

Diabetes mellitus has developed into an epidemic and represents a major public health challenge. Cardiovascular disorders are the primary cause of mortality and morbidity in diabetic patients. Diabetes-associated changes in myocardial structure and function, referred to as diabetic cardiomyopathy, can develop in the absence of overt myocardial ischemia and hypertension, or are accelerated by the presence of these comorbidities [1]. To date, a plethora of cell and molecular mechanisms have been implicated in the development and progression of diabetic cardiomyopathy, including: reduced energy production due to decreases in mitochondrial respiration and pyruvate dehydrogenase activity; oxidative stress due to over generation of reactive oxygen or nitrogen species (ROS or RNS); apoptosis; impaired autophagy; and malfunction of cardiac contractile and intracellular Ca2+ regulatory proteins such as myosin, sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), and the Na+/Ca2+ exchanger [1,2]. Despite the plethora of proposed mechanisms, the scientific community has not reached a definitive consensus about common underlying mechanisms or processes driving the functional and morphological changes associated with the pathophysiology of diabetic cardiomyopathy.
The epidermal growth factor (EGF) receptor EGFR is an 1186-anino-acid receptor, containing a single transmembrane domain, an extracellular portion involved in ligand binding, and an intracellular portion harboring the tyrosine kinase domain, and is therefore also referred to as EGF receptor tyrosine kinase [3,4]. EGFR can be activated by different ligands such as EGF and heparin-binding EGF-like proteins [3,4]. In addition, angiotensin II (Ang II), leptin, and even glucose at high concentrations also transactivate EGFR [5–8], as summarized in Fig. 1. Emerging evidence shows that the EGFR tyrosine kinase signaling pathway is an important signaling hub in regulating cell growth, proliferation, migration and differentiation in normal and pathological states such as cancer [3]. In the past two decades, EGFR was found to also play an important role in non-malignant disorders, including cardiovascular diseases [4–8]. In order to modulate EGFR tyrosine kinase signaling, more than 10 EGFR inhibitors have been developed, the most prevalent of which is AG1478, and some of these inhibitors have been used in clinic trials for cancer treatment [9]. By applying these pharmacological and genetic approaches that interfere with EGFR transactivation, it has recently been shown that EGFR transactivation plays an essential role in cardiac hypertrophy/fibrosis, vascular neointimal hyperplasia, and renal fibrosis [4–8,10].
Fig. 1.
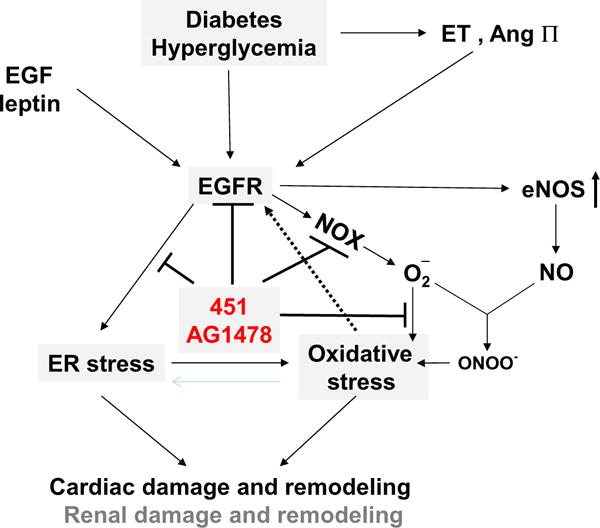
Illustration of the possible mechanisms for the diabetic effects on EGFR activation-mediated down-stream signaling pathways that in turn lead to cardiac and/or renal damage and remodeling.
EGFR expression and function are also implicated in chronic diseases, such as diabetes and diabetic complications [8,11,12]. EGFR pathway activation mediated diabetes-induced kidney damage and vascular dysfunction [11,12]. In experimental diabetes, up-regulation of EGFR signaling leads to vascular dysfunction in several tissues and is detrimental to micro-vasculature in both type 1 and type 2 diabetes [8,11,12]. Reportedly, EGFR phosphorylation is down-regulated in the liver, but up-regulated in the kidney and gastric mucosa as well as in the kidney/coronary/mesenteric bed micro-vascular system following experimentally induced diabetes [13–17]. Numerous studies focusing on the role of EGFR in the pathogenesis of diabetic nephropathy and micro-vascular dysfunction in diabetes have been carried out [11–13, 17,18], and clearly show detrimental effects of EGFR phosphorylation on the development of diabetic nephropathy and micro-vascular dysfunction, which, generally speaking, was mediated by increased endoplasmic reticulum (ER) stress and oxidative stress as well as decreased autophagy [11], as outlined in Fig. 1.
Less is known about the role of EGFR in the pathogenesis of diabetic cardiomyopathy. Surprisingly, several studies have shown the essential role of EGFR expression and phosphorylation in cardiac protection, particularly, from ischemia-induced damage [17,19–21]. For instance, one study by Akhtar et al. showed that 4 weeks after onset of streptozotocin (STZ)-induced hyperglycemia in rats, hearts were isolated and subjected to 40 min of global ischemia. Compared to hearts from age-matched control rats, diabetic hearts showed significantly impaired recovery of (cardiac contractility and hemodynamics) following ischemia, which was worsened with chronic treatment with AG1478, the selective inhibitor of EGFR kinase [21]. Consistent with this, the administration of the EGFR ligand, EGF, before or after ischemia in isolated hearts, led to significantly improved recovery of cardiac function [21]. This study supported several previous studies that directly or indirectly indicated that EGFR signaling protected the cardiac tissue against ischemic injury and was involved in cardiac preconditioning [19,20,22]. For the latter, inhibition of EGFR with AG1478 was attenuated the beneficial effects of cardiac preconditioning to ischemia-reperfusion injury, implying that activation of EGFR signaling during preconditioning is important for improving recovery following ischemia-reperfusion injury [22].
In contrast with the above studies [19,20,22], however, Galan et al. reported the critical role of the EGFR activation to ER stress in cardiac damage caused by type 1 diabetes [23]. The authors used the type 1 diabetes mouse model induced with a single dose of STZ (200 mg/kg), and found that EGFR phosphorylation and expression were up-regulated in the heart of diabetic mice, and this upregulation was associated with ER stress induction and cardiac fibrosis at 8 weeks after diabetes onset. Consistent with this, inhibition of EGFR with AG1478, improved glucose levels, body weight and reduced cardiac fibrosis and ER stress markers [23]. Given that the histopathological features of diabetic cardiomyopathy include cardiac cell death, hypertrophy, and myofibril disorganization, the target article would be more interesting if they could investigate the association of EGFR with these pathogenesis processes in diabetic mice.
The article from Liang’s group in this issue reports the detrimental role of EGFR in the pathogenesis of diabetic cardiomyopathy [24]. In this study in mice, type 1 diabetes was induced with a single dose of STZ (100 mg/kg); 8 weeks after diabetes onset, pathological examination and biochemical measurements were performed on the cardiac tissue, matching the study by Galan et al. Liang’s study confirmed the earlier study, showing that EGFR inhibition protected against cardiac damage and remodeling in the STZ-induced diabetic mouse model. It also provided evidence that diabetes induced cardiac remodeling led to significantly increased phosphorylation of EGFR, and was accompanied by cardiac myofibril disorganization, hypertrophy, and fibrosis, which, remarkably were all prevented by inhibition of EGFR phosphorylation with AG1478. These two independent studies establish the detrimental role of EGFR phosphorylation in the pathogenesis of diabetes-induced cardiac remodeling, consistent with its role in diabetic nephropathy [11,12,22], as illustrated in Fig. 1. Therefore, these studies seemingly indicate that EGFR activation could be protective in the acute damage period such as acute ischemic injury, but detrimental in chronic pathogenesis such as diabetes-induced cardiac remodeling. These studies also suggest that EGFR is essential for the physiological and structural function of the heart under normal conditions, and that sustained up-regulation or down-regulation leads to pathogenic effects, as shown in a study in which chronic treatment with EGF receptor antagonists in normal C57BL/6 J mice led to cardiac dysfunction [25].
The study by Liang et al. also provides evidence that inhibition of EGFR-mediated cardiac effect is mediated by the suppression of NADPH oxidase- (NOX-) mediated oxidative stress [24]. It is known that oxidative stress plays an important role in the development and progression of diabetes and its complications. In the present work, Liang et al. demonstrated that cardiac oxidative stress markers were significantly increased in the hearts of diabetic mice, which is associated with cardiac remodeling in vivo. In addition, increased cardiac hypertrophic markers, fibrotic markers and cell apoptotic markers (induced by high glucose concentration in vitro) were significantly attenuated by NAC, a well-studied ROS scavenger. In this study, increased cardiac oxidative stress in the diabetic model was markedly reduced by the EGFR inhibitor AG1478 and a similar result was observed in vitro using H9c2 cardiac fibroblasts, primary cardiomyocytes, and human umbilical vein endothelial cells. Knockdown of EGFR by sh-RNA also significantly reduced HG-induced ROS and H9c2 cell damage [24]. These results suggest that EGFR may mediate diabetes/HG-induced cardiac oxidative stress and ROS generation, in agreement with the study by Galan et al., where the NOX activity was significantly reduced by treatment with AG1478 in type 1 diabetic mice [23]. These studies demonstrate that the exposure of cardiac cells to HG activated EGFR/Akt-mediated NOX expression and ROS production, resulting in cardiomyocyte hypertrophy, fibrosis, and apoptosis.
However, there was some minor differences between the studies by Galan et al. and the target study: Galan et al. showed that administration with EGFR inhibitor AG1478 (10 mg/kg/day) for 2 weeks can markedly improve blood glucose level and body weight in STZ-induced type 1 diabetic mice [23], while Liang et al. did not observed any hypoglycemic effect of EGFR inhibition via AG1478 (20 mg/kg/2 day) during the 8-week period. There was a study where in type 2 diabetic db/db mice, AG1478 (10 mg/kg/day) treatment for 2 weeks had no effect on blood glucose levels [13]; therefore, further elucidation of the role of EGFR in the hypoglycemic effect of diabetes is desired.
Another interesting point from the study by Liang et al. is the use of two EGFR inhibitors [24]: one is AG1478, and the other, 451. AG1478 is a well-known EGFR-specific inhibitor [9] and is widely used in EGFR-related biological studies. Liang et al. have designed and synthesized a series of chemical analogs of AG1478, among which compound 451 was found to exhibit comparable EGFR-inhibitory activity with AG1478, as stated by the authors [24] but have an IC50 of 0.2 nM against recombinant EGFR kinase activity, which is lower than AG1478 with an IC50 of 3 nM. They demonstrated that 451 showed comparable effects with AG1478 at the same concentration on improving cardiac remodeling in diabetic mice. However, in vitro studies showed that 451 at a dose 2-fold less than AG1478 provided similar protection against cardiomyocyte injury [24].
In summary, the significance of Liang’s study is that it has independently confirmed the detrimental effect of up-regulated EGFR phosphorylation in the pathogenesis of diabetes-induced oxidative damage and remodeling (hypertrophy and fibrosis) in the single STZ-induced type 1 diabetic model [23], matching the recent report by Galan et al. These findings contradict several other studies that showed the protective effects of EGFR on ischemic cardiac damage in diabetic animals. These two studies provide consistent evidence for the detrimental effect of EGFR up-regulation on cardiac damage under chronic diabetic conditions, as summarized in Fig. 1. However, there remain several important issues that urgently need to be addressed:
Although these two studies have demonstrated the detrimental effect on the heart of type 1 diabetic hearts, these results were all obtained from STZ-induced type 1 diabetic models at only 8 weeks after diabetes onset. It would be important to know whether the improvement of the biochemical and pathological changes by inhibition of EGFR phosphorylation in the diabetic heart result in a significant improvement of cardiac dysfunction at later stages in the progression of diabetes.
Both studies were done with the model induced by a single dose of STZ, which is the model that fails to replicate the cardiac pathogenesis seen in type 1 diabetes patients. Therefore, it remains to be seen whether the findings obtained from these two studies can also be achieved with type 2 diabetic models, or even other type 1 diabetic models such as NOD and/or OVE26 mice.
The advantages and disadvantages of the newly synthesized compound 451 for the potential clinical application must be systematically investigated.
We believe that addressing these issues will provide us a better understanding of the mechanism responsible for cardiovascular disease progression, and also may potentially provide new therapeutic targets in the prevention and/or treatment of diabetic cardiomyopathy.
Acknowledgments
The author’s work was supported in part by awards from American Diabetes Association (1-11-BS-17; 7-14-BS-18) and from the National Institutes of Health (1R01DK 091338-01A1).
References
- 1.Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57:660–71. doi: 10.1007/s00125-014-3171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142:375–415. doi: 10.1016/j.pharmthera.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Dhomen NS, Mariadason J, Tebbutt N, Scott AM. Therapeutic targeting of the epidermal growth factor receptor in human cancer. Crit Rev Oncog. 2012;17:31–50. doi: 10.1615/critrevoncog.v17.i1.40. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Guerrero E, Jo SR, Chong BH, Khachigian LM. EGFR and the complexity of receptor crosstalk in the cardiovascular system. Curr Mol Med. 2013;13:3–12. [PubMed] [Google Scholar]
- 5.Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, et al. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II-mediated cardiac hypertrophy. Circ Res. 2006;99:528–36. doi: 10.1161/01.RES.0000240147.49390.61. [DOI] [PubMed] [Google Scholar]
- 6.Kawanabe Y, Masaki T, Hashimoto N. Involvement of epidermal growth factor receptor-protein tyrosine kinase transactivation in endothelin-1-induced vascular contraction. J Neurosurg. 2004;100:1066–71. doi: 10.3171/jns.2004.100.6.1066. [DOI] [PubMed] [Google Scholar]
- 7.Han L, Ma Q, Li J, Liu H, Li W, Ma G, et al. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS One. 2011;6:e27074. doi: 10.1371/journal.pone.0027074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matrougui K. Diabetes and microvascular pathophysiology: role of epidermal growth factor receptor tyrosine kinase. Diabetes Metab Res Rev. 2010;26:13–6. doi: 10.1002/dmrr.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucchini E, Pilotto S, Spada E, Melisi D, Bria E, Tortora G. Targeting the epidermal growth factor receptor in solid tumors: focus on safety. Expert Opin Drug Saf. 2014;13:535–49. doi: 10.1517/14740338.2014.904283. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, et al. EGFR signaling promotes TGFbeta-dependent renal fibrosis. J Am Soc Nephrol. 2012;23:215–24. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang MZ, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63:2063–72. doi: 10.2337/db13-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akhtar S, Almubrad T, Bron AJ, Yousif MH, Benter IF, Akhtar S. Role of epidermal growth factor receptor (EGFR) in corneal remodelling in diabetes. Acta Ophthalmol. 2009;87:881–9. doi: 10.1111/j.1755-3768.2008.01434.x. [DOI] [PubMed] [Google Scholar]
- 13.Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57:1629–37. doi: 10.2337/db07-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasayama S, Yoshimura M, Oka T. Decreased expression of hepatic epidermal growth factor receptor gene in diabetic mice. J Mol Endocrinol. 1989;3:49–56. doi: 10.1677/jme.0.0030049. [DOI] [PubMed] [Google Scholar]
- 15.Sayed-Ahmed N, Besbas N, Mundy J, Muchaneta-Kubara E, Cope G, Pearson C, et al. Upregulation of epidermal growth factor and its receptor in the kidneys of rats with streptozotocin-induced diabetes. Exp Nephrol. 1996;4:330–9. [PubMed] [Google Scholar]
- 16.Khan AJ, Fligiel SE, Liu L, Jaszewski R, Chandok A, Majumdar AP. Induction of EGFR tyrosine kinase in the gastric mucosa of diabetic rats. Proc Soc Exp Biol Med. 1999;221:105–10. doi: 10.1046/j.1525-1373.1999.d01-62.x. [DOI] [PubMed] [Google Scholar]
- 17.Benter IF, Yousif MH, Hollins AJ, Griffiths SM, Akhtar S. Diabetes-induced renal vascular dysfunction is normalized by inhibition of epidermal growth factor receptor tyrosine kinase. J Vasc Res. 2005;42:284–91. doi: 10.1159/000085904. [DOI] [PubMed] [Google Scholar]
- 18.Akhtar S, Yousif MH, Dhaunsi GS, Sarkhouh F, Chandrasekhar B, Attur S, et al. Activation of ErbB2 and downstream signalling via rho kinases and ERK1/2 contributes to diabetes-induced vascular dysfunction. PLoS One. 2013;8:e67813. doi: 10.1371/journal.pone.0067813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pareja M, Sanchez O, Lorita J, Soley M, Ramirez I. Activated epidermal growth factor receptor (ErbB1) protects the heart against stress-induced injury in mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R455–62. doi: 10.1152/ajpregu.00588.2002. [DOI] [PubMed] [Google Scholar]
- 20.Williams-Pritchard G, Knight M, Hoe LS, Headrick JP, Peart JN. Essential role of EGFR in cardioprotection and signaling responses to A1 adenosine receptors and ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2011;300:H2161–8. doi: 10.1152/ajpheart.00639.2010. [DOI] [PubMed] [Google Scholar]
- 21.Akhtar S, Yousif MH, Chandrasekhar B, Benter IF. Activation of EGFR/ERBB2 via pathways involving ERK1/2, P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from ischemia-reperfusion injury. PLoS One. 2012;7:e39066. doi: 10.1371/journal.pone.0039066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benter IF, Juggi JS, Khan I, Yousif MH, Canatan H, Akhtar S. Signal transduction mechanisms involved in cardiac preconditioning: role of Ras-GTPase, Ca2+/calmodulin-dependent protein kinase II and epidermal growth factor receptor. Mol Cell Biochem. 2005;268:175–83. doi: 10.1007/s11010-005-3895-1. [DOI] [PubMed] [Google Scholar]
- 23.Galan M, Kassan M, Choi SK, Partyka M, Trebak M, Henrion D, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60:71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang D, Zhong P, Hu J, Lin F, Qian Y, Xu Z, et al. EGFR inhibition protects cardiac damage and remodeling through attenuating oxidative stress in STZ-induced diabetic mouse model. J Mol Cell Cardiol. 2015;82:63–74. doi: 10.1016/j.yjmcc.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 25.Barrick CJ, Yu M, Chao HH, Threadgill DW. Chronic pharmacologic inhibition of EGFR leads to cardiac dysfunction in C57BL/6 J mice. Toxicol Appl Pharmacol. 2008;228:315–25. doi: 10.1016/j.taap.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]