

Abstract
A highly substrate‐general synthesis of all‐carbon‐substituted E‐ and Z‐stereodefined olefins is performed. The method comprises two sets of parallel and stereocomplementary preparations of (E)‐ and (Z)‐α,β‐unsaturated esters involving two robust and distinctive reactions: 1) stereocomplementary enol tosylations using readily available TsCl/diamine/(LiCl) base reagents, and 2) stereoretentive Negishi cross‐coupling using the catalysts [Pd(dppe)Cl2] (for E) and [Pd(dppb)Cl2] (for Z). The present parallel approach is categorized as both type I (convergent approach: 16 examples, 56–87 % yield) and type II (divergent approach: 18 examples, 70–95 % yield). The obtained (E)‐ and (Z)‐α,β‐unsaturated ester scaffolds are successfully transformed into various E‐ and Z‐stereodefined known and novel olefins (8×2 derivatization arrays). As a demonstration, application to the parallel synthesis of both (E)‐ and (Z)‐tamoxifens, a representative motif of all‐carbon‐substituted olefins, is accomplished in a total of eight steps with an overall yield of 58 % (average 93 %) and 57 % (average 93 %), respectively.
Keywords: cross-coupling reactions, enols, esters, parallel synthesis, tamoxifen
1. Introduction
Regio‐ and stereo‐controlled syntheses of E‐ and Z‐stereodefined olefins are of pivotal importance in organic chemistry, because of their wide distribution in natural products, pharmaceuticals, and in supramolecules as key structural building blocks. Among the olefins, construction of acyclic stereodefined all‐carbon‐substituted olefins remains a challenge due to their structural complexity. Considerable efforts have been invested in this over the recent decades. The impressive progress in this area has been comprehensively reviewed.1 The strategy for the synthesis of acyclic fully‐substituted olefins is generally categorized into five approaches: 1) carbometalations of alkynes using Cu, B, Sn, Mg, Pd, and so forth, followed by reactions with electrophiles, 2) acid‐induced carbonyl olefinations of unsymmetrically substituted ketones, 3) elimination reactions of tertiary alcohols, 4) cross‐metatheses between olefins, and 5) ynolate‐mediated reactions derived from α,α‐dibromoesters.
Cross‐coupling reactions with stereodefined enol sulfonate2 and phosphonate3 partners derived from β‐ketoesters, which emerged in recent decades, are considered a promising and reliable approach compared with the above‐mentioned methods, with the following advantages: 1) various starting β‐ketoester substrates are readily available,4 and 2) the E‐ and Z‐stereocomplementary enol tosylation step is robust and cost‐effective.5 E‐ and Z‐stereoretention during the cross‐coupling step is guaranteed, especially for Suzuki–Miyaura (SM) cross‐coupling. Additionally, recent developments of cross‐couplings using enol sulfonates facilitate and enhance this strategy. As depicted in Scheme 1, the current privileged protocols were adopted for the synthesis of “tri‐substituted” (R1 or R2=H) elaborated natural products and pharmacophore‐containing compounds, such as γ‐aminobutanoic acid (GABA) analogues,6 juvenile hormones 0 and Ι,7 functionalized steroids,8 madangamine A,9 and (E)‐ and (Z)‐zimelidines.10
Scheme 1.

Synthetic applications of trisubstituted (E)‐ and (Z)‐enol tosylates.
This background led us to envisage a highly substrate‐general synthesis of fully all‐carbon‐substituted E‐ and Z‐stereodefined olefins, and especially to focus on a parallel methodology. We present divergent access to a variety of acyclic stereodefined all‐carbon‐substituted olefins and the first parallel synthesis of (E)‐ and (Z)‐tamoxifens, representatives of these olefins (Figure 1).
Figure 1.

The structures of (E)‐ and (Z)‐tamoxifens.
2. Results and Discussion
Stereocontrolled synthesis of ubiquitous (E)‐ and (Z)‐α,β‐unsaturated ester scaffolds occupies a central position in organic synthesis. Due to the intrinsic higher complexity in differentiating the substituents, synthesis of all‐carbon‐substituted E‐ and Z‐stereodefined olefin precursors are not sufficiently established. Here we elaborate a plan for two distinctive parallel and stereoretentive syntheses for fully substituted (E)‐ and (Z)‐α,β‐unsaturated esters 3 and 5 starting from readily accessible β‐ketoesters 1 a and 1 b, by utilizing type I and type II strategies via dual approaches 1 and 2 (Scheme 2).
Scheme 2.

Two types of parallel and stereoretentive syntheses of fully substituted (E)‐ and (Z)‐α,β‐unsaturated esters 3 and 5.
In 2015, our group reported the synthesis of specific but substrate‐general fully substituted α,β‐diarylbut‐2‐enoic esters, utilizing a parallel approach.10 Later in 2016, the Merck process group independently disclosed asymmetric synthesis of α‐methyl‐β‐cyclopropyldihydrocinnamates via the corresponding (Z)‐enol tosylate of methyl 3‐cyclopropyl‐3‐oxopropanoate.11 Both methods utilize Suzuki–Miyaura (SM) cross‐coupling for construction of the α,β‐unsaturated esters. One key difference between the approaches of the two groups is the stereocomplementary enol tosylation reagents [our group: TsCl/N‐methylimidazole or N,N,N′,N′‐tetramethylenediamine (TMEDA)/LiCl; the Merck group: para‐toluenesulfonic anhydride/lithium bis‐ (trimethylsilyl)amide (LHMDS)].12
As part of our ongoing investigation,13 we recently observed that Negishi cross‐coupling tends to exhibit higher reactivity with lower catalyst loadings for this type of synthetic approach (unpublished results). By contrast, enol phosphonates serve as effective SM and Negishi cross‐coupling partners.14 Against this background, as a preliminary evaluation, comparable Negishi cross‐coupling experiments were examined using a 1:1 mixture of enol tosylate (E)‐4 a and enol phosphonate (E)‐6 (Scheme 3). The result indicated the superiority of (E)‐4 a as the cross‐coupling partner. Thus, for this objective we focused our attention on Negishi cross‐coupling instead of SM cross‐coupling using various enol tosylates 4.
Scheme 3.

Comparative experiment between enol tosylate (E)‐4 a and enol phosphonate (E)‐6.
Starting enol tosylates 2 and 4 were conveniently prepared by using a recently improved robust and cost‐effective method for E‐ and Z‐stereocomplementary reactions of β‐ketoesters [E: Me2N(CH2)6NMe2; Z: LiCl–TMEDA].5
The initial screening of several Pd catalysts for Negishi cross‐coupling was guided by the reaction using intentionally less‐reactive enol tosylates (E)‐4 a or (Z)‐4 a with PhMgBr/ZnCl2 (in situ generation of PhZnCl; Table 1). Among them, [Pd(dppe)Cl2] produced fruitful results in the E‐stereoretentive reaction to give (E)‐5 a‐1 with high yield (82 %) and selectivity (E/Z=96:4) in MeCN/THF (Table 1, entry 7). Notably, in contrast to the E isomer, reactions using (Z)‐4 a proceeded with nearly perfect Z‐stereoretention in all cases examined to yield (Z)‐5 a‐1 and [Pd(dppb)Cl2] afforded the best result (85 %, E/Z=2:98) in THF (Table 1, entry 5).
Table 1.
Optimization of Negishi cross‐coupling conditions.

| ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yield [%] (E/Z)[a] | |
| (E)‐5 a‐1 | (Z)‐5 a‐1 | |||
| 1 | [Pd(PPh3)4] | THF | 4 (48:52) | 74 (2:>98) |
| 2 | [Pd(PPh3)2Cl2] | 8 (49:51) | 38 (2:>98) | |
| 3 | [Pd(dppe)Cl2] | 24 (94:6) | 39 (2:>98) | |
| 4 | [Pd(dppp)Cl2] | 42 (78:22) | 43 (2:>98) | |
| 5 | [Pd(dppb)Cl2] | 47 (72:28) | 85 (2:>98) | |
| 6 | [Pd(dppf)Cl2] | 28 (75:25) | 12 (2:>98) | |
| 7 | [Pd(dppe)Cl2] | MeCN/THF (2:1) | 82 [b,c] (96:4) | – |
[a] Determined by 1H NMR spectroscopy of the crude products. [b] [Pd(dppe)Cl2] (2 mol %). [c] 80–85 °C.
Table 2 shows the successful results of the present parallel synthesis (type I) (convergent oriented approach) by using the Negishi cross‐coupling method under optimized conditions A and B (Table 1). Ar1ZnCl and Ar2ZnCl reagents containing both electron‐donating groups (p‐Me, p‐MeO) and an electron‐withdrawing (p‐Cl) group were applicable. Two pairs of Ar1‐ and Ar2‐substituted enol tosylates, (E)‐2, 2′ and (Z)‐2, 2′, were transformed into the corresponding products, (E)‐3, 3′ and (Z)‐3, 3′, respectively, through dual convergent pathways. The salient features are as follows: 1) for the four sets examined, all reactions proceeded in good to excellent yield; 2) excellent Z‐selectivity was produced in all eight cases; 3) E‐selectivity was slightly decreased in a few cases (Table 1, entries 5, 9, and 13).
Table 2.
Parallel and stereoretentive syntheses for fully substituted (E)‐ and (Z)‐α,β‐unsaturated esters 3 (type I, convergent). Method A: ArMgBr (2.0 equiv), ZnCl2 (2.0 equiv), [Pd(dppe)Cl2] (2 mol %), MeCN/THF (2:1), 60 °C, 2 h. Method B: [Pd(dppb)Cl2] (1 mol %) and THF instead of those given for method A.

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar1 | Ar2 | Substrate[a] | Method | Product | Yield [%] | E/Z [b] |
| 1 | Ph | p‐MeC6H4 | [E]‐2 a | A | [E]‐3 a | 75 | 84:16 |
| 2 | (Z)‐2 a | B | (Z)‐3 a | 84 | 2:>98 | ||
| 3 | (Z)‐2 a′ | B | (E)‐3 a | 80 | >98:2 | ||
| 4 | (E)‐2 a′ | A | (Z)‐3 a | 83 | 2:>98 | ||
| 5 | Ph | p‐MeOC6H4 | (E)‐2 a | A | (E)‐3 b | 77 | 84:16 |
| 6 | (Z)‐2 a | B | (Z)‐3 b | 82 | 2:>98 | ||
| 7 | (Z)‐2 b′ | B | (E)‐3 b | 80 | >98:2 | ||
| 8 | (E)‐2 b′ | A | (Z)‐3 b | 80 | 2:>98 | ||
| 9 | Ph | p‐ClC6H4 | (E)‐2 a | A | (E)‐3 c | 54 | 83:17 |
| 10 | (Z)‐2 a | B | (Z)‐3 c | 85 | 2:>98 | ||
| 11 | (Z)‐2 c′ | B | (E)‐3 c | 70 | >98:2 | ||
| 12 | (E)‐2 c′ | A | (Z)‐3 c | 70 | 2:>98 | ||
| 13 | p‐MeOC6H4 | p‐ClC6H4 | (E)‐2 d | A[c] | (E)‐3 d | 85 | 68:32 |
| 14 | (Z)‐2 d | B | (Z)‐3 d | 80 | 2:>98 | ||
| 15 | (Z)‐2 d′ | B | (E)‐3 d | 87 | >98:2 | ||
| 16 | (E)‐2 d′ | A[c] | (Z)‐3 d | 77 | 10:90 | ||
[a] The purities of E and Z isomers were up to >98 % based on the 1H NMR spectra. [b] Determined by 1H NMR spectroscopy of the crude products. [c] 3.0 equiv of ArZnCl was used in toluene at reflux.
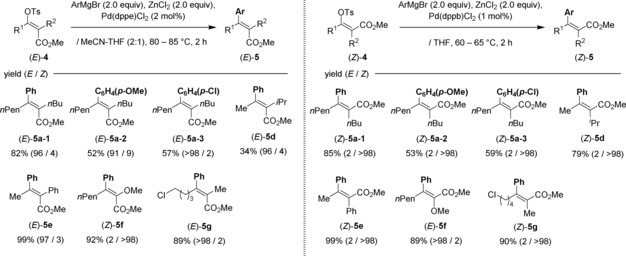
Conversely, parallel synthesis (type II, divergent‐oriented approach) was investigated and the results are shown in Table 3. The salient features are as follows: 1) in all cases, good to excellent yield and almost perfect E and Z selectivities were achieved; 2) α,β‐dimethyl enol tosylates (E)‐4 b and (Z)‐4 b were transformed into a total of six (E)‐ and (Z)‐α,β‐unsaturated ester analogues (E)‐5 b and (Z)‐5 b (Table 3, entries 1–6); 3) regioisomers (E)‐ and (Z)‐4 c and 4 c’ afforded three sets of all four stereoisomers (E)‐ and (Z)‐5 c‐1–3 and 5 c′‐1–3 (Table 3, entries 7–10, 11–14, 15–18). To further strengthen the substrate scope, seven syntheses using various (E)‐ and (Z)‐α,β‐unsaturated esters 5 a and 5 d–g are summarized in Table 4.
Table 3.
Parallel and stereoretentive syntheses for fully substituted (E)‐ and (Z)‐α,β‐unsaturated esters 5 (type II, divergent). Method A: ArMgBr (2.0 equiv), ZnCl2 (2.0 equiv), [Pd(dppe)Cl2] (2 mol %), MeCN/THF (2:1), 60–65 °C, 2 h. Method B: [Pd(dppe)Cl2] (1 mol %) and THF instead of those given for method A.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Substrate[a] | Ar | Method | Product | Yield [%] | E/Z [b] |
| 1 | Me | Me | (E)‐4 b | Ph | A | (E)‐5 b‐1 | 84 | >98:2 |
| 2 | (Z)‐4 b | B | (Z)‐5 b‐1 | 83 | 2:>98 | |||
| 3 | Me | Me | (E)‐4 b | p‐MeOC6H4 | A | (E)‐5 b‐2 | 82 | 95:5 |
| 4 | (Z)‐4 b | B | (Z)‐5 b‐2 | 95 | 2:>98 | |||
| 5 | Me | Me | (E)‐4 b | p‐ClC6H4 | A | (E)‐5 b‐3 | 93 | 98:2 |
| 6 | (Z)‐4 b | B | (Z)‐5 b‐3 | 80 | 2:>98 | |||
| 7 | Me | nBu | (E)‐4 c | Ph | A | (E)‐5 c‐1 | 85 | 98:2 |
| 8 | (Z)‐4 c | B | (Z)‐5 c‐1 | 88 | 2:>98 | |||
| 9 | Me | nBu | (E)‐4 c′ | Ph | A | (E)‐5 c′‐1 | 72 | >98:2 |
| 10 | (Z)‐4 c′ | B | (Z)‐5 c′‐1 | 70 | 2:>98 | |||
| 11 | Me | nBu | (E)‐4 c | p‐MeOC6H4 | A | (E)‐5 c‐2 | 89 | 91:9 |
| 12 | (Z)‐4 c | B | (Z)‐5 c‐2 | 91 | 2:>98 | |||
| 13 | Me | nBu | (E)‐4 c′ | p‐MeOC6H4 | A | (E)‐5 c′‐2 | 75 | >98:2 |
| 14 | (Z)‐4 c′ | B | (Z)‐5 c′‐2 | 82 | 2:>98 | |||
| 15 | Me | nBu | (E)‐4 c | p‐ClC6H4 | A | (E)‐5 c‐3 | 88 | 97:3 |
| 16 | (Z)‐4 c | B | (Z)‐5 c‐3 | 78 | 2:>98 | |||
| 17 | Me | nBu | (E)‐4 c′ | p‐ClC6H4 | A | (E)‐5 c′‐3 | 78 | >98:2 |
| 18 | (Z)‐4 c′ | B | (Z)‐5 c′‐3 | 70 | 2:>98 | |||
[a] The purities of the E and Z isomers were up to >98 % based on 1H NMR spectra. [b] Determined by 1H NMR spectroscopy of the crude products.
Table 4.
Stereocomplementary syntheses for fully substituted (E)‐ and (Z)‐α,β‐unsaturated esters 5 a and 5 d–g.
|
Our next study was focused on stereoretentive and complementary derivatizations of (E)‐5 b‐1 and (Z)‐5 b‐1 to furnish various E‐ and Z‐stereodefined fully substituted olefin scaffolds. A literature survey revealed few E‐ and Z‐stereoretentive reactions of either α‐alkyl‐ or aryl‐substituted α,β‐unsaturated esters.1 Scheme 4 shows the success of the derivatization array. Non‐marked compounds 7–10 are known, whereas compounds 11–14 are novel. The reaction conditions can be summarized as follows: [a] acid hydrolysis gave acids (E)‐7 and (Z)‐7;15 [b] DIBAL reduction afforded allyl alcohols (E)‐8 and (Z)‐8;16 [c] MnO2 allylic alcohol oxidation of (E)‐8 and (Z)‐8 yielded aldehydes (E)‐9 and (Z)‐9, respectively;16a [d] dimethyl alcohols syn‐10 and isomeric anti‐10 were obtained by the reported catalytic hydrogenation (H2–Pd/C), followed by lithium aluminum hydride (LAH) reduction.16a Conversion steps [b]–[d] were performed following the Serra group's reliable procedures; [e] Wittig methylene formation gave dienes (4E)‐11 and (4Z)‐11; [f] Horner–Wadsworth–Emmons reaction using methyl phosphonoacetate yielded (2E,4E)‐12 and (2E,4Z)‐12; [g] Knoevenagel condensation of (E)‐9 and (Z)‐9 under the Hayashi group's mild conditions17 afforded (4E)‐13 and (4Z)‐13; (h) notably, alkylations (Me and nBu) and phenylation using acetates of (E)‐8 and (Z)‐8 proceeded smoothly to afford the corresponding all‐carbon olefins (E)‐14 a–c and (Z)‐14 a–c; this finding was successfully applied for the parallel synthesis of (E)‐ and (Z)‐tamoxifens (vide infra).
Scheme 4.

Stereoretentive and complementary derivatization array of α,β‐unsaturated esters (E)‐ and (Z)‐5 b‐1. Reagents and conditions: [a] CF3CO2H‐H2O (2:1), 90 °C, 12 h; [b] DIBAL (3.0 equiv), CH2Cl2, RT, 1 h. [c] MnO4 (40 equiv), CH2Cl2, RT, 1 h; [d] i) H2/Pd‐C, AcOEt, RT, 1 h; ii) LAH (1.0 equiv), Et2O, 0 °C; [e] MeP+Ph3I− (4.0 equiv), tBuOK (4.0 equiv), CH2Cl2, RT, 1 h; [f] (MeO)2POCH2CO2Me, DBU (1.2 equiv), LiCl (1.2 equiv), MeCN, RT, 1 h; [g] H2C(CN)2 (1.0 equiv), Ti(OiPr)4 (0.5 equiv), iPrOH, RT, 24 h; [h] (E)‐ and (Z)‐8, i) Ac2O (1.2 equiv), Et3N (1.2 equiv), DMAP (5 mol %) CH2Cl2, RT, 1 h. ii) RLi (6.0 equiv), CuI (3.5 equiv), THF, RT, 1 h.
Finally, with these successful outcomes in hand, we describe the first successful fully parallel synthesis of both (E)‐ and (Z)‐tamoxifens,18 a representative of all carbon (fully)‐substituted olefins, utilizing the type I (convergent‐oriented approach) shown in Table 2. Scheme 5 (preparation of precursors) and Scheme 6 (synthesis in the final stage) illustrate this challenging task. The notable features in Scheme 5 are as follows: 1) β‐ketoester 15 was converted to enol tosylates (E)‐17 and (Z)‐17 following the reported (E)‐ and (Z)‐stereocomplementary procedures5 (conditions [a] and [b]); 2) in a similar approach, (Z)‐18 and (E)‐18 were prepared from β‐ketoester 16 (conditions [c] and [d]); 3) Negishi cross‐couplings using (E)‐17 and (Z)‐17 produced the corresponding α,β‐unsaturated ester precursors (Z)‐19 and (E)‐19 in excellent yield (95 % and 93 %) with good to almost perfect stereoretention, respectively (conditions [e] and [f]); 4) in a similar approach, (Z)‐18 and (E)‐18 were transformed to (Z)‐19 and (E)‐19, respectively in excellent yield (91 % and 81 %) with good to almost perfect stereoretention (conditions [g] and [h]).
Scheme 5.
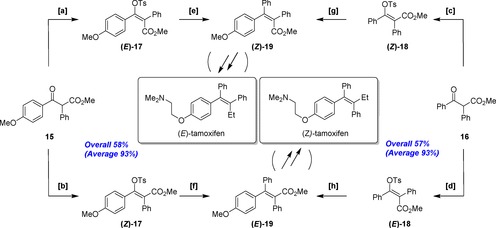
Parallel syntheses of both tamoxifen precursors (E)‐ and (Z)‐19. Reagents and conditions: [a] TsCl (1.5 equiv), Me2N(CH2)6NMe2 (1.5 equiv)/MeCN, −15 °C, 1 h and 20–25 °C, 1 h, 95 %, E/Z=66:34; pure (E)‐17 was isolated in 26 % yield (recrystallized from toluene); [b] TsCl (1.5 equiv), TMEDA (1.5 equiv), LiCl (1.5 equiv)/MeCN, 0–5 °C, 1 h and 20–25 °C, 1 h, 99 %, E/Z=2:>98; [c] The same conditions to those given for [b], 97 %, E/Z=2:98; [d] the same conditions to those given for [a], 92 %, E/Z=74:26; pure (E)‐18 was isolated in 49 % yield (recrystallized from AcOEt); [e] PhMgBr (3.0 equiv), ZnCl2 (3.0 equiv), [Pd(dppb)Cl2] (2 mol %), MeCN/THF (2:3), 60–65 °C, 2 h, 95 %, E/Z=16:84; [f] PhMgBr (2.0 equiv), ZnCl2 (2.0 equiv), Pd(OAc)2 (1 mol %), 1,4‐bis(diphenylphosphino)butane (DPPB; 2 mol %), THF, 60–65 °C, 2 h, 93 %, E/Z=98:2; [g] p‐(MeO)C6H4MgBr (2.0 equiv), ZnCl2 (2.0 equiv), Pd(OAc)2 (1 mol %), DPPB (2 mol %), THF, 60–65 °C, 2 h, 99 %, E/Z=2:98; [h] p‐(MeO)C6H4MgBr (3.0 equiv), ZnCl2 (3.0 equiv), [Pd(dppb)Cl2] (2 mol %), MeCN/THF (2:3), 60–65 °C, 2 h, 81 %, E/Z=90:10.
Scheme 6.

Parallel syntheses of both (E)‐ and (Z)‐tamoxifens. Reagents and conditions: [a] DIBAL (3.0 equiv)/CH2Cl2, 0–5 °C, 98 %, E/Z=>98:2; [b] Ac2O (1.2 equiv), Et3N (1.2 equiv), DMAP (5 mol %), CH2Cl2, 20–25 °C, 99 %, E/Z= >98:2; [c] MeLi (4.0 equiv), CuI (2.5 equiv), THF, 0–5 °C, 1 h, 82 %, E/Z= 2:>98; [d] NaSEt (10 equiv), DMF, reflux, 1 h, 97 %, E/Z=2:>98; [e] ClCH2CH2NMe2 ⋅HCl (2.0 equiv), K2CO3 (4.0 equiv), toluene/EtOH (1:1), 80–85 °C, 3 h, 93 %, E/Z=2:>98; [d] the same conditions to those given for [a], 90 % over two steps, E/Z=2:>98; [e] the same conditions to those given for [b], 80 %, E/Z=>98:2; [f] the same conditions to those given for [c], 96 % over two steps, E/Z=>98:2.
The salient features in Scheme 6 are that DIBAL reduction of precursors (E)‐19 and (Z)‐19, followed by acetylation led to allylic acetates (E)‐20 and (Z)‐20, respectively (conditions [a], [b], [f], and [g]). Utilizing the method for step [h] (Scheme 4), methylation of (E)‐20 and (Z)‐20 using MeLi/CuI proceeded smoothly to yield the corresponding fully substituted olefins (E)‐21 and (Z)‐21 (conditions [c] and [h]). A notable advantage of the present method is the reduced number of steps compared with reported transformations16b, 18c, 18k, 19 (Dess–Martin oxidation, Wittig methylene formation, and catalytic hydrogenation).
At the last stage of the synthesis, Miller and Al‐Hassan's protocol for demethylation and subsequent N,N‐dimethylethylene formation20 furnished both (E)‐ and (Z)‐tamoxifens in a total of 8 steps, with an overall 58 % (average 93 %) yield and an overall 57 % (average 93 %) yield, respectively. More than 50 syntheses of (E)‐ and/or (Z)‐tamoxifens have appeared to date and these achievements are documented in an impressive review.18a To the best of our knowledge, we have achieved the first two sets (all four) of fully‐parallel syntheses of both (E)‐ and (Z)‐tamoxifens with excellent overall yields.
3. Conclusions
We developed two sets (all four) of parallel and stereocomplementary synthetic pathway to access preparations of all‐carbon‐substituted (E)‐ and (Z)‐α,β‐unsaturated esters scaffolds. This robust and distinctive method involves stereocomplementary enol tosylations using readily available TsCl/diamine reagents and highly stereoretentive Negishi cross‐coupling reactions with fine‐tuned catalysis. The parallel approach is categorized into a pair of type I and type II pathways. Among a number of (E)‐ and (Z)‐α,β‐unsaturated esters, a set of methyl (E)‐ and (Z)‐α,β‐dimethylcinnamates was transformed into various E‐ and Z‐stereoretentive novel and known olefins with a total of 16 (8×2) derivatization arrays. As an attractive demonstration, the first parallel synthesis of both (E)‐ and (Z)‐tamoxifens, a representative compound of all‐carbon‐substituted olefins, was accomplished in a total of eight steps with both high overall yields and individual step average yields.
Experimental Section
General
Starting enol tosylates (E)‐ and (Z)‐2 a and (E)‐ and (Z)‐2 a’ are known compounds.5 Novel enol tosylates (E)‐ and (Z)‐2 b, (E)‐ and (Z)‐2 b’, (E)‐ and (Z)‐2 c, and (E)‐ and (Z)‐2 c’ were prepared according to a reported procedure (Supporting Information).5
Methyl (E)‐2‐Butyl‐3‐phenyloct‐2‐enoate [(E)‐5 a‐1]14
PhMgBr (0.92 mL, 1.00 mL; 1.09 m in THF) was added to a stirred suspension of ZnCl2 (136 mg, 1.00 mmol) in MeCN (0.50 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at the same temperature for 0.5 h. Enol tosylate (E)‐4 a 5 (191 mg, 0.50 mmol) in MeCN (0.5 mL) and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) were successively added to the mixture, which was then stirred at 60–65 °C for 2 h. After cooling to room temperature, 1 m aq. HCl solution was added to the mixture, which was then extracted twice with AcOEt. The combined organic phase was washed with aq. HCl (1 m), water, and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 column chromatography (hexane/AcOEt 100:1) to give (E)‐5 a‐1 as a colorless oil (118 mg, 82 %, E/Z=96:4). 1H NMR (300 MHz, CDCl3): δ=0.75 (t, J=7.5 Hz, 3 H), 0.82 (t, J=7.5 Hz, 3 H), 1.08–1.35 (m, 10 H), 2.07 (t, J=7.6 Hz, 2 H), 2.46 (t, J=7.6 Hz, 2 H), 3.80 (s, 3 H), 7.07–7.12 (m, 2 H), 7.24–7.30 (m, 1 H), 7.31–7.37 ppm (m, 2 H); 13C NMR (75 MHz, CDCl3): δ=13.8, 14.0. 22.3, 22.4, 27.6, 30.8, 31.2, 31.7, 36.4, 51.4, 126.8, 127.8 (2C), 128.1 (2C), 130.6, 141.4, 147.4, 170.8 ppm; IR (neat): max=2959, 1717, 1458, 1379, 1321, 1240, 1206, 1114 cm−1; HRMS (ESI): m/z: calcd for C19H28O2: 311.1987 [M+Na]+; found: 311.1987.
Methyl (Z)‐2‐Butyl‐3‐phenyloct‐2‐enoate [(Z)‐5 a‐1]14
PhMgBr (0.92 mL, 1.00 mL; 1.09 m in THF) was added to a stirred suspension of ZnCl2 (136 mg, 1.00 mmol) in THF (0.50 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at the same temperature for 0.5 h. Enol tosylate (Z)‐4 a 5 (191 mg, 0.50 mmol) in THF (0.5 mL) and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) were successively added to the mixture, which was then stirred at 60–65 °C for 2 h. After cooling to room temperature, aq. HCl (1 m) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with aq. HCl (1 m), water, and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 column chromatography (hexane/AcOEt 100:1) to give (Z)‐5 a‐1 as a colorless oil (122 mg, 85 %, E/Z=2:98).
1H NMR (300 MHz, CDCl3): δ=0.82 (t, J=6.9 Hz, 3 H), 0.96 (t, J=7.5 Hz, 3 H), 1.19–1.32 (m, 6 H), 1.34–1.48 (m, 4 H), 2.44 (t, J=7.2 Hz, 4 H), 3.33 (s, 3 H), 7.09–7.14 (m, 2 H), 7.20–7.32 ppm (m, 3 H); 13C NMR (75 MHz, CDCl3): δ=13.9, 22.4, 22.6, 27.5, 29.9, 31.1, 31.7, 34.0, 51.1, 126.9, 127.4 (2C), 127.9 (2C), 131.6, 142.7, 146.2, 171.3 ppm; IR (neat): max=2957, 2961, 1719, 1458, 1437, 1246, 1208, 1140 cm−1.
Methyl (E)‐2‐Methyl‐3‐phenyl‐3‐(p‐tolyl)acrylate [(E)‐3 a]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐2 a 5 (173 mg, 0.50 mmol) with p‐MeC6H4MgBr (0.94 mL, 1.00 mmol; 1.06 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐3 a as a colorless oil (100 mg, 75 %, E/Z=84:16).
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of tosylate (Z)‐2 a’ (188 mg, 0.50 mmol) with PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (E)‐3 a as a colorless oil (107 mg, 80 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=2.04 (s, 3 H), 2.35 (s, 3 H), 3.47 (s, 3 H), 7.02–7.07 (m, 2 H), 7.09–7.16 (m, 4 H), 7.20–7.90 (m, 3 H); 13C NMR (125 MHz, CDCl3): 18.5, 21.2, 51.4, 127.3 (2C), 127.8 (2C), 128.6 (2C), 128.7 (2C), 129.5 (2C), 137.4, 137.8, 142.6, 146.8, 171.7 ppm; IR (neat): max=3023, 2947, 1710, 1509, 1432, 1324, 1253, 1123 cm−1; HRMS (ESI): m/z: calcd for C18H18O2: 289.1204 [M+Na]+; found: 289.1186.
Methyl (Z)‐2‐Methyl‐3‐phenyl‐3‐(p‐tolyl)acrylate [(Z)‐3 a]
Method A: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 a 5 (173 mg, 0.50 mmol) using p‐MeC6H4MgBr (0.94 mL, 1.00 mmol; 1.06 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐3 a as a colorless oil (112 mg, 84 %, E/Z=2:98).
Method B: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐2 a’, 188 mg, 0.50 mmol) using PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (Z)‐3 a as a colorless oil (110 mg, 83 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=2.02 (s, 3 H), 2.32 (s, 3 H), 3.52 (s, 3 H), 6.96–7.02 (m, 2 H), 7.04–7.89 (m, 2 H), 7.14–7.86 (m, 2 H), 7.32–7.37 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=18.5, 21.2, 51.5, 127.3, 127.5, 128.0 (2C), 128.5 (2C), 128.6 (2C), 129.5 (2C), 137.8, 139.4, 141.0, 146.7, 171.7 ppm; IR (neat): max=3024, 2946, 1710, 1510, 1432, 1325, 1254, 1125 cm−1.
Methyl (E)‐3‐(4‐Methoxyphenyl)‐2‐methyl‐3‐phenylacrylate [(E)‐3 b]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐2 a (173 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐3 b as a colorless oil (109 mg, 77 %, E/Z=84:16).
[Method B]: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 b’ (181 mg, 0.50 mmol) with PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (E)‐3 b as a colorless oil (113 mg, 80 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=2.07 (s, 3 H), 3.46 (s, 3 H), 3.81 (s, 3 H), 6.83–6.89 (m, 2 H), 7.06–7.13 (m, 4 H), 7.21–7.30 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=18.5, 51.4, 55.1, 113.3 (2C), 127.0, 127.3, 127.8 (2C), 128.6 (2C), 131.0 (2C), 133.0, 142.7, 146.6, 159.0, 171.7 ppm; IR (neat): max=1709, 1605, 1508, 1443, 1325, 1304, 1288, 1277, 1175 cm−1; HRMS (ESI): m/z: calcd for C18H18O3: 305.1154 [M+Na]+; found: 305.1152.
Methyl (Z)‐3‐(4‐Methoxyphenyl)‐2‐methyl‐3‐phenylacrylate [(Z)‐3 b]
Method A: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 a (173 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (1.02 mL, 1.00 mmol; 0.98 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐3 b as a colorless oil (115 mg, 82 %, E/Z=2:98).
Method B: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐2 b’ (181 mg, 0.50 mmol) with PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (Z)‐3 b as a colorless oil (113 mg, 80 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=2.01 (s, 3 H), 3.53 (s, 3 H), 3.79 (s, 3 H), 6.77–6.81 (m, 2 H), 7.01–7.05 (m, 2 H), 7.14–7.17 (m, 2 H), 7.26–7.35 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=18.5, 51.5, 55.1, 113.3 (2C), 126.9, 127.5, 128.0 (2C), 129.6 (2C), 129.9 (2C), 134.7, 141.0, 146.2, 159.0, 171.9 ppm; IR (neat): max=2947, 2837, 1709, 1607, 1508, 1244, 1123, 1032, 775, 762, 700 cm−1.
Methyl (E)‐3‐(4‐Chlorophenyl)‐2‐methyl‐3‐phenylacrylate [(E)‐3 c]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐2 a (173 mg, 0.50 mmol) with p‐ClC6H4MgBr (0.96 mL, 1.00 mmol; 1.04 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐3 c as a colorless oil (78 mg, 54 %, E/Z=83:17).
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 c’ (183 mg, 0.50 mmol) with PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (E)‐3 c as a colorless oil (100 mg, 70 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=2.03 (s, 3 H), 2.48 (s, 3 H), 7.05–7.13 (m, 4 H), 7.23–7.38 ppm (m, 5 H); 13C NMR (125 MHz, CDCl3): δ=18.5, 51.6, 127.6 (2C), 128.0 (2C), 128.4 (2C), 128.5 (2C), 130.9 (2C), 133.6, 139.1, 141.9, 145.4, 171.3 ppm; IR (neat): max=1712, 1489, 1433, 1325, 1254, 1016 cm−1; HRMS (ESI): m/z: calcd for C17H15ClO2: 309.0658 [M+Na]+; found: 309.0665.
Methyl (Z)‐3‐(4‐Chlorophenyl)‐2‐methyl‐3‐phenylacrylate [(Z)‐3 c]
Method A: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 a (173 mg, 0.50 mmol) with p‐ClC6H4MgBr (0.96 mL, 1.00 mmol; 1.04 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐3 c as a colorless oil (122 mg, 85 %, E/Z=2:98).
Method B: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐2 c’ (183 mg, 0.50 mmol) with PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (Z)‐3 c, 100 mg, 70 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=2.03 (s, 3 H), 3.52 (s, 3 H), 7.02–7.07 (m, 2 H), 7.11–7.15 (m, 2 H), 7.21–7.25 (m, 2 H), 7.28–7.38 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=18.4, 51.6, 127.8 (2C), 128.2 (2C), 128.4 (2C), 129.4 (2C), 129.9 (2C), 133.3, 140.3, 140.8, 145.5, 171.2 ppm; IR (neat): max=1712, 1489, 1443, 1433, 1323, 1252, 1125 cm−1.
Methyl (E)‐3‐(4‐Chlorophenyl)‐3‐(4‐methoxyphenyl)‐2‐methylacrylate [(E)‐3 d]
Method A: Following the procedure for the preparation of (E)‐5 a‐1 (the reaction of enol tosylate (E)‐2 d (=2 b′) (183 mg, 0.50 mmol) with p‐ClC6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) in toluene (3 mL) at reflux gave (E)‐3 d as a colorless oil (136 mg, 85 %, E/Z=68:32).
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 d (=2 c′) (181 mg, 0.50 mmol) with p‐ClC6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (E)‐3 d as a colorless oil (122 mg, 77 %, E/Z=90:10).
1H NMR (500 MHz, CDCl3): δ=2.01 (s, 3 H), 3.23 (s, 3 H), 3.74 (s, 3 H), 6.76–6.82 (m, 2 H), 6.98–7.03 (m, 2 H), 7.07–7.12 (m, 2 H), 7.29–7.34 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=18.5, 51.5, 55.0, 113.3 (2C), 127.4, 128.9 (2C), 129.9 (2C), 131.0 (2C), 133.5, 134.1, 139.4, 144.8, 159.1, 171.5 ppm; IR (neat): max=1713, 1607, 1508, 1487, 1456, 1323, 1306, 1288, 1175 cm−1; HRMS (ESI): m/z: calcd for C18H17ClO3: 339.0764 [M+Na]+; found: 339.0762.
Methyl (Z)‐3‐(4‐Chlorophenyl)‐3‐(4‐methoxyphenyl)‐2‐methylacrylate [(Z)‐3 d]
Method A: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐2 c (=2 b′) (183 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) in toluene (3 mL) at reflux gave (Z)‐3 d as a colorless oil (127 mg, 80 %, E/Z=2:98).
Method B: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate [(E)‐2 d’ (=2 c′), 181 mg, 0.50 mmol] with p‐ClC6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐3 d’ as a colorless oil (138 mg, 87 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=2.06 (s, 3 H), 3.50 (s, 3 H), 3.82 (s, 3 H), 6.84–6.89 (m, 2 H), 7.01–7.09 (m, 4 H), 7.21–7.26 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=18.6, 51.5, 55.1, 113.5 (2C), 127.5, 128.0 (2C), 130.0 (2C), 131.0 (2C), 132.6, 133.3, 141.2, 145.5, 159.2, 171.3 ppm; IR (neat): max=1605, 1508, 1489, 1456, 1433, 1321, 1304, 1288 cm−1.
Methyl (E)‐2‐Methyl‐3‐phenylbut‐2‐enoate [(E)‐5 b‐1]20
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 b (142 mg, 0.50 mmol) with PhMgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 b‐1 as a colorless oil (80 mg, 84 %, E/Z=98:2; Ref. 19: E/Z=14:86; Ref. 19: E/Z=80:20).
1H NMR (300 MHz, CDCl3): δ=1.75 (q, J=1.4 Hz, 3 H,), 2.26 (q, J=1.4 Hz, 3 H), 3.80 (s, 3 H), 7.12–7.15 (m, 2 H), 7.27–7.38 ppm (m, 3 H); IR (neat): max=2949, 1716, 1433, 1253, 1133, 1099 cm−1.
Methyl (Z)‐2‐Methyl‐3‐phenylbut‐2‐enoate [(Z)‐5 b‐1]20a
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 b (142 mg, 0.50 mmol) with PhMgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 b‐1 as a colorless oil (79 mg, 83 %, E/Z=2:98; Ref. 19: 95 % yield, E/Z=14:86).
1H NMR (300 MHz, CDCl3): δ=2.05 (q, J=0.6 Hz, 3 H), 2.09 (q, J=0.6 Hz, 3 H), 3.39 (s, 3 H), 7.12–7.14 (m, 2 H), 7.23–7.32 ppm (m, 3 H); IR (neat): max=2947, 1714, 1433, 1316, 1243, 1139 cm−1.
Methyl (E)‐3‐(4‐Methoxyphenyl)‐2‐methylbut‐2‐enoate [(E)‐5 b‐2]20a
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 b (142 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 b‐2 as a colorless oil (90 mg, 82 %, E/Z=95:5; Ref. 19: 14 % yield, E/Z=4:96; 90 % yield, E/Z=41:59).
1H NMR (300 MHz, CDCl3): δ=1.78 (d, J=1.4 Hz, 3 H), 2.25 (d, J=1.4 Hz, 3 H), 3.79 (s, 3 H), 3.82 (s, 3 H), 6.87–6.91 (m, 2 H), 7.06–7.10 ppm (m, 2 H); IR (neat): max=2950, 1714, 1608, 1510, 1248, 1132, 1032 cm−1.
Methyl (Z)‐3‐(4‐Methoxyphenyl)‐2‐methylbut‐2‐enoate [(Z)‐5 b‐2]14
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 b (142 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (1.03 mL, 1.00 mmol; 0.97 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 b‐2 (104 mg, 95 %, E/Z=2:98).
Methyl (E)‐3‐(4‐Chlorophenyl)‐2‐methylbut‐2‐enoate [(E)‐5 b‐3]14
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 b (142 mg, 0.50 mmol) with p‐ClC6H4MgBr (1.03 mL, 1.00 mmol; 0.97 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 b‐3 (104 mg, 93 %, E/Z=98:2).
Methyl (Z)‐3‐(4‐Chlorophenyl)‐2‐methylbut‐2‐enoate [(Z)‐5 b‐3]14
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 b (142 mg, 0.50 mmol) with p‐ClC6H4MgBr (1.03 mL, 1.00 mmol; 0.97 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 b‐3 (90 mg, 80 %, E/Z=2:98).
Methyl (E)‐2‐(1‐Phenylethylidene)hexanoate [(E)‐5 c‐1]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 c (163 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 c‐1 as a colorless oil (99 mg, 85 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=0.76 (t, J=7.5 Hz, 3 H), 1.15 (sextet, J=7.5 Hz, 2 H), 1.23–1.32 (m, 2 H), 2.13 (t, J=8.0 Hz, 2 H), 2.16 (s, 3 H), 3.81 (s, 3 H), 7.11–7.15 (m, 2 H), 7.25–7.29 (m, 1 H), 7.32–7.37 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 22.3, 23.3, 30.6, 31.2, 51.4, 126.9, 127.1 (2C), 128.2 (2C), 130.6, 143.0, 143.5, 170.5 ppm; IR (neat): max=2955, 2860, 1718, 1490, 1434, 1316, 1258, 1136 cm−1.
Methyl (Z)‐2‐(1‐Phenylethylidene)hexanoate [(Z)‐5 c‐1]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 c (163 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 c‐1 as a colorless oil (88 mg, 88 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.94 (t, J=7.5 Hz, 3 H), 1.34–1.51 (m, 4 H), 2.10 (s, 3 H), 2.44 (t, J=8.0 Hz, 2 H), 3.37 (s, 3 H), 7.12–7.16 (m, 2 H), 7.21–7.25 (m, 1 H), 7.27–7.32 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.9, 20.8, 22.5, 30.4, 30.7, 51.1, 126.8 (2C), 126.9, 127.9 (2C), 131.5, 141.7, 144.0, 171.1 ppm; IR (neat): max=2955, 2871, 1714, 1492, 1433, 1318, 1240, 1139 cm−1; HRMS (ESI): m/z: calcd for C15H20O2: 255.1361 [M+Na]+; found: 255.1360.
Methyl (E)‐2‐Methyl‐3‐phenylhept‐2‐enoate [(E)‐5 c′‐1]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 c’ (163 mg, 0.50 mmol) using PhMgBr (1.04 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave the desired product (E)‐5 c′‐1 as a colorless oil (83 mg, 72 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=0.82 (t, J=6.9 Hz, 3 H), 1.21–1.34 (m, 4 H), 1.71 (s, 3 H), 2.59 (t, J=6.9 Hz, 2 H), 3.79 (s, 3 H), 7.08–7.13 (m, 2 H), 7.27–7.30 (m, 1 H), 7.33–7.38 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 17.4, 22.6, 30.9, 35.9, 51.4, 124.5, 126.9, 127.7 (2C), 128.1 (2C), 141.8, 150.0, 170.4 ppm; IR (neat): max=1717, 1441, 1433, 1246, 1190, 1134, 1105 cm−1; HRMS (ESI): m/z: calcd for C15H20O2: 255.1361 [M+Na]+; found: 255.1362.
Methyl (Z)‐2‐Methyl‐3‐phenylhept‐2‐enoate [(Z)‐5 c′‐1]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4′c (163 mg, 0.50 mmol) using PhMgBr (10.4 mL, 1.00 mmol; 0.96 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave the desired product (Z)‐5 c′‐1 as a colorless oil (79 mg, 70 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.85 (t, J=6.9 Hz, 3 H), 1.24–1.33 (m, 4 H), 2.03 (s, 3 H), 2.45 (t, J=6.9 Hz, 2 H), 3.36 (s, 3 H), 7.08–7.11 (m, 2 H), 7.22–7.31 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 15.8, 22.5, 29.6, 34.6, 51.1, 125.7, 126.8, 127.2 (2C), 127.8 (2C), 142.8, 147.7, 171.0 ppm; IR (neat): max=1713, 1433, 1315, 1240, 1136, 1084 cm−1.
Methyl (E)‐2‐[1‐(4‐Methoxyphenyl)ethylidene]hexanoate [(E)‐5 c‐2]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 c (163 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave the desired product (E)‐5 c‐2 as a colorless oil (117 mg, 89 %, E/Z=91:9).
1H NMR (500 MHz, CDCl3): δ=0.78 (t, J=7.5 Hz, 3 H), 1.17 (sextet, J=7.5 Hz, 2 H), 1.24–1.32 (m, 2 H), 2.14 (s, 3 H), 2.15 (t, J=8.0 Hz, 2 H), 3.80 (s, 3 H), 3.82 (s, 3 H), 6.86–6.90 (m, 2 H), 7.04–7.09 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 22.3, 23.4, 30.7, 31.3, 51.3, 55.1, 113.6 (2C), 128.3 (2C), 130.5, 135.3, 143.1, 158.5, 170.7 ppm; IR (neat): max=2955, 2837, 1715, 1609, 1508, 1244, 1134, 1032, 831 cm−1; HRMS (ESI): m/z: calcd for C16H22O3: 285.1467 [M+Na]+; found: 285.1466.
Methyl (Z)‐2‐[1‐(4‐Methoxyphenyl)ethylidene]hexanoate [(Z)‐5 c‐2]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 c (163 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 c‐2 as a colorless oil (119 mg, 91 %, E/Z=2:>98).
1H NMR (500 MHz, CDCl3): δ=0.93 (t, J=7.5 Hz, 3 H), 1.33–1.49 (m, 4 H), 2.07 (s, 3 H), 2.42 (t, J=7.5 Hz, 2 H), 3.41 (s, 3 H), 3.80 (s, 3 H), 6.81–6.85 (m, 2 H), 7.06–7.11 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.9, 20.8, 22.6, 30.6, 30.8, 51.2, 55.1, 113.4 (2C), 128.1 (2C), 131.1, 136.3, 141.0, 158.6, 171.6 ppm; IR (neat): max=2955, 2837, 1711, 1607, 1510, 1244, 1138, 1026, 831 cm−1.
Methyl (E)‐3‐(4‐Methoxyphenyl)‐2‐methylhept‐2‐enoate [(E)‐5 c′‐2]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 c’ (163 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 c′‐2, as a colorless oil (98 mg, 75 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=0.82 (t, J=6.9 Hz, 3 H), 1.20–1.33 (m, 4 H), 1.73 (s, 3 H), 2.57 (t, J=6.9 Hz, 2 H), 3.78 (s, 3 H), 3.82 (s, 3 H), 6.86–6.92 (m, 2 H), 7.02–7.07 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.9, 17.5, 22.6, 30.4, 36.1, 51.4, 55.2, 113.5 (2C), 124.4, 129.0 (2C), 134.0, 149.7, 158.5, 170.6 ppm; IR (neat): max=1715, 1609, 1508, 1456, 1433, 1287, 1242, 1177, 1132 cm−1; HRMS (ESI): m/z: calcd for C16H22O3: 285.1467 [M+Na]+; found: 285.1458.
Methyl (Z)‐3‐(4‐Methoxyphenyl)‐2‐methylhept‐2‐enoate [(Z)‐5 c′‐2]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4′c (163 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (0.98 mL, 1.00 mmol; 1.02 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5′c‐2 as a colorless oil (108 mg, 82 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.85 (t, J=6.9 Hz, 3 H), 1.22–1.33 (m, 4 H), 2.01 (s, 3 H), 2.43 (t, J=6.9 Hz, 2 H), 3.41 (s, 3 H), 3.80 (s, 3 H), 6.80–6.85 (m, 2 H), 7.02–7.07 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 15.9, 22.5, 29.5, 34.5, 51.1, 55.0, 113.2 (2C), 125.3, 128.4 (2C), 134.9, 147.0, 158.5, 171.3 ppm; IR (neat): max=1711, 1607, 1508, 1456, 1433, 1317, 1288 cm−1.
Methyl (E)‐2‐[1‐(4‐Chlorophenyl)ethylidene]hexanoate [(E)‐5 c‐3]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 c (163 mg, 0.50 mmol) with p‐ClC6H4MgBr (0.96 mL, 1.00 mmol; 1.04 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 c‐3, as a colorless oil (117 mg, 88 %, E/Z=97:3].
1H NMR (500 MHz, CDCl3): δ=0.77 (t, J=7.5 Hz, 3 H), 1.16 (sextet, J=7.5 Hz, 2 H), 1.23–1.30 (m, 2 H), 2.11 (t, J=8.0 Hz, 2 H), 2.13 (s, 3 H), 3.80 (s, 3 H), 7.05–7.09 (m, 2 H), 7.31–7.34 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 22.3, 23.2, 30.7, 31.2, 51.5, 128.5 (2C), 128.6 (2C), 131.3, 132.8, 141.3, 142.0, 170.3 ppm; IR (neat): max=2955, 2860, 1717, 1489, 1433, 1258, 1246, 1206, 1136, 1092, 1015, 827 cm−1; HRMS (ESI): m/z: calcd for C115H19O2Cl: 289.0971 [M+Na]+; found: 289.0966.
Methyl (Z)‐2‐[1‐(4‐Chlorophenyl)ethylidene]hexanoate [(Z)‐5 c‐3]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 c (163 mg, 0.50 mmol) with p‐ClC6H4MgBr (0.96 mL, 1.00 mmol; 1.04 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 c‐3, as a colorless oil (104 mg, 78 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.94 (t, J=6.9 Hz, 3 H), 1.33–1.49 (m, 4 H), 2.06 (s, 3 H), 2.43 (t, J=6.9 Hz, 2 H), 3.41 (s, 3 H), 7.06–7.09 (m, 2 H), 7.24–7.28 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.9, 20.8, 22.5, 30.4, 30.7, 51.2, 128.15 (2C), 128.23 (2C), 132.1, 132.7, 140.6, 142.4, 170.7 ppm; IR (neat): max=2955, 2872, 1713, 1485, 1433, 1315, 1242, 1138, 1090, 1013, 827 cm−1.
Methyl (E)‐3‐(4‐Chlorophenyl)‐2‐methylhept‐2‐enoate [(E)‐5 c′‐3]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 c’ (163 mg, 0.50 mmol) with p‐ClC6H4MgBr (0.96 mL, 1.00 mmol; 1.04 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) in toluene (3 mL) at reflux gave the (E)‐5 c′‐3 as a colorless oil (104 mg, 78 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=0.82 (t, J=6.9 Hz, 3 H), 1.21–1.31 (m, 4 H), 1.70 (s, 3 H), 2.56 (t, J=6.9 Hz, 2 H), 3.79 (s, 3 H), 7.02–7.07 (m, 2 H), 7.31–7.36 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 17.4, 22.6, 30.2, 35.8, 51.5,125.2, 128.5 (2C), 129.2 (2C), 132.8, 140.1, 148.5, 170.2 ppm; IR (neat): max=1717, 1489, 1456, 1433, 1246, 1134, 1115 cm−1; HRMS (ESI): m/z: calcd for C15H19ClO2: 289.0971 [M+Na]+; found: 289.0966.
Methyl (Z)‐3‐(4‐Chlorophenyl)‐2‐methylhept‐2‐enoate [(Z)‐5 c′‐3]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4′c (163 mg, 0.50 mmol) with p‐ClC6H4MgBr (0.96 mL, 1.00 mmol; 1.04 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 c′‐3 as a colorless oil (93 mg, 70 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.85 (t, J=6.9 Hz, 3 H), 1.22–1.32 (m, 4 H), 2.02 (s, 3 H), 2.41 (t, J=6.9 Hz, 2 H), 3.41 (s, 3 H), 7.01–7.06 (m, 2 H), 7.24–7.29 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 15.9, 22.5, 29.4, 34.6, 51.3, 126.3, 128.1 (2C), 128.6 (2C), 132.7, 141.3, 146.6, 170.6 ppm; IR (neat): max=1715, 1489, 1433, 1315, 1238, 1136 cm−1.
Methyl (E)‐2‐Butyl‐3‐(4‐methoxyphenyl)oct‐2‐enoate [(E)‐5 a‐2]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 a (191 mg, 0.50 mmol) with p‐(MeO)C6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.010 mmol) gave (E)‐5 a‐2 as a colorless oil (82 mg, 52 %, E/Z=91:9).
1H NMR (500 MHz, CDCl3): δ=0.77 (t, J=6.9 Hz, 3 H), 0.82 (t, J=6.9 Hz, 3 H), 1.11–1.34 (m, 10 H), 2.10 (t, J=8.0 Hz, 2 H), 2.43 (t, J=8.0 Hz, 2 H), 3.79 (s, 3 H), 3.82 (s, 3 H), 6.86–6.90 (m, 2 H), 7.00–7.05 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 14.0, 22.37, 22.39, 27.7, 30.8, 31.3, 31.7, 36.6, 51.3, 55.2, 113.5 (2C), 128.9 (2C), 130.6, 133.7, 147.0, 158.4, 170.9 ppm; IR (neat): max=2954, 2927, 2859, 1717, 1608, 1509, 1463, 1245 cm−1; HRMS (ESI): m/z: calcd for C20H30O3: 341.2093 [M+Na]+; found: 341.2095.
Methyl (Z)‐2‐Butyl‐3‐(4‐methoxyphenyl)oct‐2‐enoate [(Z)‐5 a‐2]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 a (191 mg, 0.500 mmol) with p‐(MeO)C6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.0050 mmol) gave (Z)‐5 a‐2 as a colorless oil (84 mg, 53 %, E/Z=2:>98).
1H NMR (500 MHz, CDCl3): δ=0.83 (t, J=6.9 Hz, 3 H), 0.93 (t, J=7.5 Hz, 3 H), 1.20–1.29 (m, 6 H), 1.33–1.46 (m, 4 H), 2.38–2.45 (m, 4 H), 3.38 (s, 3 H), 3.80 (s, 3 H), 6.80–6.84 (m, 2 H), 7.03–7.07 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.9 (2C), 22.4, 22.5, 27.5, 30.0, 31.1, 31.6, 34.0, 51.1, 55.1, 113.3 (2C), 128.5 (2C), 131.3, 134.8, 145.5, 158.5, 171.6 ppm; IR (neat): max=2955, 2871, 1710, 1607, 1509, 1462, 1323, 1244 cm−1.
Methyl (E)‐2‐Butyl‐3‐(4‐chlorophenyl)oct‐2‐enoate [(E)‐5 a‐3]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 a (191 mg, 0.50 mmol) with p‐ClC6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 a‐3 as a colorless oil (92 mg, 57 %, E/Z=>98:2).
1H NMR (500 MHz, CDCl3): δ=0.77 (t, J=7.5 Hz, 3 H), 0.82 (t, J=6.9 Hz, 3 H), 1.11–1.31 (m, 10 H), 2.06 (t, J=8.0 Hz, 2 H), 2.43 (t, J=8.0 Hz, 2 H), 3.79 (s, 3 H), 7.02–7.06 (m, 2 H), 7.30–7.34 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 13.9, 23.3, 23.4, 27.6, 30.8, 31.1, 31.6, 36.3, 51.4, 128.4 (2C), 129.2 (2C), 131.3, 132.8, 139.8, 145.8, 170.5 ppm; IR (neat): max=2956, 2929, 2860, 1720, 1489, 1462, 1433, 1204 cm−1.
Methyl (Z)‐2‐Butyl‐3‐(4‐chlorophenyl)oct‐2‐enoate [(Z)‐5 a‐3]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 a (191 mg, 0.50 mmol) with p‐ClC6H4MgBr (1.00 mL, 1.00 mmol; 1.00 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 a‐3 as a colorless oil (96 mg, 59 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.83 (t, J=6.9 Hz, 3 H), 0.93 (t, J=6.9 Hz, 3 H), 1.19–1.29 (m, 6 H), 1.34–1.46 (m, 4 H), 2.37–2.45 (m, 4 H), 3.38 (s, 3 H), 7.02–7.07 (m, 2 H), 7.23–7.28 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.9 (2C), 22.4, 22.5, 27.4, 29.9, 31.0, 31.6, 34.0, 51.2, 128.1 (2C), 128.8 (2C), 132.2, 132.7, 141.1, 145.0, 170.9 ppm; IR (neat): max=2956, 2860, 1715, 1488, 1466, 1432, 1242, 1139 cm−1; HRMS (ESI): m/z: calcd for C19H27O2Cl: 345.1597 [M+Na]+; found: 345.1611.
Methyl (E)‐2‐Isopropyl‐3‐phenylbut‐2‐enoate [(E)‐5 d]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 d (156 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 d as a colorless oil (37 mg, 34 %, E/Z=96:4].
1H NMR (500 MHz, CDCl3): δ=1.00 (d, J=7.5 Hz, 6 H), 1.99 (s, 3 H), 2.54 (septet, J=7.5 Hz, 1 H), 3.82 (s, 3 H), 7.14–7.17 (m, 2 H), 7.25–7.30 (m, 1 H), 7.33–7.38 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=21.4 (2C), 23.3, 29.8, 51.2, 126.9, 127.2 (2C), 128.3 (2C), 136.8, 137.3, 142.2, 170.5 ppm; IR (neat): max=2964, 1723, 1599, 1492, 1433, 1301, 1243, 1143 cm−1.
Methyl (Z)‐2‐Isopropyl‐3‐phenylbut‐2‐enoate [(Z)‐5 d]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 d (156 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 d as a colorless oil (86 mg, 79 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=1.17 (d, J=7.5 Hz, 6 H), 2.08 (s, 3 H), 2.94 (hept, J=7.5 Hz, 1 H), 3.36 (s, 3 H), 7.16–7.31 ppm (m, 5 H); 13C NMR (125 MHz, CDCl3): δ=19.6, 20.8 (2C), 29.3, 50.8, 126.9, 127.0 (2C), 128.0 (2C), 136.7, 137.8, 143.7, 170.4 ppm; IR (neat): max=2968, 1717, 1431, 1385, 1304, 1239, 1138, 1014 cm−1; HRMS (ESI): m/z: calcd for C14H18O2: 219.1385 [M+H]+; found: 219.1384.
Methyl (E)‐2,3‐Diphenylbut‐2‐enoate [(E)‐5 e]14
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 e (180 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (E)‐5 e, 126 mg, >99 %, E/Z=97:3).
Methyl (Z)‐2,3‐Diphenylbut‐2‐enoate [(Z)‐5 e]14, 21
Method A: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 e (180 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 e (125 mg, 99 %, E/Z=2:98).
Methyl (Z)‐2‐Methoxy‐3‐phenyloct‐2‐enoate [(Z)‐5 f]
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (Z)‐4 f (178 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave (Z)‐5 f as a colorless oil (121 mg, 92 %, E/Z=2:98).
1H NMR (500 MHz, CDCl3): δ=0.80–0.86 (m, 3 H), 1.19–1.40 (m, 6 H), 2.67–2.73 (m, 2 H), 3.34 (s, 3 H), 3.85 (s, 3 H), 7.24–7.38 ppm (m, 5 H); 13C NMR (125 MHz, CDCl3): δ=13.9, 22.3, 28.0, 31.6, 32.8, 51.6, 59.5, 127.3, 127.9 (2C), 128.0 (2C), 138.9, 140.6, 142.1, 165.3 ppm; IR (neat): max=2929, 2859, 1718, 1442, 1254, 1200, 1137, 1089 cm−1.
Methyl (E)‐2‐Methoxy‐3‐phenyloct‐2‐enoate [(E)‐5 f]
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (E)‐4 f (178 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.0050 mmol) gave (E)‐5 f as a colorless oil (117 mg, 89 %, E/Z=98:2).
1H NMR (500 MHz, CDCl3): δ=0.81–0.87 (m, 3 H), 1.21–1.33 (m, 6 H), 2.52 (t, J=8.0 Hz, 2 H), 3.49 (s, 3 H), 3.65 (s, 3 H), 7.09–7.14 (m, 2 H), 7.24–7.34 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=13.9, 22.3, 17.0, 31.6, 32.5, 51.3, 59.0, 127.1, 127.6 (2C), 127.8 (2C), 138.6, 139.4, 142.9, 164.9 ppm; IR (neat): max=2928, 1720, 1631, 1434, 1318, 1259, 1205, 1144 cm−1; HRMS (ESI): m/z: calcd for C16H22O3: 285.1467 [M+Na]+; found: 285.1461.
Methyl (E)‐7‐Chloro‐2‐methyl‐3‐phenylhept‐2‐enoate [(E)‐5 g]14
Method A: Following the procedure for the preparation of (E)‐5 a‐1, the reaction of enol tosylate (E)‐4 g (180 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppe)Cl2] (5.8 mg, 0.01 mmol) gave the desired product (E)‐5 g (119 mg, 89 %, E/Z=98:2).
Methyl (Z)‐7‐Chloro‐2‐methyl‐3‐phenylhept‐2‐enoate [(Z)‐5 g]14
Method B: Following the procedure for the preparation of (Z)‐5 a‐1, the reaction of enol tosylate (Z)‐4 g (180 mg, 0.50 mmol) with PhMgBr (0.92 mL, 1.00 mmol; 1.09 m in THF), ZnCl2 (136 mg, 1.00 mmol), and [Pd(dppb)Cl2] (3.0 mg, 0.005 mmol) gave (Z)‐5 g (120 mg, 90 %, E/Z=2:98).
(E)‐2‐Methyl‐3‐phenylbut‐2‐enoic acid [(E)‐7] (Scheme 4)
(E)‐5 b‐1 (102 mg, 0.5 mmol) in CF3CO2H (1.0 mL) and H2O (0.5 mL) was heated at 90–95 °C under an Ar atmosphere for 12 h. The reaction mixture was cooled to room temperature, then aq. NaOH (3 m, 3.0 mL) was added. The reaction mixture was then washed with Et2O. Aq. HCl (1 m) was added to the combined water phase, which was re‐extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated to give (E)‐7 as pale yellow crystals (72 mg, 82 %, E/Z=98:2), which did not require purification. M.p. 105–107 °C; 1H NMR (500 MHz, CDCl3): δ=1.80 (s, 3 H), 2.38 (s, 3 H), 7.11–7.18 (m, 2 H), 7.27–7.33 (m, 1 H), 7.34–7.41 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=17.3, 23.7, 123.8, 126.9 (2C), 127.2, 128.4 (2C), 143.7, 150.3, 175.5 ppm; IR (neat): max=2961, 2621, 1660, 1489, 1438, 1404, 1261, 1155, 1096, 937, 727, 700 cm−1.
(E)‐2‐Methyl‐3‐phenylbut‐2‐en‐1‐yl acetate [(E)‐8′] (Scheme 4)
DIBAL (19.2 mL, 19.2 mmol, 1.0 m in toluene) was added to a stirred solution of (E)‐5 b‐1 (1.22 g, 6.4 mmol) in CH2Cl2 (12.8 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at the same temperature for 1 h. Saturated aq. Rochelle salt (12.0 mL) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated to give the desired allyl alcohol (E)‐8 as a colorless oil (935 mg, 90 %, E/Z=>98:2), which did not require purification. 1H NMR (500 MHz, CDCl3): δ=1.37 (s, 1 H), 1.68 (s, 3 H), 2.04 (s, 3 H), 4.31 (d, J=4.6 Hz, 2 H), 7.09–7.16 (m, 2 H), 7.20–7.25 (m, 1 H), 7.29–7.36 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=17.8, 20.3, 63.6, 126.2, 127.9 (2C), 128.0 (2C), 130.3, 134.1, 144.4 ppm; IR (neat): max=3329, 2914, 1653, 1599, 1491, 1441, 1371, 993, 764, 700 cm−1.
Ac2O (613 mg, 6.0 mmol) was added to a stirred solution of (E)‐8 (811 mg, 5.0 mmol), Et3N (607 mg, 6.0 mmol), and 4‐(dimethylamino)pyridine (DMAP, 31 mg, 0.25 mmol) in CH2Cl2 (10.0 mL) at 20–25 °C under an Ar atmosphere, then the reaction mixture was stirred at the same temperature for 1 h. Water was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane/AcOEt 20:1) to give (E)‐8′ as a colorless oil (868 mg, 85 %, E/Z=>98:2). 1H NMR (500 MHz, CDCl3): δ=1.62 (s, 3 H), 2.04 (s, 3 H), 2.11 (s, 3 H), 4.76 (s, 2 H), 7.11–7.17 (m, 2 H), 7.20–7.26 (m, 1 H), 7.30–7.37 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=18.0, 20.6, 20.9, 65.3, 125.8, 126.4, 127.8 (2C), 128.0 (2C), 136.5, 144.0, 171.2 ppm; IR (neat): max=3021, 1736, 1493, 1441, 1373, 1225, 1022, 984, 766, 700 cm−1.
(E)‐(3‐Methylpent‐2‐en‐2‐yl)benzene [(E)‐14 a] (Scheme 4)
MeLi (3.37 mL, 3.0 mmol; 0.89 m in Et2O) was added dropwise to a stirred suspension of CuI (333 mg, 1.75 mmol) in THF (0.75 mL) at 0–5 °C under an Ar atmosphere. (E)‐8′ (102 mg, 0.5 mmol) in THF (0.75 mL) was added to the mixture at the same temperature, and the reaction mixture was stirred for 1 h. The mixture was reversely quenched into saturated aq. NH4Cl and filtered through Celite. The obtained filtrate was extracted twice with Et2O. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane) to give (E)‐14 a as a colorless oil (64 mg, 80 %, E/Z=>98:2). 1H NMR (500 MHz, CDCl3): δ=1.06 (t, J=7.5 Hz, 3 H), 1.56 (s, 3 H), 1.95 (s, 3 H), 2.19 (q, J=7.5 Hz, 2 H), 7.08–7.14 (m, 2 H), 7.15–7.21 (m, 1 H), 7.26–7.33 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=12.5, 19.4, 20.2, 27.4, 125.7, 127.9 (2C), 128.4 (2C), 129.7, 132.9, 145.5 ppm; IR (neat): max=2965, 1599, 1491, 1441, 1371, 1132, 1072, 1026, 763, 700 cm−1.
(E)‐(2‐Methylbut‐2‐ene‐1,3‐diyl)dibenzene [(E)‐14 b] (Scheme 4)
In a similar procedure for the preparation of (E)‐14 a, the reaction of (E)‐8′ (102 mg, 0.5 mmol) with PhMgBr (2.94 mL, 3.00 mmol; 1.02 m in THF) using CuI (333 mg, 1.75 mmol) gave (E)‐14 b as a colorless oil (82 mg, 54 %, E/Z=>98:2, containing ≈20 % of biphenyl). 1H NMR (500 MHz, CDCl3): δ=1.52 (s, 3 H), 2.09 (s, 3 H), 3.56 (s, 2 H), 7.13–7.27 (m, 6 H), 7.28–7.38 ppm (m, 4 H); 13C NMR (125 MHz, CDCl3): δ=20.0, 21.1, 40.0, 125.9, 126.0, 128.0 (2C), 128.3 (2C), 128.37 (2C), 128.40 (2C), 129.9, 132.3, 140.4, 145.0 ppm; IR (neat): max=3026, 2914, 1599, 1493, 1441, 1371, 1072, 1028, 737, 696, 627 cm−1.
(E)‐(3‐Methyloct‐2‐en‐2‐yl)benzene [(E)‐14 c] (Scheme 4)
In a similar procedure for the preparation of (E)‐14 a, the reaction of (E)‐8’ (102 mg, 0.5 mmol) with nBuLi (1.88 mL, 3.00 mmol; 1.60 m in hexane) using CuI (333 mg, 1.75 mmol) gave (E)‐14 c as a colorless oil (43 mg, 43 %, E/Z=>98:2). 1H NMR (500 MHz, CDCl3): δ=0.93 (t, J=6.9 Hz, 3 H), 1.28–1.42 (m, 4 H), 1.42–1.52 (m, 2 H), 1.55 (s, 3 H), 1.95 (s, 3 H), 2.16 (t, J=6.9 Hz, 2 H), 7.09–7.14 (m, 2 H), 7.16–7.22 (m, 1 H), 7.27–7.34 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=14.1, 19.9, 20.5, 22.7, 27.8, 31.9, 34.3, 125.7, 127.9 (2C), 128.4 (2C), 130.2, 131.6, 145.6 ppm; IR (neat): max=2924, 1599, 1491, 1441, 1373, 1072, 1024, 764, 700 cm−1.
(E)‐2‐Methyl‐3‐phenylbut‐2‐enal [(E)‐9] (Scheme 4)
A mixture of (E)‐8 (316 mg, 2.0 mmol) and MnO2 (6.96 g, 80 mmol) in CH2Cl2 (40 mL) was stirred at 20–25 °C under an Ar atomosphere for 12 h. The mixture was filtered through Celite, and the filtrate was concentrated to give (E)‐9 as a colorless oil (316 mg, 99 %, E/Z=>98:2), which did not need further purification. 1H NMR (500 MHz, CDCl3): δ=1.67 (s, 3 H), 2.47 (s, 3 H), 7.15–7.23 (m, 2 H), 7.30–7.37 (m, 1 H), 7.37–7.45 (m, 2 H), 10.35 ppm (s, 1 H); 13C NMR (125 MHz, CDCl3): δ=13.1, 19.5, 126.9 (2C), 128.0, 128.5 (2C), 133.3, 142.9, 155.7, 192.0 ppm; IR (neat): max=2864, 1620, 1597, 1493, 1373, 1308, 1277, 1217, 972 cm−1; HRMS (ESI): m/z: calcd for C11H12O: 183.0786 [M+Na]+; found: 183.0781.
(E)‐(3‐Methylpenta‐2,4‐dien‐2‐yl)benzene [(E)‐11] (Scheme 4)
A mixture of MeP+Ph3I− (808 mg, 2.0 mmol) and tBuOK (224 mg, 2.0 mmol) in THF (1.0 mL) was stirred for 0.5 h at 0–5 °C under an Ar atmosphere. (E)‐9 (80 mg, 0.5 mmol) in THF (1.0 mL) was added to the mixture at the same temperature, and the reaction mixture was stirred for 1 h. The mixture was poured into water, and the obtained mixture was extracted twice with Et2O. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane) to give (E)‐11 (52 mg, 66 %, E/Z=>98:2). Colorless oil; 1H NMR (500 MHz, CDCl3): δ=1.69 (s, 3 H), 2.12 (s, 3 H), 5.16 (d, J=10.9 Hz, 1 H), 5.28 (d, J=17.2 Hz, 1 H), 6.99 (dd, J=10.9, 17.2 Hz, 1 H), 7.10–7.17 (m, 2 H), 7.19–7.26 (m, 1 H), 7.29–7.40 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=15.6, 20.7, 113.5, 126.4, 128.2 (2C), 128.3 (2C), 128.9, 135.8, 136.3, 145.2 ppm; IR (neat): max=3022, 2920, 1603, 1491, 1441, 1373, 1109, 986 cm−1.
Methyl (2E,4Z)‐4‐Methyl‐5‐phenylhexa‐2,4‐dienoate [(E)‐12] (Scheme 4)
(MeO)2P(O)CH2CO2Me (137 mg, 0.75 mmol) and 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU, 91 mg, 0.6 mmol) were successively added to a stirred solution of LiCl (25 mg, 0.6 mmol) in MeCN (0.5 mL) at 20–25 °C under an Ar atmosphere. (E)‐9 (80 mg, 0.5 mmol) in THF (0.5 mL) was added to the mixture, which was stirred at the same temperature for 1 h. Saturated aq. NH4Cl was added to the mixture, which was extracted twice with Et2O. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane/AcOEt 20:1) to give (4E)‐12 as yellow crystals (98 mg, 91 %, E/Z=>98:2) M.p. 88–89 °C; 1H NMR (500 MHz, CDCl3): δ=1.71 (s, 3 H), 2.24 (s, 3 H), 3.78 (s, 3 H), 5.94 (d, J=15.5 Hz, 1 H), 7.09–7.17 (m, 2 H), 7.22–7.30 (m, 1 H), 7.31–7.38 (m, 2 H), 8.00 ppm (d, J=15.5 Hz, 1 H); 13C NMR (125 MHz, CDCl3): δ=16.1, 21.4, 51.6, 117.6, 127.1, 127.7, 127.8 (2C), 128.3 (2C), 143.5, 144.1, 145.0, 168.3 ppm; IR (neat): max=2947, 1713, 1614, 1491, 1433, 1377, 1298, 1269, 1167 cm−1; HRMS (ESI): m/z: calcd for C14H16O2: 239.1048 [M+Na]+; found: 236.1047.
(E)‐2‐(2‐Methyl‐3‐phenylbut‐2‐en‐1‐ylidene)malononitrile [(E)‐13] (Scheme 4)
Ti(OiPr)4 (0.073 mL, 0.25 mmol) was added to a stirred solution of (E)‐9 (80 mg, 0.5 mmol) and malononitrile (33 mg, 0.5 mmol) in iPrOH (0.5 mL) at 20–25 °C under an Ar atmosphere, followed by being stirred at the same temperature for 24 h. Aqueous HCl (1 m) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude solid was purified by SiO2 gel column chromatography (hexane/AcOEt 20:1) to give (4E)‐13 as yellow crystals (84 mg, 81 %, E/Z=97:3). M.p. 83–86 °C; 1H NMR (500 MHz, CDCl3): δ=2.07 (s, 3 H), 2.33 (s, 3 H), 7.12–7.17 (m, 2 H), 7.34–7.39 (m, 1 H), 7.39–7.44 (m, 2 H) 7.97 ppm (s, 1 H); 13C NMR (125 MHz, CDCl3): δ=17.6, 22.7, 81.8, 113.2, 115.1, 127.2 (2C), 128.3, 128.6, 128.7 (2C), 142.0, 157.4, 158.1 ppm; IR (neat): max=2962, 2924, 2218, 1580, 1443, 1377, 1323, 1234, 1063 cm−1.
(Z)‐2‐Methyl‐3‐phenylbut‐2‐enoic acid [(Z)‐7] (Scheme 4)
In a procedure similar to the preparation of (E)‐7, the reaction of (Z)‐5 b‐1 (204 mg, 1.00 mmol) in CF3CO2H (1.0 mL) and H2O (0.5 mL) gave (Z)‐7; as pale yellow crystals (105 mg, 60 %, E/Z=2:>98). M.p. 105–107 °C; 1H NMR (500 MHz, CDCl3): δ=2.01 (s, 3 H), 2.09 (s, 3 H), 7.02–7.17 (m, 2 H), 7.18–7.37 (m, 3 H), 9.32 ppm (br s, 1 H); 13C NMR (125 MHz, CDCl3): δ=16.2, 22.6, 124.6, 126.7 (2C), 127.1, 128.0 (2C), 143.6, 146.4, 175.3 ppm; IR (neat): max=2999, 2669, 1657, 1506, 1435, 1308, 1279, 1206, 1153 cm−1; HRMS (DART): m/z: calcd for C11H12O2: 177.0916 [M+H]+; found: 177.0927.
(Z)‐2‐Methyl‐3‐phenylbut‐2‐en‐1‐yl acetate [(Z)‐8′, Scheme 4]
In a procedure similar to the preparation of (E)‐8, the reaction of (Z)‐5 b‐1 (1.43 g, 7.0 mmol) with DIBAL (20.6 mL, 21.0 mmol; 1.02 m in toluene) gave (Z)‐8 as a colorless oil (1.11 g, 98 %, E/Z=2:>98). 1H NMR (500 MHz, CDCl3): δ=1.19 (s, 1 H), 1.91 (s, 3 H), 2.00 (s, 3 H), 3.95 (s, 2 H), 7.09–7.16 (m, 2 H), 7.20–7.25 (m, 1 H), 7.28–7.35 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=16.2, 21.1, 64.7, 126.4, 127.96 (2C), 128.03 (2C), 130.4, 134.7, 143.7 ppm; IR (neat): max=3335, 2918, 1599, 1491, 1441, 1242, 997, 763, 700 cm−1; HRMS (DART): m/z: calcd for C11H13: 145.1017 [M−OAc]+; found: 145.1003.
In a procedure similar to the preparation of (E)‐8′, the reaction of (Z)‐8 (744 mg, 4.59 mmol) using Ac2O (562 mg, 5.51 mmol), DMAP (28 mg, 5 mol %), and Et3N (557 mg, 5.51 mmol) gave (Z)‐8′ as a colorless oil (849 mg, 91 %, E/Z=2:>98). 1H NMR (500 MHz, CDCl3): δ=1.85 (s, 3 H), 2.03 (s, 3 H), 2.04 (s, 3 H), 4.42 (s, 2 H), 7.07–7.15 (m, 2 H), 7.20–7.25 (m, 1 H), 7.27–7.34 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=16.3, 20.9, 21.2, 66.7, 125.8, 126.6, 127.9 (2C), 128.1 (2C), 137.3, 143.1, 171.0 ppm; IR (neat): max=2918, 1738, 1491, 1441, 1375, 1229, 1022, 766, 702 cm−1; HRMS (DART): m/z: calcd for C11H13 145.1017 [M−OAc]+; found: 145.1003.
(Z)‐(3‐Methylpent‐2‐en‐2‐yl)benzene [(Z)‐14 a, Scheme 4]
In a procedure similar to the preparation of (E)‐14 a, the reaction of (Z)‐8′ (102 mg, 0.5 mmol) using MeLi (3.37 mL, 3.00 mmol; 0.89 m in Et2O) and CuI (333 mg, 1.75 mmol) gave (E)‐14 a as a colorless oil (72 mg, 90 %, E/Z=2:>98). 1H NMR (500 MHz, CDCl3): δ=0.92 (t, J=7.5 Hz, 3 H), 1.78 (s, 3 H), 1.90 (q, J=7.5 Hz, 2 H), 1.93 (s, 3 H), 7.06–7.15 (m, 2 H), 7.16–7.22 (m, 1 H), 7.26–7.33 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.3, 17.3, 21.1, 28.3, 125.7, 128.0 (2C), 128.1 (2C), 130.0, 132.7, 145.4 ppm; IR (neat): max=2964, 1599, 1491, 1440, 1371, 1271, 1113, 1070, 1026, 764, 700 cm−1.
(Z)‐(2‐Methylbut‐2‐ene‐1,3‐diyl)dibenzene [(Z)‐14 b, Scheme 4]
In a procedure similar to the preparation of (Z)‐14 a, the reaction of (Z)‐2‐methyl‐3‐phenylbut‐2‐en‐1‐yl acetate (102 mg, 0.5 mmol) with PhMgBr (2.94 mL, 3.00 mmol; 1.02 m in THF) using CuI (333 mg, 1.75 mmol) gave (Z)‐14 b as a colorless oil (94 mg, 63 %, E/Z=>98:2, containing 26 % of biphenyl). 1H NMR (500 MHz, CDCl3): δ=1.71 (s, 3 H), 2.03 (s, 3 H), 3.30 (s, 2 H), 7.06–7.12 (m, 2 H), 7.13–7.38 ppm (m, 8 H); 13C NMR (125 MHz, CDCl3): δ=17.9, 21.3, 41.2, 125.6, 126.0, 128.2 (6C), 128.6 (2C), 129.4, 132.6, 140.9, 145.0 ppm; IR (neat): max=2914, 2361, 1599, 1493, 1483, 1450, 1072, 1026, 779, 764, 737 cm−1.
(Z)‐(3‐Methyloct‐2‐en‐2‐yl)benzene [(Z)‐14 c, Scheme 4]
In a procedure similar to the preparation of (E)‐14 a, the reaction of (Z)‐8′ (102 mg, 0.5 mmol) with nBuLi (1.88 mL, 3.00 mmol; 1.60 m in hexane) and CuI (333 mg, 1.75 mmol) gave (Z)‐14 c as a colorless oil (82 mg, 81 %, E/Z=2:>98). 1H NMR (500 MHz, CDCl3): δ=0.81 (t, J=7.5 Hz, 3 H), 1.03–1.23 (m, 4 H), 1.28–1.41 (m, 2 H), 1.77 (s, 3 H), 1.87 (t, J=7.5 Hz, 2 H), 1.93 (s, 3 H), 7.04–7.13 (m, 2 H), 7.16–7.22 (m, 1 H), 7.26–7.33 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=14.1, 17.9, 21.3, 22.6, 28.3, 31.8, 35.3, 125.8, 128.0 (2C), 128.4 (2C), 130.6, 131.6, 145.6 ppm; IR (neat): max=2922, 1599, 1491, 1440, 1379, 1026, 763, 700 cm−1.
(Z)‐2‐Methyl‐3‐phenylbut‐2‐enal [(Z)‐9, Scheme 4]
In a procedure similar to the preparation of (E)‐9, the reaction of (Z)‐8 (162 mg, 1.0 mmol) with MnO2 (3.48 g, 40 mmol) gave (Z)‐9 as a colorless oil (157 mg, 98 %, E/Z=2:>98). 1H NMR (500 MHz, CDCl3): δ=1.92 (s, 3 H), 2.28 (s, 3 H), 7.16–7.24 (m, 2 H), 7.29–7.44 (m, 3 H), 9.46 ppm (s, 1 H); 13C NMR (125 MHz, CDCl3): δ=11.3, 23.5, 128.3 (3C), 128.9 (2C), 134.4, 140.2, 157.6, 193.6 ppm; IR (neat): max=2850, 1662, 1442, 1391, 1300, 1228, 764, 700 cm−1.
(Z)‐(3‐Methylpenta‐2,4‐dien‐2‐yl)benzene [(Z)‐11, Scheme 4]
In a procedure similar to the preparation of (E)‐11, the reaction of (Z)‐9 (80 mg, 0.5 mmol) with MeP+Ph3I− (808 mg, 2.0 mmol) and tBuOK (224 mg, 2.0 mmol) gave (Z)‐11 as a colorless oil (52 mg, 66 %, E/Z=>98:2). 1H NMR (500 MHz, CDCl3): δ=1.93 (s, 3 H), 2.10 (s, 3 H), 4.86 (d, J=10.9 Hz, 1 H), 5.13 (d, J=17.2 Hz, 1 H), 6.43 (dd, J=10.9, 17.2 Hz, 1 H), 7.10–7.17 (m, 2 H), 7.20–7.27 (m, 1 H), 7.28–7.37 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 21.9, 111.5, 126.3, 128.0 (2C), 128.8 (2C), 129.4, 136.8, 137.6, 144.1 ppm; IR (neat): max=3022, 2920, 1603, 1491, 1441, 1109, 986, 897, 764, 700 cm−1.
Methyl (2E,4E)‐4‐Methyl‐5‐phenylhexa‐2,4‐dienoate [(E)‐12, Scheme 4]
In a procedure similar to the preparation of (E)‐12, the reaction of (Z)‐9 (80 mg, 0.5 mmol) using (MeO)2P(O)CH2CO2Me (137 mg, 0.75 mmol), DBU (91 mg, 0.6 mmol), and LiCl (25 mg, 0.6 mmol) gave (Z)‐12 as yellow crystals (93 mg, 86 %, E/Z=2:>98). M.p. 88–89 °C; 1H NMR (500 MHz, CDCl3): δ=1.97 (s, 3 H), 2.19 (s, 3 H), 3.66 (s, 3 H), 5.85 (d, J=15.5 Hz, 1 H), 7.11–7.15 (m, 2 H), 7.27–7.31 (m, 1 H), 7.33–7.38 (m, 2 H), 7.43 ppm (d, J=15.5 Hz, 1 H); 13C NMR (125 MHz, CDCl3): δ=14.5, 22.7, 51.3, 116.0, 127.2, 128.1 (3C), 128.3, 128.6 (2C), 142.7, 145.7, 168.3 ppm; IR (neat): max=2947, 2363, 1713, 1614, 1489, 1433, 1269, 1165, 731, 702 cm−1.
(Z)‐2‐(2‐Methyl‐3‐phenylbut‐2‐en‐1‐ylidene)malononitrile [(Z)‐13, Scheme 4]
In a procedure similar to the preparation of (E)‐13, the reaction of (4Z)‐9 (80 mg, 0.5 mmol) with malononitrile (33 mg, 0.5 mmol) and Ti(OiPr)4 (0.073 mL, 0.25 mmol) gave (Z)‐13 as yellow crystals (80 mg, 77 %, E/Z=3:97). M.p. 89–90 °C; 1H NMR (500 MHz, CDCl3): δ=2.07 (s, 3 H), 2.33 (s, 3 H), 7.07–7.17 (m, 2 H), 7.35 (s, 1 H), 7.39–7.46 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=15.6, 23.6, 79.9, 113.4, 115.0, 128.7 (2C), 128.8 (2C), 129.1, 129.3, 141.1, 159.1, 160.8 ppm; IR (neat): max=2926, 2218, 1557, 1441, 1356, 1246, 764, 698 cm−1.
Methyl (Z)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylacrylate [(Z)‐19,14 Scheme 5 [e]]
PhMgBr (0.78 mL, 0.75 mmol; 0.96 m in THF) was added to a stirred suspension of ZnCl2 (102 mg, 0.75 mmol) in MeCN (0.25 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at that temperature for 0.5 h. Methyl (E)‐3‐(4‐methoxyphenyl)‐2‐phenyl‐3‐(tosyloxy)acrylate [(E)‐17; 102 mg, 0.25 mmol] and [Pd(dppb)Cl2] (3 mg, 0.005 mmol) in MeCN (0.25 mL) were successively added to the mixture, which was then stirred at 60–65 °C for 2 h. After cooling to room temperature, aq. HCl (3 m) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane/AcOEt 10:0–3:1) to give (Z)‐19 as colorless crystals (82 mg, 95 %, E/Z=16:84). M.p. 130–131 °C (Ref. 14: 130–131 °C); 1H NMR (500 MHz, CDCl3): δ=3.60 (s, 3 H), 3.82 (s, 3 H), 6.82–6.87 (m, 2 H), 6.97–7.02 (m, 2 H), 7.22–7.05 ppm (m, 10 H); 13C NMR (125 MHz, CDCl3): δ=52.0, 55.2, 113.6 (2C), 127.2, 127.6, 127.8 (2C), 128.2 (2C), 129.8 (2C), 130.4 (2C), 131.0 (2C), 132.4, 134.7, 137.7, 140.7, 146.0, 159.5, 171.2 ppm; IR (neat): max=3020, 2950, 2838, 1714, 1606, 1509, 1248, 1216, 1177, 1150 cm−1.
Methyl (E)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylacrylate [(E)‐19,18b Scheme 5 [f]]
PhMgBr (10.4 mL, 10.0 mmol; 0.96 m in THF) was added to a stirred suspension of ZnCl2 (13.6 g, 10.0 mmol) in THF (5.0 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at that temperature for 0.5 h. Methyl (Z)‐3‐(4‐methoxyphenyl)‐2‐phenyl‐3‐(tosyloxy)acrylate [(Z)‐17, 2.19 g, 5.00 mmol], Pd(OAc)2 (11 mg, 0.05 mmol), and 1,4‐bis(diphenylphosphino)butane (DPPB; 43 mg, 0.10 mmol) in THF (5.0 mL) were successively added to the mixture, which was then stirred at 60–65 °C for 2 h. After cooling to room temperature, aq. HCl (3 m) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product (1.84 g) was purified by SiO2 gel column chromatography (hexane/AcOEt 10:0–3:1) to give (E)‐19 as colorless crystals (1.60 g, 93 %, E/Z=98:2). M.p. 120–124 °C (Ref. 2: 118–120 °C); 1H NMR (500 MHz, CDCl3): δ=3.52 (s, 3 H), 3.74 (s, 3 H), 6.61–6.68 (m, 2 H), 6.86–6.93 (m, 2 H), 7.09–7.14 (m, 2 H), 7.14–7.23 (m, 3 H), 7.24–7.29 (m, 2 H), 7.29–7.37 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=51.9, 55.1, 113.2 (2C), 127.2, 128.1 (3C), 128.3 (2C), 129.1 (2C), 129.8 (2C), 132.3, 132.4 (2C), 132.7, 137.9, 142.7, 146.3, 159.1, 171.2 ppm; IR (neat): max=3020, 2929, 2837, 1715, 1605, 1508, 1247, 1217, 1176, 1149 cm−1; HRMS (ESI): m/z: calcd for C23H20O3: 367.1295 [M+Na]+; found: 367.1310.
Methyl (Z)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylacrylate [(Z)‐19,14 Scheme 5 [g]]
p‐(MeO)C6H4MgBr (17.6 mL, 18.7 mmol; 1.06 m in THF) was added to a stirred suspension of ZnCl2 (2.55 g, 18.7 mmol) in THF (9.4 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at that temperature for 0.5 h. Methyl (Z)‐2,3‐diphenyl‐3‐(tosyloxy)acrylate [(Z)‐18; 3.82 g, 9.35 mmol], Pd(OAc)2 (21 mg, 0.09 mmol) and DPPB (80 mg, 0.19 mmol) in THF (9.4 mL) were successively added to the mixture, which was then stirred at 60–65 °C for 2 h. After cooling to room temperature, aq. HCl (3 m) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane/AcOEt 100:1–3:1) to give (Z)‐19 (3.18 g, 99 %, E/Z=2:98).
Methyl (E)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylacrylate [(E)‐19], Scheme 5 [h]]
p‐(MeO)C6H4MgBr (0.71 mL, 0.75 mmol; 1.06 m in THF) was added to a stirred suspension of ZnCl2 (102 mg, 0.75 mmol) in MeCN (0.25 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at that temperature for 0.5 h. Methyl (E)‐2,3‐diphenyl‐3‐(tosyloxy)acrylate [(E)‐18, 102 mg, 0.25 mmol) and [Pd(dppb)Cl2] (3 mg, 0.005 mmol) in MeCN (0.25 mL) were successively added to the mixture, which was then stirred at 60–65 °C for 2 h. After cooling down, aq. HCl (3 m) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane/AcOEt 100:1–3:1) to give (E)‐19 (70 mg, 81 %, E/Z=90:10).
(E)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylallyl Acetate [(E)‐20,23 Scheme 6 [a] and [b]]
DIBAL (90 mL, 90 mmol, 1.0 m in toluene) was added to a stirred solution of methyl (E)‐3‐(4‐methoxyphenyl)‐2,3‐diphenylacrylate [(E)‐19; 10.30 g, 30 mmol] in CH2Cl2 (60 mL) at 0–5 °C under an Ar atmosphere, and the mixture was stirred at that temperature for 1 h. Saturated aq. Rochelle salt solution (20 mL) was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated to give the desired alcohol as colorless crystals (9.29 g, 98 %, E/Z=98:2), which was of sufficient purity for the next step. M.p. 115–116 °C; 1H NMR (500 MHz, CDCl3): δ=1.53 (s, 1 H), 3.68 (s, 3 H), 4.44 (s, 2 H), 6.51–6.65 (m, 2 H), 6.76–6.87 (m, 2 H), 7.08–7.50 ppm (m, 10 H); 13C NMR (125 MHz, CDCl3): δ=55.0, 65.1, 112.9 (2C), 126.7, 127.2, 128.2 (2C), 128.2 (2C), 129.7 (2C), 129.9 (2C), 131.9 (2C), 134.5, 137.5, 140.6, 142,3, 142.4, 158.0 ppm; IR (neat): max=3552, 1607, 1508, 1252, 1177, 1028, 833, 791, 700 cm−1; HRMS (ESI): m/z: calcd for C22H20O2 :339.1361 [M+Na]+; found: 339.1378.
Ac2O (0.11 mL, 1.2 mmol) was added to a stirred solution of the (E)‐alcohol (316 mg, 1.00 mmol), Et3N (0.17 mL, 1.2 mmol), and DMAP (6 mg, 0.05 mmol) in CH2Cl2 (2.0 mL) at 20–25 °C under an Ar atmosphere, which was then stirred at that temperature for 1 h. Water was added to the mixture, which was extracted twice with AcOEt. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude solid was purified by SiO2 gel column chromatography (hexane/AcOEt 4:1) to give (E)‐20 as colorless crystals (332 mg, 99 %, E/Z=98:2). M.p. 101–102 °C; 1H NMR (500 MHz, CDCl3): δ=1.94 (s, 3 H), 3.70 (s, 3 H), 4.89 (s, 2 H), 6.48–6.66 (m, 2 H), 6.72–6.92 (m, 2 H), 7.06–7.45 ppm (m, 10 H); 13C NMR (125 MHz, CDCl3): δ=20.9, 55.0, 66.8, 112.9 (2C), 126.6, 127.4, 128.0 (2C), 128.2 (2C), 129.5 (2C), 129.7 (2C), 131.9 (2C), 132.9, 134.2, 140.3, 142.2, 144.9, 158.2, 170.9 ppm; IR (neat): max=1732, 1605, 1508, 1223, 1175, 1028, 951, 833, 826, 770, 702 cm−1; HRMS (ESI): m/z: calcd for C22H19O: 299.1436 [M−OAc]+; found: 299.1418.
(Z)‐[1‐(4‐Methoxyphenyl)but‐1‐ene‐1,2‐diyl]dibenzene [(Z)‐21,21 Scheme 6 [c]]
MeLi (71 mL, 80 mmol; 1.13 m in Et2O) was added dropwise to a stirred suspension of CuI (9.52 g, 50 mmol) in THF (30 mL) at 0–5 °C under an Ar atmosphere. (E)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylallyl acetate [(E)‐20, 6.69 g, 20.0 mmol] in THF (30 mL) was added to the mixture, which was then stirred at the same temperature for 1 h. The mixture was reversely quenched into saturated aq. NH4Cl, and then filtered through Celite. The filtrate was extracted twice with Et2O. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2 gel column chromatography (hexane/AcOEt 5:1) to give (Z)‐21 as colorless crystals (5.13 g, 82 %, E/Z=2:98). M.p. 120–122 °C (Ref. 21: 116–119 °C); 1H NMR (500 MHz, CDCl3): δ=0.92 (t, J=7.5 Hz, 3 H,), 2.45 (q, J=7.5 Hz, 2 H), 3.67 (s, 3 H), 6.49–6.58 (m, 2 H), 6.73–6.82 (m, 2 H), 7.07–7.20 (m, 5 H), 7.21–7.29 (m, 3 H), 7.30–7.39 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.6, 29.0, 54.9, 112.7 (2C), 126.0, 126.5, 127.9 (2C), 128.1 (2C), 129.4 (2C), 129.7 (2C), 131.9 (2C), 135.4, 138.2, 141.3, 142.4, 143.8, 157.5 ppm; IR (neat): max=1504, 1439, 1234, 1171, 1028, 822, 768, 700 cm−1; HRMS (ESI): m/z: calcd for C23H22O: 337.1204 [M+Na]+; found: 337.1217.
(Z)‐2‐(4‐(1,2‐Diphenylbut‐1‐en‐1‐yl)phenoxy)‐N,N‐dimethylethan‐1‐amine [(Z)‐tamoxifen,21 Scheme 6 [d] and [e]]
A mixture of (Z)‐[1‐(4‐methoxyphenyl)but‐1‐ene‐1,2‐diyl]dibenzene [(Z)‐21; 314 mg, 1.00 mmol] and NaSEt (841 mg, 10.0 mmol) in DMF (5.0 mL) was heated at refluxed for 1 h under an Ar atmosphere. After cooling to room temperature, aq. HCl (3 m) was added to the mixture, which was extracted twice with Et2O. The combined organic phase was washed four times with aq. HCl (3 m) and brine, then dried (Na2SO4) and concentrated. The obtained crude solid was washed with hexane (2 mL×3) to give the desired phenol as colorless crystals (292 mg, 97 %, E/Z=2:98). M.p. 128–132 °C; 1H NMR (500 MHz, CDCl3): δ=0.92 (t, J=7.5 Hz, 3 H), 2.45 (q, J=7.5 Hz, 2 H), 4.44 (s, 1 H), 6.43–6.50 (m, 2 H), 6.71–6.77 (m, 2 H), 7.08–7.14 (m, 3 H), 7.15–7.20 (m, 2 H), 7.21–7.30 (m, 3 H), 7.31–7.37 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.6, 29.0, 114.3 (2C), 126.0, 126.5, 127.8 (2C), 128.1 (2C), 129.4 (2C), 129.7 (2C), 132.0 (2C), 135.7, 138.1, 141.4, 142.4, 143.7, 153.3 ppm; IR (neat): max=3418, 1607, 1506, 1441, 1234, 1173, 824, 772, 702 cm−1; HRMS (ESI): m/z: calcd for C22H20O: 299.1436 [M−H]−; found: 299.1435.
A mixture of the (Z)‐phenol (75 mg, 0.25 mmol), 2‐chloro‐N,N‐dimethylethan‐1‐amine hydrochloride (72 mg, 0.50 mmol), and K2CO3 (138 mg, 1.0 mmol) in EtOH (2.5 mL) and toluene (2.5 mL) was stirred at 80–85 °C for 3 h under an Ar atmosphere. Saturated aq. NH4Cl was added to the mixture, which was extracted twice with Et2O. The combined organic phase was washed with water and brine, then dried (Na2SO4) and concentrated to give (Z)‐tamoxifen as colorless crystals (300 mg, 93 %, E/Z=2:98], of sufficient purity without the need for purification. M.p. 92–94 °C (Ref. 21: 95–97 °C); 1H NMR (500 MHz, CDCl3): δ=0.92 (t, J=7.5 Hz, 3 H), 2.28 (s, 6 H), 2.45 (q, J=7.5 Hz, 2 H), 2.64 (t, J=5.7 Hz, 2 H), 3.92 (t, J=5.7 Hz, 2 H), 6.52–6.60 (m, 2 H), 6.72–6.80 (m, 2 H), 7.07–7.13 (m, 3 H), 7.14–7.20 (m, 2 H), 7.21–7.29 (m, 3 H), 7.31–7.38 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 29.1, 46.0 (2C), 58.4, 65.8, 113.5 (2C), 126.1, 126.6, 128.0 (2C), 128.2 (2C), 129.6 (2C), 129.8 (2C), 132.0 (2C), 135.6, 138.4, 141.4, 142.5, 143.9, 156.9 ppm; IR (neat): max=1609, 1510, 1443, 1287, 1244, 1175, 1030, 966, 818, 775, 768, 698 cm−1; HRMS (ESI): m/z: calcd for C26H29NO: 372.2327 [M+Na]+; found: 372.2315.
(Z)‐3‐(4‐Methoxyphenyl)‐2,3‐diphenylallyl acetate [(Z)‐20, Scheme 6 [f] and [g]]
In a procedure similar to the preparation of (E)‐20, the reaction of methyl (Z)‐3‐(4‐methoxyphenyl)‐2,3‐diphenylacrylate [(Z)‐19; 3.18 g, 9.22 mmol] with DIBAL (1.0 m in toluene, 27.7 mL, 27.7 mmol) gave the desired alcohol as colorless crystals (2.71 g, 93 %, E/Z=2:>98). M.p. 159–160 °C (Ref. 18c: 161 °C); 1H NMR (500 MHz, CDCl3): δ=1.49 (t, J=5.7 Hz, 1 H), 3.83 (s, 3 H), 4.51 (d, J=5.7 Hz, 2 H), 6.85–6.96 (m, 4 H), 7.00–7.08 (m, 3 H), 7.10–7.22 (m, 4 H), 7.23–7.30 ppm (m, 3 H); 13C NMR (125 MHz, CDCl3): δ=55.2, 65.1, 113.5 (2C), 126.4, 126.7, 127.4 (2C), 128.1 (2C), 129.9 (2C), 130.7 (2C), 130.9 (2C), 134.6, 137.9, 140.5, 142.5, 142.6, 158.8 ppm; IR (neat): max=3536, 1603, 1574, 1506, 1439, 1358, 1306, 1236, 1180 cm−1.
In a procedure similar to the preparation of (E)‐20, the reaction of (Z)‐3‐(4‐methoxyphenyl)‐2,3‐diphenylprop‐2‐en‐1‐ol (316 mg, 1.00 mmol) with Ac2O (123 mg, 1.20 mmol) using DMAP (6 mg, 0.05 mmol) and Et3N (121 mg, 1.20 mmol) gave (Z)‐20 as colorless crystals (325 mg, 97 %, E/Z=2:>98], which were of sufficient purity without the need for purification. M.p. 104–105 °C; 1H NMR (500 MHz, CDCl3): δ=1.96 (s, 3 H), 3.83 (s, 3 H), 4.94 (s, 2 H), 6.85–6.94 (m, 4 H), 7.01–7.08 (m, 3 H), 7.09–7.22 ppm (m, 7 H); 13C NMR (125 MHz, CDCl3): δ=20.9, 55.2, 66.9, 113.6 (2C), 126.56, 126.58, 127.5 (2C), 127.9 (2C), 129.8 (2C), 130.6 (2C), 130.8 (2C), 133.4, 134.3, 140.3, 142.2, 145.2, 159.0, 170.9 ppm; IR (neat): max=1730, 1605, 1506, 1442, 1377, 1356, 1225, 1171, 955, 835, 779, 762, 698 cm−1.
(E)‐1‐(4‐Methoxyphenyl)but‐1‐ene‐1,2‐diyl)dibenzene [(E)‐21,21 Scheme 6 [h]]
In a procedure similar to the preparation of (Z)‐21, the reaction of (Z)‐3‐(4‐methoxyphenyl)‐2,3‐diphenylallyl acetate [(Z)‐20; 2.92 g, 8.73 mmol], MeLi (33.6 mL, 34.9 mmol; 1.04 m in Et2O) and CuI (4.16 g, 21.8 mmol) gave (E)‐21 as colorless crystals (2.19 g, 80 %, E/Z=98:2). M.p. 100–101 °C (Ref. 21: 99–101 °C); 1H NMR (500 MHz, CDCl3): δ=0.95 (t, J=7.5 Hz, 3 H), 2.51 (q, J=7.5 Hz, 2 H), 3.82 (s, 3 H), 6.83–6.91 (m, 4 H), 6.94–7.03 (m, 3 H), 7.06–7.11 (m, 3 H), 7.12–7.20 ppm (m, 4 H); 13C NMR (125 MHz, CDCl3): δ=13.8, 29.2, 55.3, 113.6 (2C), 125.8, 126.2, 127.4 (2C), 127.9 (2C), 129.9 (2C), 130.7 (2C), 131.0 (2C), 136.1, 138.6, 142.0, 142,5, 143.5, 158.4 ppm; IR (neat): max=1506, 1439, 1242, 1171, 1078, 1034, 845, 824, 773, 752, 702 cm−1.
(E)‐2‐[4‐(1,2‐Diphenylbut‐1‐en‐1‐yl)phenoxy]‐N,N‐dimethylethan‐1‐amine [(E)‐tamoxifen, Scheme 6 [i] and [j]]24
In a procedure similar to the preparation of (Z)‐tamoxifen, the reaction of (E)‐[1‐(4‐methoxyphenyl)but‐1‐ene‐1,2‐diyl]dibenzene [(E)‐21; 260 mg, 0.83 mmol] and NaSEt (698 mg, 8.30 mmol) gave (E)‐4‐(1,2‐diphenylbut‐1‐en‐1‐yl)phenol as colorless crystals (246 mg, 99 %, E/Z=98:2). M.p. 130–132 °C; 1H NMR (500 MHz, CDCl3): δ=0.94 (t, J=7.5 Hz, 3 H), 2.50 (q, J=7.5 Hz, 2 H), 4.69 (br s, 1 H), 6.78–6.83 (m, 2 H), 6.85–6.90 (m, 2 H), 6.95–7.03 (m, 3 H), 7.06–7.18 ppm (m, 7 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 29.1, 115.1 (2C), 125.8, 126.2, 127.4 (2C), 127.9 (2C), 129.8 (2C), 130.9 (4C), 130.9, 136.3, 138.4, 142.1, 142.5, 143.4, 154.3 ppm; IR (neat): max=3383, 1609, 1508, 1443, 1221, 1169, 1078, 818, 770, 752, 692 cm−1.
The reaction of (E)‐4‐(1,2‐diphenylbut‐1‐en‐1‐yl)phenol (300 mg, 1.00 mmol) with 2‐chloro‐N,N‐dimethylethan‐1‐amine hydrochloride (288 mg, 2.00 mmol) using K2CO3 (553 mg, 4.00 mmol) gave (E)‐tamoxifen as colorless crystals (360 mg, 97 %, E/Z=98:2). M.p. 72–73 °C (Ref. 23: 70–72 °C); 1H NMR (500 MHz, CDCl3): δ=0.94 (t, J=7.5 Hz, 3 H), 2.35 (s, 6 H), 2.50 (q, J=7.5 Hz, 2 H), 2.74 (t, J=5.7 Hz, 2 H), 4.08 (t, J=5.7 Hz, 2 H), 6.84–6.92 (m, 4 H), 6.94–7.03 (m, 3 H), 7.05–7.19 ppm (m, 7 H); 13C NMR (125 MHz, CDCl3): δ=13.7, 29.1, 46.1 (2C), 58.5, 66.0, 114.2 (2C), 125.8, 126.1, 127.4 (2C), 127.9 (2C), 129.8 (2C), 130.7 (2C), 130.9 (2C), 136.2, 138.5, 142.0, 142.5, 143.5, 157.7 ppm; IR (neat): max=1506, 1474, 1454, 1242, 1167, 1034, 820, 802, 768, 750, 696 cm−1.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was partially supported by Grant‐in‐Aids for Scientific Research on Basic Areas (B) “18350056”, Priority Areas (A) “17035087” and “18037068”, (C) 15K05508, and Exploratory Research “17655045” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Y.T. offers his warmest congratulations to Professor Ben L. Feringa (University of Groningen, The Netherlands) on being awarded the 2016 Nobel Prize in Chemistry.
Y. Ashida, A. Honda, Y. Sato, H. Nakatsuji, Y. Tanabe, ChemistryOpen 2017, 6, 73.
Contributor Information
Dr. Hidefumi Nakatsuji, Email: nakatsuji@kwansei.ac.jp
Prof. Dr. Yoo Tanabe, Email: tanabe@kwansei.ac.jp.
References
- 1.For representative reviews, see:
- 1a. Flynn A. B., Ogilvie W. W., Chem. Rev. 2007, 107, 4698; [DOI] [PubMed] [Google Scholar]
- 1b. Polák P., Váňová H., Dvořák D., Tobrman T., Tetrahedron Lett. 2016, 57, 3684. [Google Scholar]
- 2.For a representative review, and the concept on cross-couplings using enol tosylates and phosphates, see: Lindhardt A. T., Skrydstrup T., Chem. Eur. J. 2008, 14, 8756, and relevant references cited therein.18506859 [Google Scholar]
- 3.For a representative review, see: Sellars J. D., Steel P. G., Chem. Soc. Rev. 2011, 40, 5170. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Smith M. T., March's Advanced Organic Chemistry, 6th ed., Wiley, New York, 2007, p. 624, 1355, 1452; [Google Scholar]
- 4b. Clayden J., Greeves N., Warren S., Wothers P., Organic Chemistry, Oxford University, New York, 2001, p. 728; [Google Scholar]
- 4c. Kürti L., Czakó B., Strategic Applications of Named Reactions in Organic Synthesis Elsevier, Burlington, 2005, p. 86. [Google Scholar]
- 5. Ashida Y., Sato Y., Honda A., Nakatsuji H., Tanabe Y., Synthesis 2016, 48, 4072. [Google Scholar]
- 6. Baxter J. M., Steinhuebel D., Palucki M., Davies I. W., Org. Lett. 2005, 7, 215. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Manabe A., Ohfune Y., Shinada T., Synlett 2012, 23, 1213; [Google Scholar]
- 7b. Totsuka Y., Ueda S., Kuzuyama T., Shinada T., Bull. Chem. Soc. Jpn. 2015, 88, 575. [Google Scholar]
- 8. Li H., Mazet C., J. Am. Chem. Soc. 2015, 137, 10720. [DOI] [PubMed] [Google Scholar]
- 9. Yanagita Y., Suto T., Matsuo N., Kurosu Y., Sato T., Chida T. N., Org. Lett. 2015, 17, 1946. [DOI] [PubMed] [Google Scholar]
- 10. Ashida Y., Sato Y., Suzuki T., Ueno K., Kai K., Nakatsuji H., Tanabe Y., Chem. Eur. J. 2015, 21, 5934. [DOI] [PubMed] [Google Scholar]
- 11. Christensen M., Nolting A., Shevlin M., Weisel M., Maligres P. E., Lee J., Orr R. K., Plummer C. W., Tudge M. T., Campeau L.-C., Ruck R. T., J. Org. Chem. 2016, 81, 824. [DOI] [PubMed] [Google Scholar]
- 12.Use of the TsCl/LHMDS reagent generally yielded α-chlorinated β-ketoester byproducts as is described in Klapars A., Campos K. R., Chen C., Volante R., Org. Lett. 2005, 7, 1185.15760170 [Google Scholar]
- 13.
- 13a. Nakatsuji H., Ueno K., Misaki T., Tanabe Y., Org. Lett. 2008, 10, 2131; [DOI] [PubMed] [Google Scholar]
- 13b. Nakatsuji H., Nishikado H., Ueno K., Tanabe Y., Org. Lett. 2009, 11, 4258; [DOI] [PubMed] [Google Scholar]
- 13c. Nishikado H., Nakatsuji H., Ueno K., Nagase R., Tanabe Y., Synlett 2010, 2087. [Google Scholar]
- 14. Nakatsuji H., Ashida Y., Hori H., Sato Y., Honda A., Taira M., Tanabe Y., Org. Biomol. Chem. 2015, 13, 8205. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Calvin J. R., Frederick M. O., Laird D. L. T., Remacle J. R., May S. A., Org. Lett. 2012, 14, 1038; [DOI] [PubMed] [Google Scholar]
- 15b. Abe M., Nishikawa K., Fukuda H., Nakanishi K., Tazawa Y., Taniguchi T., Park S.-y., Hiradate S., Fujii Y., Okuda K., Shindo M., Phytochemistry 2012, 84, 56. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Fronza G., Fuganti C., Serra S., Eur. J. Org. Chem. 2009, 6160; [Google Scholar]
- 16b. Tessier P. E., Penwell A. J., Souza F. E. S., Fallis A. G., Org. Lett. 2003, 5, 2989; [DOI] [PubMed] [Google Scholar]
- 16c. Hülskämper L., Weyerstahl P., Chem. Ber. 1981, 114, 746. [Google Scholar]
- 17. Yamashita K., Tanaka T., Hayashi T., Tetrahedron 2005, 61, 7981. [Google Scholar]
- 18.For a review on the synthesis, see
- 18a. Kasiotis K. M., Haroutounian S. A., Curr. Org. Chem. 2012, 16, 335; for recent representative (Z)-selective syntheses, see [Google Scholar]
- 18b. Matsumoto K., Shindo M., Adv. Synth. Catal. 2012, 354, 642; [Google Scholar]
- 18c. He Z., Kirchberg S., Froehlich R., Studer A., Angew. Chem. Int. Ed. 2012, 51, 3699; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3759; [Google Scholar]
- 18d. Takemoto Y., Yoshida H., Takaki K., Chem. Eur. J. 2012, 18, 14841; [DOI] [PubMed] [Google Scholar]
- 18e. Corpet M., Bai X. Z., Gosmini C., Adv. Synth. Catal. 2014, 356, 2937; [Google Scholar]
- 18f. Zhou Y., You W., Smith K. B., Brown M. K., Angew. Chem. Int. Ed. 2014, 53, 3475; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3543; [Google Scholar]
- 18g. Cahiez G., Moyeux A., Poizat M., Chem. Commun. 2014, 50, 8982; [DOI] [PubMed] [Google Scholar]
- 18h. Ganapathy D., Sekar G., Org. Lett. 2014, 16, 3856; [DOI] [PubMed] [Google Scholar]
- 18i. Pichette Drapeau M., Fabre I., Grimaud L., Ciofini I., Ollevier T., Taillefer M., Angew. Chem. Int. Ed. 2015, 54, 10587; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10733; [Google Scholar]
- 18j. Xue F., Zhao J., Hor T. S. A., Hayashi T., J. Am. Chem. Soc. 2015, 137, 3189; [DOI] [PubMed] [Google Scholar]
- 18k. Nagao K., Ohmiya H., Sawamura M., Org. Lett. 2015, 17, 1304. [DOI] [PubMed] [Google Scholar]
- 19. Shimizu K., Takimoto M., Mori M., Sato Y., Synlett 2006, 18, 3182. [Google Scholar]
- 20.
- 20a. Sano S., Takehisa T., Ogawa S., Yokoyama K., Nagao Y., Chem. Pharm. Bull. 2002, 50, 1300; [DOI] [PubMed] [Google Scholar]
- 20b. Shindo M., Sato Y., Yoshikawa T., Koretsune R., Shishido K., J. Org. Chem. 2004, 69, 3912. [DOI] [PubMed] [Google Scholar]
- 21. Miller R. B., Al-Hassan M. I., J. Org. Chem. 1985, 50, 2121. [Google Scholar]
- 22. Tsuda T., Yoshida T., Saegusa T., J. Org. Chem. 1988, 53, 607. [Google Scholar]
- 23. Suzuki M., Ijuin R., Doi H., Koyama H., PCT. Int. Appl., 2014014067, 2014.
- 24.
- 24a. Bedford G. R., Richardson D. N., Nature 1966, 212, 733; [Google Scholar]
- 24b. AI-Hassan M. I., Synthesis 1987, 816. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary