small quantity of DMSO) at a concentration of 20 mg L−1 24 h before insects were introduced. Non‐manipulated plants grown in the same nutrient solution with a small quantity of DMSO but without compounds were used as controls. The basal stem of each plant was confined within a glass cylinder (diameter 4 cm, height 8 cm, with 48 small holes, diameter 0.8 mm), into which 11 newly hatched BPH nymphs were introduced. The numbers of surviving BPH nymphs each day on each plant were recorded until they emerged as adults. The experiment was replicated six times.
Direct Effects of Chemical Elicitors on N. lugens Nymphs
Survival rates of newly‐hatched BPH nymphs on an artificial diet42 containing one of these chemicals at a concentration of 20 mg L−1 were determined using 30 mL glass cylinders, each side containing diet covered with a thin layer of parafilm into which 11 BPH nymphs were introduced and allowed to feed for 6 days. The number of BPH nymphs alive in each glass cylinder was recorded every day. The experiment was replicated six times. To further determine if these candidate elicitors have toxicity upon direct contact with insects, newly hatched BPH nymphs were put in Petri dishes lined with filter papers and sprayed with one of these elicitors in solution (20 mg L−1). The control group of BPH nymphs was sprayed with water only. As soon as the insects were dry, they were all transferred onto rice seedlings, each containing 11 nymphs. The number of BPH nymphs that were alive on each plant was recorded every day. The experiment was replicated six times.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The study was jointly sponsored by the Special Fund for Agro‐scientific Research in the Public Interest (201403030), the National Natural Science Foundation of China (No.31471807), the Special Fund for Agro‐scientific Research in the Public Interest of Zhejiang (2014C22004).
X. He, X. Chen, S. Lin, X. Mo, P. Zhou, Z. Zhang, Y. Lu, Y. Yang, H. Gu, Z. Shang, Y. Lou, J. Wu, ChemistryOpen 2017, 6, 102.
Contributor Information
Prof. Zhicai Shang, Email: shangzc@zju.edu.cn
Prof. Yonggen Lou, Email: yglou@zju.edu.cn
Prof. Jun Wu, Email: wujunwu@zju.edu.cn
References
- 1.
- 1a. Silverman F. P., Petracek P. D., Heiman D. F., Fledderman C. M., Warrior P., J. Agric. Food Chem. 2005, 53, 9775–9780; [DOI] [PubMed] [Google Scholar]
- 1b. Sticher L., Mauch-Mani B., Métraux J. P., Annu. Rev. Phytopathol. 1997, 35, 235–270. [DOI] [PubMed] [Google Scholar]
- 2. Cohen Y. R., Plant Dis. 2002, 86, 448–457. [DOI] [PubMed] [Google Scholar]
- 3. Sobhy I. S., Erb M., Lou Y., Turlings T. C., Philos. Trans. R. Soc. London Ser. B 2014, 369, 20120283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Lachance H., Wetzel S., Kumar K., Waldmann H., J. Med. Chem. 2012, 55, 5989–6001; [DOI] [PubMed] [Google Scholar]
- 4b. Cordier C., Morton D., Murrison S., Nelson A., O'Leary-Steele C., Nat. Prod. Rep. 2008, 25, 719–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Camp D., Davis R. A., Campitelli M., Ebdon J., Quinn R. J., J. Nat. Prod. 2012, 75, 72–81; [DOI] [PubMed] [Google Scholar]
- 5b. Larsson J., Gottfries J., Muresan S., Backlund A., J. Nat. Prod. 2007, 70, 789–794; [DOI] [PubMed] [Google Scholar]
- 5c. Rimando A. M., Duke S. O., Natural Products for Pest Management, ACS Symposium Series, Washington, 2006, pp. 2–21. [Google Scholar]
- 6. Kulkarni B. A., Roth G. P., Lobkovsky E., Porco J. A., J. Comb. Chem. 2002, 4, 56–72. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Schreiber S. L., Science 2000, 287, 1964–1969; [DOI] [PubMed] [Google Scholar]
- 7b. Lenci E., Menchi G., Guarna A., Trabocchi A., J. Org. Chem. 2015, 80, 2182–2191; [DOI] [PubMed] [Google Scholar]
- 7c. Tan D. S., Nat. Chem. Biol. 2005, 1, 74–84. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Bauer R. A., Wenderski T. A., Tan D. S., Nat. Chem. Biol. 2013, 9, 21–29; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Kopp F., Stratton C. F., Akella L. B., Tan D. S., Nat. Chem. Biol. 2012, 8, 358–365; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Beckmann H. S. G., Nie F., Hagerman C. E., Johansson H., Tan Y. S., Wilcke D., Spring D. R., Nat. Chem. 2013, 5, 861–867. [DOI] [PubMed] [Google Scholar]
- 9. Maffei M. E., Arimura G., Mithofer A., Nat. Prod. Rep. 2012, 29, 1288–1303. [DOI] [PubMed] [Google Scholar]
- 10. Liu Y.-P., Lai R., Yao Y.-G., Zhang Z.-K., Pu E.-T., Cai X.-H., Luo X.-D., Org. Lett. 2013, 15, 4940–4943. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Hirai Y., Doe M., Kinoshita T., Morimoto Y., Chem. Lett. 2004, 33, 136–137; [Google Scholar]
- 11b. Hagele B., Rowell-Rahier M., J. Evol. Biol. 2000, 13, 131–142. [Google Scholar]
- 12. Jiao M., Ding C., Zhang A., Tetrahedron Lett. 2015, 56, 2799–2802. [Google Scholar]
- 13.
- 13a. Patil R. B., Sawant S. D., Der Pharma Chemica 2015, 7, 26–37; [Google Scholar]
- 13b. Valizadeh H., Shockravi A., J. Heterocycl. Chem. 2006, 43, 763–765. [Google Scholar]
- 14. Ranga Rao R., Tiwari A. K., Prabhakar Reddy P., Suresh Babu K., Suresh G., Ali A. Z., Madhusudana K., Agawane S. B., Badrinarayan P., Narahari Sastry G., Madhusudana Rao J., Med. Chem. Res. 2012, 21, 760–774. [Google Scholar]
- 15. Khanapur M., Pinna N. K., Badiger J., Med. Chem. Res. 2015, 24, 2656–2669. [Google Scholar]
- 16. Li Z., Ngojeh G., DeWitt P., Zheng Z., Chen M., Lainhart B., Li V., Felpo P., Tetrahedron Lett. 2008, 49, 7243–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vasselin D. A., Westwell A. D., Matthews C. S., Bradshaw T. D., Stevens M. F., J. Med. Chem. 2006, 49, 3973–3981. [DOI] [PubMed] [Google Scholar]
- 18. Cui L.-J., Guo J., Gong G.-H., Quan Z.-S., Asian J. Chem. 2014, 26, 2553–2556. [Google Scholar]
- 19.
- 19a. Valizadeh H., Shockravi A., J. Heterocycl. Chem. 2006, 43, 763–765; [Google Scholar]
- 19b. Kitamura T., Otsubo K., J. Org. Chem. 2012, 77, 2978–2982; [DOI] [PubMed] [Google Scholar]
- 19c. Lee Y. R., Tetrahedron 1995, 51, 3087–3094; [Google Scholar]
- 19d. Dixit M., Tripathi B. K., Tamrakar A. K., Srivastava A. K., Kumar B., Goel A., Bioorg. Med. Chem. 2007, 15, 727–734; [DOI] [PubMed] [Google Scholar]
- 19e. Lee Y. R., A. T. Morehead, Jr. , Tetrahedron 1995, 51, 4909–4922. [Google Scholar]
- 20. Xin Z., Yu Z., Erb M., Turlings T. C., Wang B., Qi J., Liu S., Lou Y., New Phytol. 2012, 194, 498–510. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Gopalan A., Magnus P., J. Org. Chem. 1984, 49, 2317–2321; [Google Scholar]
- 21b. Stetter H., Lauterbach R., Angew. Chem. 1959, 71, 673–673. [Google Scholar]
- 22.
- 22a. Satyavani S. R., Kanjilal S., Rao M. S., Prasad R. B. N., Murthy U. S. N., Med. Chem. Res. 2015, 24, 842–850; [Google Scholar]
- 22b. Lee Y. R., Kim D. H., Synthesis 2006, 603–608; [Google Scholar]
- 22c. Izawa Y., Pun D., Stahl S. S., Science 2011, 333, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee Y. R., Morehead A. T., Cheminform 1995, 26, DOI: 10.1002/chin.199535304. [Google Scholar]
- 24. Kurt B. Z., Gazioglu I., Sonmez F., Kucukislamoglu M., Bioorg. Chem. 2015, 59, 80–90. [DOI] [PubMed] [Google Scholar]
- 25. Li J., Li X., Wang S., Chin. J. Struct.Chem. 2012, 7, 1003–1007. [Google Scholar]
- 26. Sagrera G., Bertucci A., Vazquez A., Seoane G., Bioorg. Med. Chem. 2011, 19, 3060–3073. [DOI] [PubMed] [Google Scholar]
- 27. Li S., Pan M.-H., Lai C.-S., Lo C.-Y., Dushenkov S., Ho C.-T., Bioorg. Med. Chem. 2007, 15, 3381–3389. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Oyama K.-i., Kawaguchi S., Yoshida K., Kondo T., Tetrahedron Lett. 2007, 48, 6005–6009; [Google Scholar]
- 28b. Bennett C. J., Caldwell S. T., McPhail D. B., Morrice P. C., Duthie G. G., Hartley R. C., Bioorg. Med. Chem. 2004, 12, 2079–2098. [DOI] [PubMed] [Google Scholar]
- 29. Baker W., J. Chem. Soc. 1933, 1381–1389. [Google Scholar]
- 30. Allan J., Robinson R., J. Chem. Soc. Trans. 1924, 125, 2192–2195. [Google Scholar]
- 31. Von Strandtmann M., Klutchko S., Cohen M. P., Shavel J., J. Heterocycl. Chem. 1972, 9, 171–172. [Google Scholar]
- 32. Gammill R. B., J. Org. Chem. 1981, 46, 3340–3342. [Google Scholar]
- 33.
- 33a. Gammill R., Synthesis 1979, 901–903; [Google Scholar]
- 33b. Li G., Zhang Z. T., Dai L. Y., Du Y. L., Xue D., Helv. Chim. Acta 2012, 95, 989–997. [Google Scholar]
- 34. Liu L., Zhang Y., Wang Y., J. Org. Chem. 2005, 70, 6122–6125. [DOI] [PubMed] [Google Scholar]
- 35. Dobson C. M., Nature 2004, 432, 824–828. [DOI] [PubMed] [Google Scholar]
- 36. Ibbeson B. M., Laraia L., Alza E., CJ O. C., Tan Y. S., Davies H. M., McKenzie G., Venkitaraman A. R., Spring D. R., Nat. Commun. 2014, 5, 3155. [DOI] [PubMed] [Google Scholar]
- 37. Bauer R. A., Wurst J. M., Tan D. S., Curr. Opin. Chem. Biol. 2010, 14, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jie D., Cao D.-S., Miao H.-Y., Shao L., Deng B.-C., Yun Y.-H., Wang N.-N., Lu A.-P., Zeng W.-B., Chen A. F., J. Cheminf. 2015, 7, DOI: 101186/s13321-015-0109-z. [Google Scholar]
- 39.
- 39a. Walsh T. A., Pest Manage. Sci. 2007, 63, 1165–1171; [DOI] [PubMed] [Google Scholar]
- 39b. Jin G., Lee S., Choi M., Son S., Kim G.-W., Oh J.-W., Lee C., Lee K., Eur. J. Med. Chem. 2014, 75, 413–425. [DOI] [PubMed] [Google Scholar]
- 40. He X., Yu Z., Jiang S., Zhang P., Shang Z., Lou Y., Wu J., Bioorg. Med. Chem. Lett. 2015, 25, 5601–5603. [DOI] [PubMed] [Google Scholar]
- 41. Yoshida S., Forno D. A., Cock J. H., Gomez K. A., Laboratory Manual for Physiological Studies of Rice , 3rd ed., International Rice Research Institute, Los Baños, 1976, pp. 61–62. [Google Scholar]
- 42. Fu Q., Zhang Z., Hu C., Lai F., Sun Z., Applied Entomology & Zoology 2001, 36, 111–116; Zoology 2001, 36, 111–116. [Google Scholar]
- 43.CCDC 1444188, 1444189, 1444187 and 1444190 (for 11 s, 12 c, 14 h and 16 a, respectively) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary