

Abstract
The unique structure‐directing properties of DNA origami nanostructures (DONs) show great potential to specifically manipulate intracellular processes. We report an innovative concept to selectively activate the transcription of a single gene in the developing zebrafish embryo. We reason that engineering a designer transcription factor in which a rigid DON imposes a fixed distance between the DNA‐binding domain (DBD) and the transactivation domain (TAD) would allow the selective activation of a gene harboring the same distance between the corresponding transcription factor binding site and the core promoter. As a test case, a rigid tubular DON was designed to separate the DBD of the GAL4 transcription factor and the VP16 viral protein as a TAD. This construct was microinjected in the yolk of one‐cell‐stage zebrafish embryos, together with a reporter plasmid to assess its functionality. The large DON was efficiently distributed to cells of the developing embryo and showed no signs of toxicity. However, because the DON showed only a cytosolic localization, it did not activate transcription of the reporter gene. Although this work clearly demonstrates that DON microinjection enables the intracellular distribution of multi‐protein architectures in most of the cells of the developing zebrafish embryo, further refinements are necessary to enable selective gene activation in vivo.
Keywords: DNA, microfluidics, nanostructures, self-assembly, targeting
1. Introduction
A key issue in biomedicine and biotechnology is to control processes in cells with the highest specificity without unwanted side effects of the intervening agents. Most processes in our body are regulated through the differential transcription of genes. Gene transcription is universally controlled by structural elements within the gene, cis‐acting regulatory elements, which specifically bind transcription factors.1 Among these cis‐acting elements, the core promoters bind general transcription factors of the general transcription machinery, whereas enhancer elements and proximal promoter elements bind sequence‐specific transcription factors, which activate transcription by stimulating transcription initiation at the core promoter. Targeting these enhancer and proximal promoter elements with, for example, synthetic sequence‐specific transcription factors, represents a way to activate the transcription of only the genes harboring these elements. Yet, many genes share the transcription factor binding sites present within these elements. However, unlike enhancer elements, which act independently of their distance to the core promoter, the effect of proximal promoter elements depends on the distance between the transcription factor binding site and the core promoter.2, 3 Transcription factors are modular proteins composed of a DNA binding domain (DBD), which specifically recognizes the response element, and a transactivation domain (TAD), which directly or indirectly interacts with the general transcription machinery at the core promoter. Therefore, if one could engineer an artificial transcription factor with a fixed distance between the DBD and the TAD that matches the distance between the proximal promoter element and the core promoter of a particular gene (Figure 1), one would potentially have a much more specific tool to selectively activate the transcription of this gene.
Figure 1.
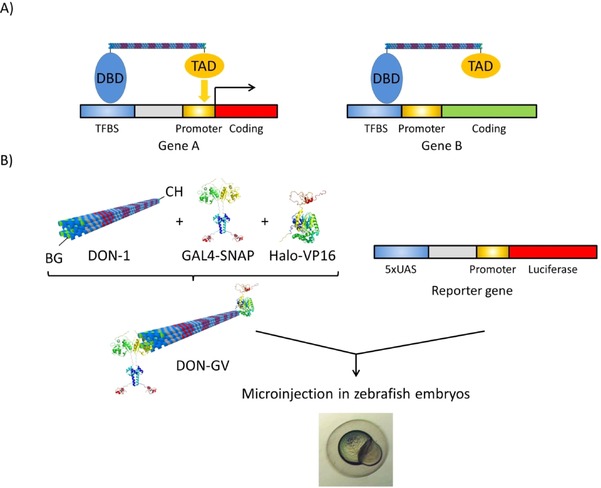
Concept and strategy for DON‐mediated selective gene activation. A) A DNA‐binding domain (DBD) and transactivation domain (TAD) are separated by a rigid DON of a length matching the distance between the transcription factor binding site (TFBS) and the core promoter of gene A. Upon binding of the DBD moiety to the TFBS, the TAD would be ideally positioned to activate the transcription of gene A. The length of the DON does not match the distance between the TFBS and core promoter in gene B. Therefore, gene B is predicted not to be activated by the construct. B) As a test case, a rigid, tubular DON (DON‐1) derivatized with the suicide ligands benzylguanine (BG) at one end and chlorohexane (CH) at the other end is functionalized with a BG‐reactive, SNAP‐tagged version of the DBD of the yeast GAL4 transcription factor and with a CH‐reactive, Halo‐tagged version of the VP16 TAD to generate DON–GV. DON–GV is then microinjected in zebrafish embryos together with a luciferase reporter gene construct, in which five copies of the consensus binding site for GAL4 (5×UAS) are separated from the core promoter by a spacer sequence predicted to match the length of the DON.
The so‐called “scaffolded DNA origami” nanostructures4 have the potential to serve as such tools. They are readily available by folding a long single‐stranded DNA (ssDNA) molecule with a designed set of short synthetic “staple‐strand” oligonucleotides.5, 6, 7 DNA origami nanostructures (DONs) have typical dimensions in the 10–100 nm regime, which perfectly match those of large supramolecular protein complexes, such as the molecular machinery involved in gene regulation, cell signaling, or cell division.8 More importantly, these dimensions are within the range of the typical distances between proximal promoter elements and core promoters. Furthermore, DONs have unique structure‐directing properties, because they can be used as scaffolds for the precise arrangement of non‐nucleic acid components such as proteins or colloidal nanoparticles.9, 10 It is, therefore, not surprising that there is increasing activity to explore DONs as tools to manipulate and analyze cellular functions.11 Along this line, efforts are underway to study the stability and function of DONs under in vitro and in vivo conditions. For instance, it has been demonstrated that DONs are stable reagents under cell culturing conditions.12, 13, 14 DONs are functional as immune‐activating programmable adjuvants,15, 16 and they can be used as drug‐delivery vehicles to circumvent drug resistance.17, 18, 19, 20 These studies have suggested that intracellular DON uptake is usually dependent on endocytosis.18 Furthermore, DONs can bind to the outside of the cells to stimulate cellular transmembrane receptors.21, 22, 23
Despite these exciting reports, the exploitation of DONs for the analysis and manipulation of cells is still in its infancy, and there is a strong demand for further insights into the complex interactions of these synthetic biomacromolecules with the machinery of living systems. Towards this goal, we report here the microinjection of a protein‐functionalized DON construct into zebrafish embryos (Figure 1).
Our working hypothesis is that using a DON to control the distance between a DBD and a TAD in order to match the distance between a proximal promoter element and a core promoter would enable the selective activation of a downstream gene. As a test system, we took advantage of the GAL4‐UAS system. This binary expression system has been widely used in many model species to drive the expression of genes. The first component of the binary system consists of a synthetic transcription factor composed of the DBD of the yeast GAL4 transcription factor fused to the TAD of the Herpes simplex VP16 protein. The second component is comprised of a synthetic gene with a tandem array of the DNA binding sites for the GAL‐VP16 chimera, called the upstream activating sequence or UAS (CGG AGTAC T GTCCT CCG),24 upstream of a minimal core promoter. This UAS promoter controls the expression of downstream cloned genes with dependence on the GAL4‐VP16 chimeric transcription factor.25 It is known that GAL4‐VP16 fusion proteins generate robust expression from the UAS responder genes in transient and stable activation assays in zebrafish.26 To allow for a quantitative assessment of transcription, we chose the firefly luciferase reporter gene as a regulated gene. To test the effect of the distance between the UAS and the core promoter in the reporter plasmid, they were separated from each other by a variable spacer (grey region of the reporter gene, Figure 1). The reporter plasmid was constructed by Gibson cloning27 by sequentially aligning five copies of the UAS, a spacer, a minimal CMV (cytomegalie virus) promoter and the firefly reporter gene28 (detailed plasmid map shown in Figure S1). We reasoned that, upon injection into zebrafish embryo, the level of expression of the firefly luciferase reporter should then depend on the distance between the DBD (GAL4) and the TAD (VP16) physically connected to each other through a DON spacer in the supramolecular construct DON–GV (Figure 1).
2. Results and Discussion
To prepare the DON–GV construct, we assembled a three‐dimensional 8×11×148 nm3 rigid‐rod tubular DON‐1 (Figure 2 A) by using the single‐stranded 5438 nt template 109Z5, prepared as previously described.29
Figure 2.
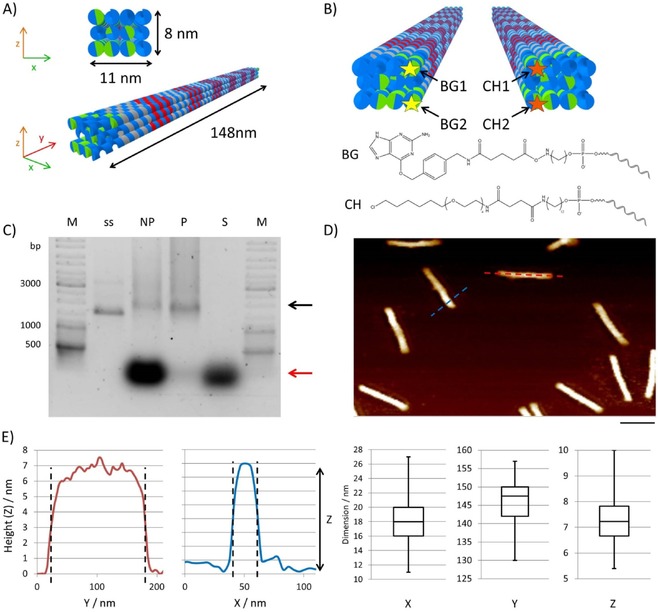
Design and analysis of DNA origami nanostructures. A) Schematic representation of the 3D DON‐1 design. B) Position (top) and chemical structure (bottom) of the staple strands used for the incorporation of the suicide ligands benzylguanine (BG) and chlorohexane (CH) at 5 ’ ends of the selected staple oligonucleotides. C) Ethidium bromide‐stained 0.75 % agarose gel of DON samples before (not purified, NP) and after (purified, P) PEG purification. M: GeneRuler DNA Ladder Mix. The supernatant (S) removed after precipitation contains the non‐incorporated staple strands (red arrow). The assembled DONs are indicated by the black arrow; ss: single‐stranded scaffold. D) Representative AFM image of purified DON‐1, indicating monodispersed rod‐shaped particles. Scale bar: 100 nm. E) Height profiles obtained from AFM images along the cross‐sections are marked in blue and red in panel (D). The box‐whisker plots show the results of the statistical analysis of 60 particles to determine their dimensions along their axes. The values for x and y were determined from the cross‐sections, whereas the z values were determined from the particle's heights.
To enable the selective attachment of the GAL4‐DBD and VP16 protein at the two ends of the rod, we incorporated at the appropriate positions pairs of the small‐molecule suicide ligands benzylguanine (BG) and chlorohexane (CH), to which SNAP‐ and Halo‐tagged proteins, respectively, can be ligated.30 The BG‐ and CH‐modified staple strands were prepared from NHS‐activated precursors, as previously described,30, 31 and characterized by using gel electrophoresis (Figure S2). The assembly of DON‐1 was achieved by temperature‐dependent annealing. In brief, the single‐stranded DNA plasmid and the staple strands at a 1:20 molar ratio were heated to 95 °C and the annealing was performed by decreasing the temperature from 85 to 65 °C at −1 °C per cycle with each step held for 10 min and, subsequently, from 65 to 25 °C at −1 °C per cycle with each step held for 15 min. The assembled DON‐1 samples were purified through polyethylene glycol precipitation32 and their integrity and correct folding was confirmed by gel electrophoresis (Figure 2 C), FRET‐dependent annealing studies (Figure S3), and AFM analysis (Figure 2 D). Statistical analysis of the AFM images (Figure 2 E) revealed that the observed length (146±5 nm), thickness (17±3 nm), and height (7±1 nm) were in agreement with the theoretically calculated values of 148 nm length, 11 nm thickness, and 8 nm height for the rods, as depicted in Figure 2 A.
To synthesize the regulatory protein‐decorated construct DON–GV (Figure 1), the required proteins were produced by recombinant expression. We cloned GAL4‐DBD C‐terminally fused to the SNAP‐tag through a flexible (GGGGS)3 linker (Figure S4). The corresponding protein GAL4‐SNAP was overexpressed in E. coli and purified to homogeneity by fast protein liquid chromatography (FPLC, Figure S5). As native GAL4 functions as a homodimer, it was important to confirm that also the novel GAL4‐SNAP fusion can dimerize. This was achieved by using size‐exclusion chromatography (Figure S6). Furthermore, the functionality of the novel GAL4‐SNAP fusion protein to bind the UAS recognition sequence was demonstrated by using a microbead‐based pull‐down assay (Figure S7). The VP16 TAD was cloned with the Halo‐tag fused to its N‐terminus (Figure S4). The corresponding Halo‐VP16 protein was overexpressed in E. coli, purified to homogeneity by FPLC, and its identity was verified with Western blot analysis (Figure S8).
Having both regulatory proteins available as pure and bioconjugatable reagents (Figure 3 A), we then assembled the bifunctional protein–DNA construct, DON–GV. To this end, a Cy5‐labeled DON‐1 (DON‐1Cy5) was prepared as described above and mixed with 25 molar equivalents of GAL4‐SNAP and Halo‐VP16. Several controls, which included DON‐2Cy5, a construct identical to DON‐1Cy5 but lacking the BG and CH ligands, and samples lacking one of the two proteins were prepared in a similar manner. The reaction products were characterized by using gel electrophoresis (Figure 3 B), which clearly indicated that DON–GV could be successfully assembled as expected. The experiments also indicated the absence of unspecific binding between GAL4‐SNAP, Halo‐VP16, and the DON.
Figure 3.
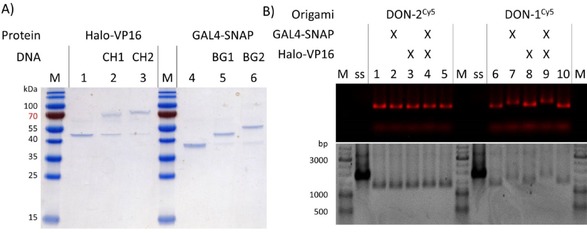
Assembly of DON–GV. A) Electrophoretic analysis of the conjugation of the two CH‐ and BG‐modified staple strands with the recombinant proteins Halo‐VP16 and GAL4‐SNAP to be used as TAD and DBD, respectively, in DON–GV (see Figure 2 B). This is a 16 % SDS gel, 20 V cm−1, 1 h, stained with Coomassie; M: Page Ruler pre‐stained protein ladder. The appearance of a second band with lower electrophoretic mobility upon incubation with the ligand‐conjugated DNA oligonucleotides (sample 2, 3, 5, and 6) indicates the successful conjugation of the protein with the respective staples. M: molecular weight marker. B) Halo‐VP16 and GAL4‐SNAP do not bind to DON‐2Cy5, a construct lacking CH‐ and BG‐modified staple strands (lanes 1–5). In contrast, DON‐1Cy5 containing the CH and BG modifications can bind both of the proteins, as indicated from the shift of the bands in lanes 7–9. Note that the conjugation with Halo‐VP16 (lane 8) induces only slight electrophoretic mobility shifts, presumably owing to net negative charge of VP16 (pI=4.4). This is a 0.75 % agarose gel, 70 V, 3 h, visualized by Cy5 fluorescence imaging (top) and ethidium bromide staining (bottom). M: GeneRuler DNA Ladder; ss: scaffold strand.
We then started to analyze the behavior of the DONs in vivo in the zebrafish embryo. To this end, we used a zebrafish line stably expressing GFP‐fused histone H2A33 that labels the cells’ nuclei and, thereby, facilitates the determination of the DONs’ subcellular localization. DON‐1Cy5 (approximately 500 pL, 250 nm) was injected into the yolk of one‐cell‐stage embryos and the embryos were imaged 6 h later (Figure 4 A).
Figure 4.
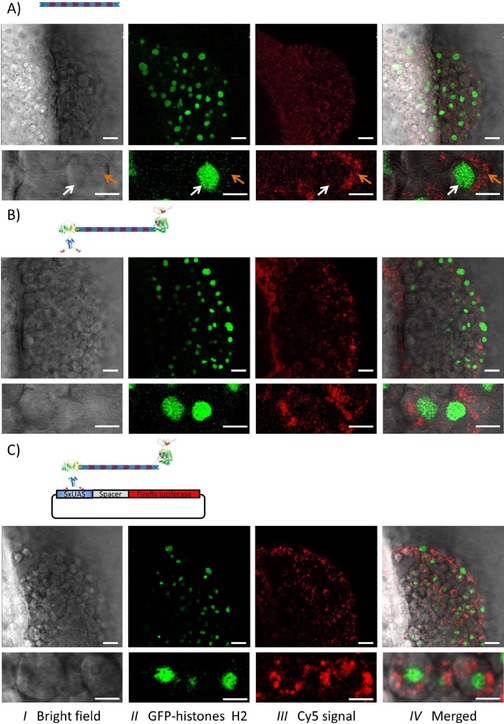
Localization of the DONs in the developing zebrafish embryos. One‐cell stage embryos from a zebrafish line stably expressing GFP‐fused histone H2A were used,33 which allows the direct localization of the cell nuclei (green channel). The Cy5‐labeled constructs (red channel) were microinjected into the yolk with DON‐1Cy5 alone (A), the protein‐decorated DON (DON‐1Cy5‐GV; B) or DON‐1Cy5‐GV along with the reporter plasmid (C). Then, at 6 h post‐fertilization, the embryos were embedded in agarose and analyzed by using confocal fluorescence microscopy. Three channels were recorded: the bright‐field image (I), the signal from the GFP‐histone H2 in green (II), and the Cy5 signal from the DONs in red (III). The high‐intensity signal in the red channel shows that the large DNA constructs were internalized at a high rate from the yolk. In addition, the high magnification (bottom images) clearly indicates that the internalized DONs are mostly cytosolic (orange arrow), with very little to no nuclear localization (white arrow). Note that the presence of neither the proteins nor the reporter plasmid influenced the internalization or the subcellular distribution of the DONs. Scale bars: 30 μm and 10 μm for low and high magnification, respectively.
The injected embryos survived and developed normally, suggesting that the introduced nanomaterial was well tolerated. The large Cy5‐labeled DNA construct was detected within many cells of the developing embryo. This result shows that DON‐1 has no obvious deleterious effects on early embryonic development and is indeed efficiently internalized from the yolk (Figure 4 A, panel III). However, comparison of the nuclei position (GFP signal, panel II) with the DON fluorescent signal (panel III) indicates that the DON localizes mainly to the cytosol. Thus, “naked” DONs do not accumulate in the nucleus.
We then studied the intracellular localization of the bifunctional protein–DNA construct DON–GV injected alone or together with the reporter plasmid (Figures 4 B and 4 C, respectively). The DON–GV also had no obvious effect on early embryonic development and was internalized by the cells. Furthermore, it was only detected in the cytosol, irrespective of the presence of the reporter plasmid (Figures 4 B and 4 C). Consistent with the absence of nuclear localization, the DON–GV had no effect on the expression of the reporter gene, as compared to the DON‐1 (data not shown). Spherical particles with a diameter below 26 nm freely diffuse into the nucleus, whereas larger particles require a nuclear localization signal (NLS) to actively translocate through the nuclear pore complex, with an upper size limit of about 39 nm.34 The GAL4‐DBD used to functionalize the DON–GV harbors an intrinsic nuclear localization signal.35 However, it was apparently not sufficient to drive the DON–GV to the nucleus. Recently, it was shown that functionalization with an NLS increases the nuclear uptake of 5–10 nm×100–300 nm rod‐shaped polymeric nanoparticles.36 Therefore, the absence of nuclear accumulation of the DON–GV is unlikely because of its size, but rather to a low efficiency of the GAL4‐DBD NLS in the context of the construct. Thus, functionalizing the DON with a “stronger” NLS as a peptide covalently attached to the DON might increase its nuclear uptake.
3. Conclusions
We report here on an ambitious concept for controlling gene transcription in a distance‐dependent manner in the developing zebrafish embryo, taking advantage of designed protein–DNA origami nanostructures. Although we could clearly demonstrate the successful synthesis of our target construct, applications in vivo require further developments. Nonetheless, we believe that our study represents an important contribution to the exploitation of DONs for the analysis and manipulation of cells in vivo. We demonstrated, for the first time, that microinjection in the yolk of the zebrafish embryo enables intracellular distribution in most of the cells of large origami constructs harboring active proteins precisely arranged in a 3D fashion. Furthermore, we demonstrated that the DON constructs are not toxic for the embryos and allowed normal development, at least in the early stages. We foresee that this technology could be applied to deliver complex multi‐protein constructs to the cytosol and maybe also to the nuclei by means of strong nuclear localization signals.
Experimental Section
For experimental details, see the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported in part through GRK 2039 founded by the German Research Foundation (DFG) and by the Helmholtz programme BioInterfaces in Technology and Medicine. We acknowledge support by DFG and Open Access Publishing Fund of Karlsruhe Institute of Technology. We thank Dr. Thomas Dickmeis for kindly supplying the luciferase‐containing plasmid, Dr. Kersten Rabe for help with the cloning and fruitful discussions, Dr. Eliana Stanganello for help with the in vivo imaging of zebrafish embryos, Cornelia Ziegler and Anke Dech for practical assistance in protein expression and DNA scaffold preparation, and Dr. Masanari Takamiya for kindly supplying the wild‐type and transgenic zebrafish lines and experimental help.
A. Angelin, O. Kassel, S. Rastegar, U. Strähle, C. M. Niemeyer, ChemistryOpen 2017, 6, 33.
References
- 1. Andersson R., Sandelin A., Danko C. G., Trends Genet. 2015, 31, 426. [DOI] [PubMed] [Google Scholar]
- 2. Tansey W. P., Schaufele F., Heslewood M., Handford C., Reudelhuber T. L., Catanzaro D. F., J. Biol. Chem. 1993, 268, 14906. [PubMed] [Google Scholar]
- 3. Smith K. P., Liu B., Scott C., Sharp Z. D., J. Biol. Chem. 1995, 270, 4484. [DOI] [PubMed] [Google Scholar]
- 4. Rothemund P. W., Nature 2006, 440, 297. [DOI] [PubMed] [Google Scholar]
- 5. Nangreave J., Han D., Liu Y., Yan H., Curr. Opin. Chem. Biol. 2010, 14, 608. [DOI] [PubMed] [Google Scholar]
- 6. Shih W. M., Lin C., Curr. Opin. Struct. Biol. 2010, 20, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Castro C. E., Kilchherr F., Kim D. N., Shiao E. L., Wauer T., Wortmann P., Bathe M., Dietz H., Nat. Methods 2011, 8, 221. [DOI] [PubMed] [Google Scholar]
- 8. Meyer R., Faesen A., Vogel K., Jeganathan S., Musacchio A., Niemeyer C. M., Small 2015, 11, 2669. [DOI] [PubMed] [Google Scholar]
- 9. Saccà B., Niemeyer C. M., Angew. Chem. Int. Ed. 2012, 51, 58; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 60. [Google Scholar]
- 10. Simmel F. C., Curr. Opin. Biotechnol. 2012, 23, 516. [DOI] [PubMed] [Google Scholar]
- 11. Kearney C. J., Lucas C. R., O'Brien F. J., Castro C. E., Adv. Mater. 2016, 28, 5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mei Q., Wei X., Su F., Liu Y., Youngbull C., Johnson R., Lindsay S., Yan H., Meldrum D., Nano Lett. 2011, 11, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perrault S. D., Shih W. M., ACS Nano 2014, 8, 5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hahn J., Wickham S. F., Shih W. M., Perrault S. D., ACS Nano 2014, 8, 8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schüller V. J., Heidegger S., Sandholzer N., Nickels P. C., Suhartha N. A., Endres S., Bourquin C., Liedl T., ACS Nano 2011, 5, 9696. [DOI] [PubMed] [Google Scholar]
- 16. Sellner S., Kocabey S., Nekolla K., Krombach F., Liedl T., Rehberg M., Biomaterials 2015, 53, 453. [DOI] [PubMed] [Google Scholar]
- 17. Jiang Q., Song C., Nangreave J., Liu X., Lin L., Qiu D., Wang Z. G., Zou G., Liang X., Yan H., Ding B., J. Am. Chem. Soc. 2012, 134, 13396. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y. X., Shaw A., Zeng X., Benson E., Nystrom A. M., Hogberg B., ACS Nano 2012, 6, 8684. [DOI] [PubMed] [Google Scholar]
- 19. Halley P. D., Lucas C. R., McWilliams E. M., Webber M. J., Patton R. A., Kural C., Lucas D. M., Byrd J. C., Castro C. E., Small 2016, 12, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Halley P. D., Lucas C. R., McWilliams E. M., Webber M. J., Patton R. A., Kural C., Lucas D. M., Byrd J. C., Castro C. E., Small 2016, 12, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Douglas S. M., Bachelet I., Church G. M., Science 2012, 335, 831. [DOI] [PubMed] [Google Scholar]
- 22. Shaw A., Lundin V., Petrova E., Fordos F., Benson E., Al-Amin A., Herland A., Blokzijl A., Hogberg B., Teixeira A. I., Nat. Methods 2014, 11, 841. [DOI] [PubMed] [Google Scholar]
- 23. Angelin A., Weigel S., Garrecht R., Meyer R., Bauer J., Kumar R. K., Hirtz M., Niemeyer C. M., Angew. Chem. Int. Ed. 2015, 54, 15813; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16039. [Google Scholar]
- 24. Brand A. H., Perrimon N., Development 1993, 118, 401. [DOI] [PubMed] [Google Scholar]
- 25. Davison J. M., Akitake C. M., Goll M. G., Rhee J. M., Gosse N., Baier H., Halpern M. E., Leach S. D., Parsons M. J., Dev. Biol. 2007, 304, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Köster R. W., Fraser S. E., Dev. Biol. 2001, 233, 329. [DOI] [PubMed] [Google Scholar]
- 27. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison 3rd C. A., Smith H. O., Nat. Methods 2009, 6, 343. [DOI] [PubMed] [Google Scholar]
- 28. Weger B. D., Weger M., Nusser M., Brenner-Weiss G., Dickmeis T., ACS Chem. Biol. 2012, 7, 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erkelenz M., Bauer D. M., Meyer R., Gatsogiannis C., Raunser S., Sacca B., Niemeyer C. M., Small 2014, 10, 73. [DOI] [PubMed] [Google Scholar]
- 30. Saccà B., Meyer R., Erkelenz M., Kiko K., Arndt A., Schroeder H., Rabe K. S., Niemeyer C. M., Angew. Chem. Int. Ed. 2010, 49, 9378; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9568. [Google Scholar]
- 31. Meyer R., Niemeyer C. M., Small 2011, 7, 3211. [DOI] [PubMed] [Google Scholar]
- 32. Stahl E., Martin T. G., Praetorius F., Dietz H., Angew. Chem. Int. Ed. 2014, 53, 12735; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12949. [Google Scholar]
- 33. Pauls S., Geldmacher-Voss B., Campos-Ortega J. A., Dev. Genes Evol. 2001, 211, 603. [DOI] [PubMed] [Google Scholar]
- 34. Panté N., Kann M., Mol. Biol. Cell 2002, 13, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silver P. A., Keegan L. P., Ptashne M., Proc. Natl. Acad. Sci. USA 1984, 81, 5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.E. Hinde, K. Thammasiraphop, H. T. T. Duong, J. Yeow, B. Karagoz, C. Boyer, J. J. Gooding, K. Gaus, Nat. Nanotechnol 2016, DOI: 10.1038/nnano.2016.160. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary