

ABSTRACT
Race 1 isolates of Fusarium oxysporum f. sp. lycopersici (FOL) are characterized by the presence of AVR1 in their genomes. The product of this gene, Avr1, triggers resistance in tomato cultivars carrying resistance gene I. In FOL race 2 and race 3 isolates, AVR1 is absent, and hence they are virulent on tomato cultivars carrying I. In this study, we analyzed an approximately 100-kb genomic fragment containing the AVR1 locus of FOL race 1 isolate 004 (FOL004) and compared it to the sequenced genome of FOL race 2 isolate 4287 (FOL4287). A genomic fragment of 31 kb containing AVR1 was found to be missing in FOL4287. Further analysis suggests that race 2 evolved from race 1 by deletion of this 31-kb fragment due to a recombination event between two transposable elements bordering the fragment. A worldwide collection of 71 FOL isolates representing races 1, 2, and 3, all known vegetative compatibility groups (VCGs), and five continents was subjected to PCR analysis of the AVR1 locus, including the two bordering transposable elements. Based on phylogenetic analysis using the EF1-α gene, five evolutionary lineages for FOL that correlate well with VCGs were identified. More importantly, we show that FOL races evolved in a stepwise manner within each VCG by the loss of function of avirulence genes in a number of alternative ways.
IMPORTANCE Plant-pathogenic microorganisms frequently mutate to overcome disease resistance genes that have been introduced in crops. For the fungus Fusarium oxysporum f. sp. lycopersici, the causal agent of Fusarium wilt in tomato, we have identified the nature of the mutations that have led to the overcoming of the I and I-2 resistance genes in all five known clonal lineages, which include a newly discovered lineage. Five different deletion events, at least several of which are caused by recombination between transposable elements, have led to loss of AVR1 and overcoming of I. Two new events affecting AVR2 that led to overcoming of I-2 have been identified. We propose a reconstruction of the evolution of races in FOL, in which the same mutations in AVR2 and AVR3 have occurred in different lineages and the FOL pathogenicity chromosome has been transferred to new lineages several times.
KEYWORDS: crops, disease resistance, evolution, fungus, pathogen
INTRODUCTION
In many plant-pathogen interactions, a pair of matching genes determines resistance of the host to a specific pathogen. One is a plant resistance (R) gene and the other a pathogen avirulence (AVR) gene (1, 2). Plants that express an R gene are resistant to the pathogens that express the matching AVR gene. This concept is known as the gene-for-gene model (1, 2). The interaction between the products of these two matching genes, either directly or indirectly, prevents further growth of the invading pathogen in the host plant (3). However, many pathogens can evolve to evade R gene-mediated resistance by loss of function of its matching AVR gene or by employing new virulence factors. In turn, the host can develop novel R genes to recognize newly evolved pathogen effectors. Knowledge about the mechanisms underlying loss of function of avirulence genes is of critical importance to understand how pathogens overcome R gene-mediated resistance in host plants. In many pathogens, loss of function of AVR genes has been associated with point mutations or transposon insertions into the promoters or coding sequences of avirulence genes, as well as with deletion of the entire gene (4–10).
Fusarium oxysporum f. sp. lycopersici (FOL) is a soilborne fungus that causes wilt disease in tomato plants (11, 12). Three physiological races of FOL have been identified based on their pathogenicity to tomato cultivars carrying dominant, race-specific resistance genes. Race 1 had been the prevalent race since the late 19th century, when Fusarium wilt was first described, until the discovery and introgression of the first R gene against race 1 (13); race 2 was first reported in 1945 in Ohio (14), whereas race 3 was first reported in 1978 in Australia (15). The interaction between tomato and races of FOL fits the so-called gene-for-gene model (1). Tomato R genes that confer race-specific resistance to FOL are known as immunity (I) genes. To date, three I genes, namely, I (or I-1), I-2, and I-3, have been identified in wild tomatoes and have been introgressed into tomato cultivars (16, 17).
The three I genes confer resistance against FOL races containing the three matching avirulence genes AVR1, AVR2, and AVR3, respectively (18–20). AVR1 is present in all FOL race 1 isolates but is absent in race 2 and 3 isolates (21), with two exceptions: a race 2 isolate and a race 3 isolate from Japan in which the AVR1 gene was independently interrupted by a transposon (22, 23). The absence of AVR1 enables race 2 and 3 isolates to infect tomato cultivars carrying I. AVR2 is present in all FOL isolates, albeit that I-2 breaking (race 3) isolates contain an AVR2 allele with a point mutation leading to an amino acid change in the protein (20). So far, three AVR2 alleles have been described, with the amino acid changes V41→M, R45→H, and R46→P. All three amino acid changes lead to loss of the avirulence function of Avr2, whereas its virulence function remains unaffected (20). Two AVR3 alleles have been described, one encoding a protein with a glutamic acid (E) at position 165 and one with a lysine (K) residue at this position. Both proteins still act as avirulence factors, but they differ in the degrees of virulence they confer (18, 24).
As a (predominantly) asexual organism, F. oxysporum is thought to evolve mostly by means of mutations and DNA rearrangements and potentially through the process of parasexual recombination (25). Heterokaryon formation is a prerequisite for parasexual recombination and is thought to be limited to strains that are vegetatively compatible. Strains that can form heterokaryons with one another are assigned to the same vegetative compatibility group (VCG), and those that cannot are assigned to different VCGs (26, 27). For F. oxysporum f. sp. lycopersici, four VCGs (VCG0030, -0031, -0033, and -0035) have been reported so far (28–30). VCG0030 includes isolates formerly assigned to VCG0032 (31).
Previously, results from restriction fragment length polymorphism (RFLP), random amplified polymorphic DNA (RAPD), and isozyme analyses demonstrated that isolates within a VCG are genetically more similar to each other than to isolates of the same race in another VCG. This suggests that each VCG represents a clonal population (28, 31, 32). To determine further phylogenetic relationships within and among formae speciales of F. oxysporum DNA sequences of the mitochondrial small-subunit (mtSSU) rRNA gene, the ribosomal DNA (rDNA) intergenic spacer (IGS) region and the translation elongation factor (EF1-α) gene have been used (33–35). Such studies confirmed multiple evolutionary origins for many formae speciales of F. oxysporum. For example, phylogenetic analysis based on rDNA IGS, mating type locus (MAT) 1, and polygalacturonase (Pgs) 1 revealed evolutionary lineages for FOL that correlate with VCGs (36).
In several studies it has been suggested that deletion of AVR1 from race 1 isolates resulted in the emergence of race 2 (avirulence genotype – AVR2 AVR3) and that a point mutation in AVR2 of race 2 brought about the birth of race 3 (– avr2 AVR3) (37). However, the molecular mechanism underlying the loss of AVR1 has not been determined. In this study, a molecular analysis that reveals a likely mechanism underlying the deletion of AVR1 in FOL race 1 isolates that led to the emergence of race 2 is presented.
RESULTS
Sequencing and annotation of the AVR1 genomic region.
In a previous study we determined the primary structure of a 2.8-kb genomic fragment (GenBank accession number AM234064.2) representing the AVR1 locus of F. oxysporum f. sp. lycopersici (FOL) race 1 isolate 004 (FOL004) (19). To analyze a larger genomic fragment containing the AVR1 locus, we needed to obtain the regions flanking this 2.8-kb fragment. To this end, a FOL004-bacterial artificial chromosome (BAC) library (38) was screened using AVR1-specific primers (numbers 1091 and 1033 [see Table S1 in the supplemental material]). This resulted in the identification of four clones with inserts ranging from approximately 65 kb to 100 kb. Since restriction analysis (see Fig. S1A in the supplemental material) revealed that at least the two smallest inserts (2G2 and 9C1) were covered completely by the largest one (9G3), these two were excluded from further analysis. The positions of the inserts of the four clones relative to each other are shown in Fig. S1B in the supplemental material. Paired-end sequencing of clones 9G3 and 14I-2 made clear that the latter did not carry additional information either. Therefore, further analysis was restricted to the approximately 100-kb insert of clone 9G3. Details of the assembly of clone 9G3 are given in Fig. S2 in the supplemental material. Briefly, the length of the full 9G3 insert was found to be 98,694 nucleotides in total. The coding sequence for Avr1 is located between nucleotides 52222 and 53014 and is interrupted by one intron (nucleotides 52376 to 52439).
Besides the Helitron transposons mentioned in the supplemental material (HelA and HelB), the 9G3 insert contains a large number of other transposable elements (TEs) from both class I (retrotransposons) and class II (DNA transposons). TEs are classified based on the system proposed by Wicker and colleagues (39). Within class I we identified members of the Copia and Gypsy superfamilies as well as solo long terminal repeat retrotransposons, LINE transposable elements, and SINE transposable elements (Fig. 1; see Table S2 in the supplemental material). Most of the class II elements are members of the Tc1-mariner superfamily and include Pogo transposable elements, hAT transposable elements, and MITE transposable elements (Fig. 1; Table S2). Altogether, the TEs occupy 58.9% (58,342 bp in total size) of the insert of BAC clone 9G3.
FIG 1.

The absence of AVR1 in the reference strain FOL4287 can be explained by recombination between Helitron transposons. (A) Genetic organization of the 9G3 BAC insert (100 kb) of FOL004 containing AVR1 and its comparison to a genomic region in supercontig 2.22 (sc2.22) of chromosome 14 of FOL4287. Conserved regions in both sequences are shown in gray boxes. Comparison revealed that a 43-kb genomic region containing AVR1 and including HelA and HelB in the insert has been replaced by HelAB and an adjacent Hop3 transposable element in FOL4287. Genomic maps are drawn to scale. (B) Schematic representations of FOL004 HelA, FOL004 HelB, and FOL4287 HelAB. FOL004 HelB contains an extra 15 bp (TCCCCTGAGCGTGGA) compared to FOL004 HelA and FOL4287 HelAB. Moreover, two polymorphisms were found in HelB (4718A/- and 4909A/G) compared to corresponding positions in FOL004 HelA and FOL4287 HelAB. A Hop3 insertion at the 5′ end of the FOL4287 HelAB is indicated.
Besides AVR1, eight non-TE open reading frames (ORFs) were identified in the 9G3 insert, ranging from 280 to 6,114 bp in length (Fig. 1; see Table S3 in the supplemental material). ORF1, -2, -5, -6, -7, and -8 have been annotated in the reference genome (FOXG_14233, FOXG_14234, FOXG_14236, FOXG_14237, FOXG_14238, and FOXG_14240, respectively). ORF3 and -4 are located within the 31-kb fragment that is absent in the reference genome (see below). ORF3 encodes a protein of 2,037 amino acids with a rolling-circle (RC) replication initiator (rep) and a DNA helicase (hel) domain, the main hallmarks of Helitron proteins (40, 41). However, except for the conserved residues in the rep and hel domains, there is virtually no homology with the HelA and HelB proteins. A BLAST search of the ORF3 sequence against the genome sequence of FOL strain 4287 (FOL4287) identified four copies (sequence identity over 99%) belonging to FoHeli3 (Table S3), one of the Helitron families that have been identified in F. oxysporum (48). Moreover, analysis of the flanking sequences of ORF3 confirmed that it possesses terminal structural features specific for FoHeli3. ORF4 encodes a 275-amino-acid protein of unknown function. Three homologs of ORF4 were identified in the FOL4287 genome (Table S3).
Loss of AVR1 in FOL4287 can be explained by homologous recombination between Helitron transposons.
The genome of FOL4287 has been sequenced, assembled, and annotated (www.broadinstitute.org) and is used here as reference genome for FOL004. FOL4287 is a race 2 isolate and lacks AVR1. Sequence identity of the 9G3 insert with the reference genome is over 99.9%, except for a fragment containing AVR1 found to be absent in FOL4287; the 9G3 sequence starting with the 5′ end of HelA and ending with the 3′ end of HelB is reduced to one Helitron copy (HelAB) in the reference genome (Fig. 1). HelAB shows 100% sequence identity with HelA, except for the insertion of a Hop3 transposable element between the fourth and fifth nucleotides of the 5′ end of HelAB (Fig. 1). Hop3 is 2,941 bp in length, including 98-bp terminal inverted repeats, and is flanked by an 11-bp direct repeat representing a target site duplication (42). These observations confirm the absence of a 31-kb (from the 3′ end of HelA to the 5′ end of HelB) genomic fragment containing AVR1 in race 2 isolate FOL4287 compared to race 1 isolate FOL004, and they strongly suggest that this absence is the result of a homologous recombination event between HelA and HelB. The site of recombination is downstream of the A/G polymorphism (Fig. 1), unless this mutation occurred later in time than the recombination event leading to the emergence of race 2. In the latter case, the point of recombination should be positioned upstream of the mutation. The same line of reasoning applies for the deletion and duplication found in HelB (Fig. 1).
An alternative mechanistic explanation for the evolvement of races could be that race 2 represents an ancestral FOL strain without AVR1 and that a genomic fragment containing AVR1 and HelB has been acquired later. However, in insert 9G3, HelA has been inserted into a nonautonomous version of transposable element YahAT7, due to which the latter element is split into two fragments of 529 bp and 71 bp. In FOL4287, the 71-bp fragment of the YahAT7 element is well conserved just upstream of HelAB, but downstream of this Helitron transposon the 529-bp fragment is not present. Acquisition by race 2 of a genomic fragment with AVR1 and HelB in a way that joins the 529-bp fragment of YahAT7 to the 3′ end of HelA, which already has the 71-bp YahAT7 fragment at its 5′ end, seems very unlikely. Consequently, we hypothesize that race 2 evolved from race 1 by the deletion of the AVR1 locus, most probably due to a homologous recombination event between the Helitron transposons that border this genomic region (HelA and HelB). We also hypothesize that the Hop3 insertion at the 5′ end of HelAB in FOL4287 is a later evolutionary event.
The presence or absence of HelB divides race 1 isolates into two groups.
In FOL4287, downstream of HelAB, there is yet another Helitron copy, HelC, with 100% sequence identity with HelA (Fig. 1). This suggests that the genome of FOL004 contains one Helitron transposon (HelA) 17.3 kb upstream of AVR1 and two Helitron transposons downstream at distances of 12.9 kb (HelB) and 88 kb (HelC). To analyze the genomic region containing HelA and HelC in different FOL strains, 10 sets of primers (A to J) that would give unique PCR products of approximately 400 to 2,000 bp were designed (Fig. 2A; Table S1). Products brought about by primer pairs A and J correspond to FOL004 sequences 5,158 bp upstream of HelA and 824 bp downstream of HelC, respectively. PCR fragments produced with primer pairs B, D, and H are indicative for the presence in the genome of the 5′ ends of HelA, HelB, and HelC, respectively. Likewise, fragments generated using pairs C, E, and I point to the presence of the 3′ ends of these elements. PCR fragments produced with primer pairs F and G correspond to sequences 2.3 kb and 14 kb downstream of HelB, respectively. These primer sets were used to “scan” a 144-kb genomic region in a collection of 71 FOL isolates, including 26 race 1, 28 race 2, and 17 race 3 isolates (listed in Table 1).
FIG 2.

Differences in the AVR1-containing genomic region between FOL strains. (A) Schematic representation of 144 kb around AVR1 in FOL004 deduced from a FOL004 BAC insert containing AVR1 (100 kb) and the FOL4287 reference genome. A to J indicate primer pairs used to scan this region in a collection of FOL isolates originating from different geographical areas. A, primer set 4539/4540; B, primer set 4298/4297; C, primer set 4242/4241; D, primer set 4345/4297; E, primer set 4242/4340; F, primer set 4355/4354; G, primer set 4309/4306; H, primer set 4395/4297; I, primer set 4242/4371; J, primer set 4541/4542. (B) Example of PCR experiments showing amplification of fragments from the genomic DNA of race 1 isolates with primer pairs A to J. The absence of PCR products with primer pairs D and E indicates the absence of Helitron B (HelB). (C) Examples of PCR experiments showing fragments amplified from genomic DNA from race 2 and race 3 isolates with primer pairs A to J. The lack of PCR products with primer pairs C and D indicates the absence of a genomic region between HelA and HelB. The absence of PCR products with primer pairs C to H indicates the absence of a genomic region between HelA and HelC.
TABLE 1.
Molecular diversity in the AVR1 genomic region and AVR2 and AVR3 genes in FOLa
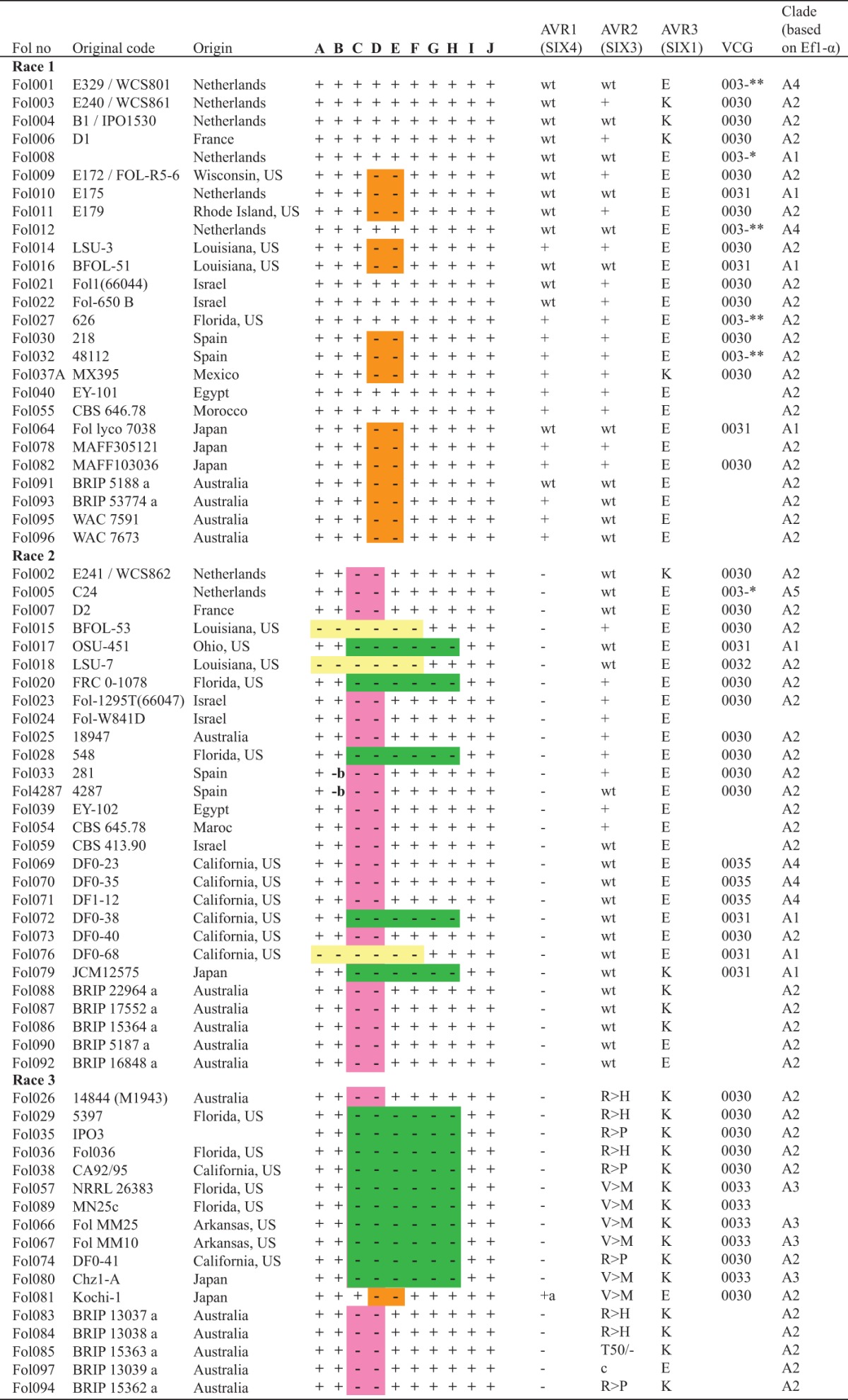
a, AVR1 has been truncated by Hormin insertion; b, a Hop 3 insertion at the 5′ end of HelA; c, deletion of entire AVR2 gene. Orange indicates absence of HelB, pink indicates deletion of the AVR1 genomic region between HelA and HelB (31 kb), green indicates deletion of the AVR1 genomic region between HelA and HelC (approximately 102 kb), and yellow indicates deletion of AVR1 genomic region including HelA and HelB. +, positive PCR result; −, negative PCR result; wt, wild-type sequence confirmed by sequencing. E and K represent different AVR3 alleles. R→H, amino acid change in Avr2 protein at 45th position; R→P, amino acid change in Avr2 protein at 46th position; V→M, amino acid change in Avr2 protein at 41th position; T50/-, amino acid deletion in Avr2 protein at 50th position. *, self-compatible; **, self-incompatible (31). FOL064 and FOL067 were provided by Bart Lievens (Belgium), FOL074 by Corby H. Kistler (USA), FOL078 by Makoto Kawase (Japan), and FOL079 by Tsutomu Arie (Japan). FOL054 was obtained from the CBS Fungal Biodiversity Centre (The Netherlands). Donors of all other isolates are mentioned in reference 24. WCS, Willie Commelin Scholten culture collection, The Netherlands; IPO, Instituur voor Plantenziektenkundig Onderzoek culture collection, The Netherlands; BRIP, Plant Pathology Herbarium culture collection, Queensland, Australia; MAFF, Ministry of Agriculture, Forestry and Fisheries culture collection, Japan; WAC, Western Australian Plant Pathology culture collection, Australia; LSU, Louisiana State University, Baton Rouge, LA, USA; CBS, Centraalbureau voor Schimmelcultures-Fungal Biodiversity Center, Utrecht, The Netherlands; OSU, Ohio State University, Columbus, OH, USA; FRC, Fusarium Research Center, The Pennsylvania State University, State College, PA, USA; JCM, Japan Collection of Microorganism, RIKEN BioResource Center, Japan; NRRL, Agricultural Research Service Culture Collection of the U.S. Department of Agriculture, Peoria, IL, USA.
In all race 1 isolates, each primer pair (A to J) gave a PCR product of the expected length except for pairs D and E (Fig. 2B). This demonstrates that all 26 race 1 isolates tested contain HelA and HelC in their genomes (Fig. 2B; Table 1). Interestingly, primer pairs D and E, together indicative for the presence of HelB, gave no PCR product in 15 race 1 isolates (Fig. 2B; Table 1). In further analysis, a 1.8-kb fragment could be amplified from the latter 15 race 1 isolates using primers corresponding to the 5′ and 3′ flanking sequences of HelB (see Fig. S3 in the supplemental material, 4345 and 4340 [Table S1]). Sequencing this 1.8-kb fragment confirmed the absence of HelB. These results suggest that race 1 isolates can be divided into two groups, one with and one without HelB (Fig. 2B).
Race 2 and race 3 isolates can also be divided in two groups.
To test the hypothesis that race 2 evolved from race 1 by the deletion of AVR1 through homologous recombination between the two Helitron transposons bordering the AVR1 locus, we analyzed the AVR1 loci of 28 race 2 isolates (Table 1) using the primer sets A to J (Fig. 2A). In 20 race 2 isolates, a PCR product was obtained with primer pairs A and B and E to J, but no product was obtained with sets C and D, indicative for deletion of the AVR1 genomic region between HelA and HelB (Fig. 2C; Table 1). This supports the hypothesis that race 2 isolates evolved by the deletion of the 31-kb genomic region with AVR1 due to homologous recombination between two adjacent Helitron transposons (HelA and HelB). However, in two race 2 isolates, FOL4287 (see Fig. S4A in the supplemental material) and FOL033 (data not shown), primer pair B did not amplify a PCR fragment. As mentioned above, in FOL4287, a Hop3 transposable element was inserted into the 5′ end of HelAB (Fig. 1). The lack of a PCR product using primer pair B could be due to the conditions used in this experiment, which were not suited to amplify a product of 3.7 kb including a full Hop3 element. To confirm this hypothesis, additional PCR experiments were conducted using primers specific for this region. Primers 4304 and 4298 (Table S1) specifically bind to the 3′ end of Hop3 and to its downstream flanking region, respectively. Using these primers, a PCR product of 624 bp could be amplified from FOL4287 and FOL033 DNA only (see Fig. S4B in the supplemental material). Similarly, using primers 4303 and 4297, which correspond to the 5′ end of Hop3 and the 5′ end of HelAB, respectively, a PCR product of 723 bp was amplified from FOL4287 and FOL033 DNA only (see Fig. S4C in the supplemental material). Sequencing of these PCR products confirmed that in FOL4287 and FOL033, a Hop3 element has been inserted into the 5′ end of HelAB. These experiments also support the hypothesis that the Hop3 insertion is a late evolutionary event after the emergence of race 2 isolates from race 1 by the deletion of AVR1. Interestingly, FOL4287 and FOL033 are from the same region in Spain and hence might be closely related to each other.
In five race 2 isolates, PCR products were produced with primer pairs A, B, I, and J but not with primer pairs C to H, indicating a deletion of an approximately 102-kb region between HelA and HelC (Fig. 2C; Table 1). Using primers corresponding to the 5′ flanking sequence of FOL004 HelA and 3′ flanking sequence of FOL004 HelC (4298 and 4371 [Table S1]), a fragment of approximately 6.5 kb could be amplified from FOL017, a representative of the group of race 2 isolates with the 102-kb deletion; no product was obtained when either FOL004 or FOL4287 DNA was used as the template (data not shown). Sequencing this 6.5-kb PCR product revealed a Helitron transposon, suggesting that the genomic region in the latter group of race 2 isolates was deleted due to a homologous recombination event between HelA and HelC. Hence, the Helitron transposon amplified from FOL017 using primer pair 4298 and 4371 was designated Helitron AC (HelAC). No sequence polymorphisms were identified between FOL004 HelA, FOL004 HelC, and FOL017 HelAC.
Unexpectedly, in three race 2 isolates, FOL076, FOL015, and FOL018, the PCR results were negative for primer sets A to F (Table 1). This suggests that the genomes of these isolates carry a large deletion including AVR1, HelA, and HelB. To investigate the extent of the deletion in these isolates, we took the same approach as described above and “scanned” a genomic region corresponding to the approximately 660-kb region upstream the position of HelC in the FOL4287 genome. Fourteen primer sets (G, H, and K to V [Table S1]) were used, and isolate FOL004 was used as a control. The relative positions of the primer sets on the 660-kb fragment as well as the positions of some relevant ORFs are shown in Fig. 3A. The results of the “scan” are shown in Fig. 3B. In FOL076, primer sets G, H, K to O, U, and V gave positive PCR results, whereas with sets P to T no fragments were amplified (Fig. 3B). This suggests that a fragment between primer pairs O and U is absent in the genome of this isolate. In FOL4287, an NHT2-like retrotransposon (43) is present just downstream of both primer set O and HelAB. Primer set U corresponds to the 3′ end of this retrotransposon and its flanking region. A primer (5049 [Table S1]) that corresponds to a unique sequence upstream of the NHT2-like element close to primer set O was designed. Furthermore, the reverse primer (4626 [Table S1]) of set U corresponds to a unique sequence downstream of the second copy of the NHT2-like element. Using these two primers, we were able to amplify an approximately 5.7-kb fragment from FOL076 genomic DNA but not from FOL4287 genomic DNA (Fig. 3B). Sequencing confirmed the presence of an NHT2-like element in the fragment. This observation suggests that deletion of AVR1 from the FOL076 genome is due to a homologous recombination event between two copies of an NHT2-like element. Compared to the genome of FOL004, a 488-kb fragment with the AVR1 locus has been deleted in FOL076. Compared to the reference genome, the deletion is 31 kb shorter, thus being approximately 457 kb (Fig. 3A).
FIG 3.

Large deletions lead to loss of AVR1 in FOL076, FOL018, and FOL015. (A) Schematic representation of an approximate 667-kb genomic region in supercontig 2.22 (sc2.22) of chromosome 14 of FOL4287 (positions 96651 to 764000). HelAB indicates the location of the AVR1 genomic region (31 kb) in FOL004. K to V, G, and H indicate primer pairs used to scan this region in FOL076, FOL018, and FOL015. Dashed lines indicate deleted regions in the genomes of FOL076, FOL018, and FOL015 compared to FOL4287, with approximate sizes. (B) Left, PCR experiments showing amplicons from FOL076, FOL018, and FOL015 with primer pairs K to V, G, and H. FOL004 genomic DNA was used as a positive control. Right, amplification of a 5.7-kb PCR product from FOL076 using primers 5049 and 4626. FOL4287 genomic DNA was used as a control. Lanes: 1, GeneRuler 1-kb DNA ladder (0.25 to 10 kb); 2, water control; K, primer set 4866/4867; L, primer set 4890/4891; M, primer set 4967/4968; N, primer set 5023/5024; O, primer set 5025/5026; P, primer set 4874/4875; Q, primer set 4749/4750; R, primer set 4880/4881; S, primer set 4751/4752; T, primer set 4618/4732; U, primer set 4733/4626; V, primer set 4309/4306; G, primer set 4616/4617; H, primer set 4395/4297.
In FOL018, primer pairs K, L, U, V, G, and H gave positive PCR results, whereas sets M to T gave no fragments (Fig. 3B). Since primer sets L and U are approximately 600 kb apart from each other in the FOL4287 genome, we estimate that in isolate FOL018 an approximately 631-kb fragment containing AVR1 is missing compared to in FOL004. The genome sequence of FOL4287 has a sequence gap just downstream of primer set L. In an attempt to the bridge this gap in the reference genome, we tried to PCR amplify sequences using primers corresponding to the ends of the two contigs flanking the gap (5052 and 4968 [Table S1]). Unfortunately, no DNA was amplified. Like in FOL076, an NHT2-like retrotransposon is present near or at the right boundary of the deletion. With the reverse primer of set U and a primer corresponding to the contig end upstream of the gap (5052 and 4626 [Table S1]), we were unable to amplify a fragment from FOL018. The exact length of the deletion in this strain therefore remains undetermined.
In FOL015, primer sets G, H, K to R, and V gave positive PCR results, whereas primer sets S to U gave negative results (Fig. 3B). This indicates a deletion between primer sets R and V and that an approximately 168-kb genomic region containing AVR1 is absent in FOL015 compared to FOL004; compared to FOL4287, the deletion is approximately 137 kb (Fig. 3A). Unfortunately, due to sequence gaps in the corresponding genomic region of reference strain FOL4287, we could not determine the exact boundaries of the deletion in this strain either.
Previous studies have indicated that FOL race 3 evolved from race 2 by a point mutation in AVR2. If this is true, one would expected two groups of race 3 isolates as well, one with the 31-kb deletion and one with a 102-kb deletion. To test this hypothesis, we extended our PCR analysis of the AVR1 locus to 17 race 3 isolates. Of the isolates analyzed, 10 carried the 102-kb genomic fragment deletion with AVR1 and six the deletion of the 31-kb fragment (Fig. 2C; Table 1). The Japanese Kochi-1 isolate (FOL081) was the only exception. PCR results for this isolate were positive for primer sets A to J except for D and E (Table 1), indicating that this isolate contains the AVR1 genomic region with HelA and HelC. This is fully in line with the observations by Inami and coworkers, who have shown that in this FOL race 3 isolate, AVR1 is inactivated by the insertion of a Hormin transposon (22). We assume that Kochi-1 evolved from a race 1 isolate without HelB into a race 2 isolate by the insertion of the Hormin element into AVR1 and subsequently evolved into a race 3 isolate by a point mutation in AVR2. However, we cannot exclude the possibility that the mutation in AVR2 occurred prior to the Hormin insertion.
In conclusion, except for three race 2 isolates and one race 3 isolate, all FOL race 2 and race 3 isolates can be divided into two groups based on the deletion events that led to the loss of the AVR1 locus. One group lacks 102 kb of the AVR1 locus, most probably due to homologous recombination between HelA and HelC, and the other lacks the 31-kb AVR1 genomic region, most probably due to a homologous recombination event between HelA and HelB.
Sequence polymorphisms in the FOL AVR genes.
Using a PCR approach with specific primers (1091 and 1033 [Table S1]), all 26 FOL race 1 isolates were tested for the presence of AVR1. In line with their classification as race 1, all gave rise to a PCR fragment of the expected length (“+” in Table 1). Fourteen fragments were sequenced and compared to AVR1 of FOL004 (“wt”); no sequence variation was observed (Table 1).
As expected, all 54 race 1 and 2 isolates contained AVR2. Thirty AVR2 amplicons from race 1 and race 2 strains were sequenced and found to be identical (Table 1). FOL race 3 isolates are believed to have evolved from race 2 by a point mutation in AVR2 leading to an amino acid change in the protein (20). So far, three AVR2 alleles have been described, each with one of the amino acid changes V41→M, R45→H, and R46→P. All three amino acid changes lead to the loss of the avirulence function of Avr2. Out of the 17 race 3 isolates analyzed here (Table 1), six contain the V41→M allele, five the R45→H allele, and four the R46→P allele. Interestingly, one race 3 isolate from Australia (FOL085) contains a hitherto-unknown AVR2 allele, one with a deletion of the triplet encoding the threonine (T) residue at position 50 of Avr2. The virulence of this isolate on I-2 lines indicates that this deletion has also led to the loss of the avirulence function of AVR2 (Table 1 and data not shown). One isolate (FOL097) failed to produce an AVR2-specific PCR fragment with primers 2934 and 964 (Table S1). The absence of AVR2 in this isolate suggests that either AVR2 is absent or PCR amplification has failed, for example, due to an insertion into AVR2 of a transposable element. To test this, additional PCR experiments were carried out on genomic DNA of FOL097. Like the original AVR2-specific primer set, the sets corresponding to either a region 950 bp upstream (1723 and 1236 [Table S1]) or a region 700 bp downstream (1237 and 1238 [Table S1]) of the AVR2 ORF and inside the ORF (962 and 963 [Table S1; Fig. S1]) failed to amplify a product from FOL097. This strongly suggests that the genomic region containing AVR2 is missing in FOL097. Another FOL effector gene, that for Six5, is located ∼1 kb upstream of AVR2 in the opposite orientation. We tested whether this gene is absent from FOL097 as well. Interestingly, PCR amplification using gene-specific primers corresponding to a region inside the Six5 ORF produced no amplicon in FOL097 (Fig. S1). In summary, one new mutation in AVR2 was identified in addition to the three already known. Remarkably, all four mutations that abolish avirulence occur in the same region of the Avr2 protein, between amino acids 40 and 51 (Fig. 4). Hence, this region appears to be recognized, directly or indirectly, by I-2. Furthermore, the first FOL isolate with the AVR2 locus deleted was identified. This isolate also lacks Six5.
FIG 4.

Novel mutation in AVR2 in FOL race 3 isolate FOL085. A sequence alignment of wild-type (FOL4287) Avr2 with four mutated versions of Avr2 (V41→/M, R45→/H, R46→/P, and T50/-) in four race 3 strains (FOL085, -074, -026, and -057) is shown. Amino acid changes are highlighted in green.
So far only a single polymorphism has been reported in FOL AVR3, resulting in either a glutamate (E) or a lysine (K) residue at position 164 (AVR3E or AVR3K) (24). In this study, AVR3 from all FOL isolates in our collection was sequenced. All carry either the AVR3E or the AVR3K allele (Table 1).
Comparison of EF1-α gene sequences reveals a hitherto-unknown clonal line of FOL.
Vegetative compatibility has often been used to estimate the diversity within the formae speciales of F. oxysporum. Isolates within a VCG are genetically very similar, and generally it is believed that VCGs represent clonal populations (28, 31, 32). To assess to what VCG a particular isolate belongs requires the creation of mutants and testing these against tester isolates. Another way to assess genetic relationships between individual isolates is the comparison of conserved sequences in the genome. Examples of such sequences are the internal transcribed spacer (ITS) sequence and intergenic spacer (IGS) sequences of rRNA genes, mating type locus (MAT) sequences, endopolygalacturonase (pg1) and exopolygalacturonase (pgx4) genes, and the elongation factor 1-α (EF1-α) gene. Table 1 indicates to which VCG each individual FOL isolate belongs as far as this is known. To investigate the genetic relationship between the FOL isolates used in this study, the sequence of approximately 565 bp corresponding to the EF1-α gene was retrieved from public databases or determined for all isolates. In the analysis, EF1-α gene sequences of representative isolates from other formae speciales of F. oxysporum were included. Fusarium commune served as outgroup (44). A maximum-likelihood tree revealed five groups (clades A1 to A5) for FOL isolates (Fig. 5). Within each clade, the EF1-α gene sequences are 99.8 to 100% identical.
FIG 5.

Maximum-likelihood (ML) tree inferred from EF1-α gene sequences of Fusarium oxysporum isolates. Fusarium commune strain NRRL28387 (GenBank accession number AF246832.1) was used as an outgroup. Bootstrap values calculated in ML analysis are shown beside branches. Five lineages (A1 to A5) for FOL inferred from ML analysis are indicated in different colors. The EF1-α gene sequences of FOL isolates indicated by asterisks were determined in a previous study (21).
All seven FOL isolates known to belong to VCG0031 group into clade A1 (clade numbering is the same as used by Kawabe and coworkers [36]), and both races 1 and 2 are represented. Moreover, clade A1 also contains isolates of F. oxysporum f. sp. batatas and F. oxysporum f. sp. erythroxili, suggesting that these isolates are closely related to FOL isolates of VCG0031. F. oxysporum isolates usually have a very narrow host range (45); hence, we assume that F. oxysporum f. sp. batatas and F. oxysporum f. sp. erythroxili are pathogenic only on sweet potato and cocoa plant, respectively, and do not have the effector repertoire that is required for virulence on tomato and is encoded on chromosome 14 in FOL4287 (45).
All isolates known to belong to VCG0030 group into clade A2, and all three FOL races are represented. In addition, clade A2 contains five self-incompatible FOL isolates, an isolate of F. oxysporum f. sp. melonis (FOM) (NRRL26406) belonging to FOM VCG0136 and five isolates of F. oxysporum f. sp. radicis-lycopersici (FORL) belonging to FORL VCG0090. This suggests that FOL VCG0030, FOM VCG0136, and FORL VCG0090 are closely related. Clade A3 contains all four known VCG0033 isolates; these are all race 3.
Clade A4 contains two FOL race 1 isolates (VCG not known) and three race 2 isolates of VCG0035 (29), all reported from a single site in California, USA. Based on VCG tests, Mes and coworkers proposed that the Dutch race 1 isolates FOL001 and FOL012 do not belong to either VCG0030 or VCG0031 (31). Here we show that the two isolates from The Netherlands share identical EF1-α gene sequences with the American VCG0035 race 2 isolates, positioning them in clade A4. This suggests that FOL001 and FOL012 belong to VCG0035 as well.
Clade A5 contains only one FOL isolate, FOL005, a race 2 isolate from The Netherlands. In a vegetative compatibility test, this isolate did not show compatibility with any other isolate belonging to a known VCG (31). Moreover, in our phylogenetic tree, FOL005 groups with FOM and F. oxysporum f. sp. cubense isolates and with one F. oxysporum isolate with unknown host specificity (BRIP53842). Isolates within this clade share 99.8 to 100% identical EF1-α gene sequences and at least five polymorphic sites compared to isolates from other clades. Together, these observations suggest that FOL005 belongs to a hitherto-unknown FOL VCG.
Taken together, this phylogenetic study based on EF1-α gene sequences shows that FOL comprises at least five evolutionary lineages. Among these, four correlate with known VCGs of FOL (VCG0030, VCG0031, VCG0033, and VCG0035). The coincidence of FOL VCGs with distinct phylogenetic lineages is also supported by previous phylogenetic studies based on the ribosomal DNA intergenic spacer region (36). The present extended analysis further confirms that FOL's ability to cause disease on tomato has emerged multiple times independently within the F. oxysporum complex (33), probably through horizontal chromosome transfer (46).
DISCUSSION
The genome of the sequenced isolate FOL4287 consists of 11 core and four lineage-specific (LS) chromosomes. The LS chromosomes are devoid of housekeeping genes, encode the so-called Six (secreted in xylem) proteins, and possess many transposable elements (42, 46). The 100-kb insert of clone 9G3 from a FOL004 BAC library shows all of the characteristics of an LS chromosome: almost 60% of it contains TE sequences, it encodes a Six protein (Six4 or Avr1), and it is devoid of housekeeping genes. Comparison of the 100-kb insert sequence to the FOL4287 reference genome revealed that, except for a 31-kb fragment containing AVR1, the entire insert aligns to a region in LS chromosome 14 of FOL4287 (Fig. 1). This suggests that FOL004 AVR1, like AVR2 (SIX3) and AVR3 (SIX1), is located on chromosome 14. However, whether the AVR1 locus is really located on this chromosome remains unclear. Inami and coworkers examined two Japanese FOL race 1 isolates by pulse-field gel electrophoresis and Southern analysis and found AVR1 in one isolate on the same chromosome as AVR2 and AVR3, whereas in the other isolate, AVR1 appeared to be located on a different chromosome (22). This indicates that in race 1 isolates, AVR1 is not always on the same chromosome as AVR2 and AVR3.
Besides AVR1 and the FoHeli3 Helitron (ORF3), the 31-kb deletion contains only one ORF (ORF4) (Fig. 1), which encodes a protein with unknown function. In FOL4287, three copies of this ORF are present at different locations in the genome (see Table S3 in the supplemental material). FOL007 (a race 2 isolate with the 31-kb deletion) is virulent on the general susceptible tomato line C32 (19). This suggests that the 31-kb deletion including AVR1 and ORF4 is not crucial for virulence of FOL, although loss of ORF4 in the 31-kb region might have been compensated for by other ORF4 copies in the genome. The 102-kb fragment lost in other race 2 isolates contains, in addition to AVR1, ORF3, and ORF4, a putative oxidoreductase gene (ORF5), two genes encoding RecQ helicases (ORF6 and -7), three ORFs encoding proteins with unknown function (FOXG_14241, FOXG_14242, and FOXG_14244), and one ORF encoding an aspartic protease (FOXG_14243) (Fig. 1). Three genes encoding a highly similar oxidoreductase and four genes encoding a highly similar aspartic protease are present in the genome of FOL4287 as well. FOL029, which possesses the 102-kb deletion, is highly virulent and does not have any copies of FOXG_14241, FOXG_14242, and FOXG_14244 elsewhere in the genome (19). This suggests that FOXG_14241, FOXG_14242, and FOXG_14244 are also not required for pathogenicity of FOL on tomato.
In the 26 FOL race 1 isolates analyzed, AVR1 is positioned in the genome between two copies of a Helitron transposable element. In 15 isolates, one copy (HelA) is located 17.2 kb upstream of AVR1, and the other (HelC) resides 88 kb downstream of the avirulence gene. In the 11 other isolates, a third copy (HelB) is present 12.9 kb downstream of AVR1 (69 kb upstream of HelC). Hence, two groups of race 1 isolates can be distinguished: one with HelB located between AVR1 and HelC and one without HelB (Fig. 2B). We assume that at some point in time HelB was inserted into the AVR1 locus upstream of HelC. However, the possibility that HelB was deleted from the AVR1 locus cannot be ruled out. So far nothing is known about a potential excision mechanism of Helitron transposons and whether such an excision event would leave footprints in the DNA. Sequencing a 1.8-kb fragment (see Fig. S3 in the supplemental material) of the AVR1 locus from which HelB could have been excised and comparing the sequence with the HelB flanking sequences did not reveal any potential excision footprint. Helitron transposons are found throughout the genome of FOL4287 but are most abundant on chromosome 14 (7 copies, of which 6 share >97% sequence identity) (42, 48). No Helitron transposons are found near AVR2, but one Helitron copy is located ∼20 kb upstream and another 3,455 bp downstream of AVR3 (42, 48).
Except for the pathway for the emergence of the Kochi-1 isolate, all FOL race 2 isolates appear to have evolved from race 1 by the loss of part of the AVR1 genomic region in at least five independent deletion events, of which three were observed only once. In three events, the deletion of the AVR1 genomic fragment was likely due to homologous recombination between identical transposable elements flanking this genomic fragment. In two of these cases, homologous recombination had occurred between two identical Helitron transposable elements, resulting in either a 102-kb or a 31-kb deletion (Fig. 1, 2, and 6) (40). In an another event, an AVR1-containing genomic region of approximately 457 kb was found to be deleted, most probably due to homologous recombination between two NHT2-like retrotransposons (Fig. 3) (43). These results suggest that transposable elements, particularly Helitron transposons, played a major role in the evolution of races within FOL.
FIG 6.

A model for the evolution of FOL race 2 and race 3 isolates from race 1. Two lineages (I and II) are indicated by arrows. Green boxes indicate AVR1, and orange boxes indicate Helitron transposons. Blue indicates regions conserved in the FOL isolates.
Helitron transposons form a separate class of DNA transposons that has been identified in many eukaryotic species, including fungi (40). Even though we propose that recombination between Helitron transposons is responsible for the deletion of the AVR1 locus, we do not have direct evidence for Helitron recombination. FOL004 HelA and FOL4287 HelAB are 100% identical, except for the insertion of transposable element Hop3 at the 5′ end of FOL4287 HelAB. Since FOL004 HelB differs from FOL004 HelA by a duplication of a 15-bp sequence, a single-base-pair deletion, and one A/G polymorphism (Fig. 1), we assume that recombination between HelA and HelB occurred at any position downstream of nucleotide 4909 (Fig. 1). No sequence polymorphisms were identified among HelA, HelC, and HelAC, and hence it is impossible to determine the precise location of this recombination event.
Clonal lineages within FOL.
Five evolutionary lineages (clades A1 to A5) were identified within FOL in a phylogenetic analysis using the sequences of the EF1-α genes from 71 FOL isolates that represent the world population (Fig. 5). Within each lineage, the EF1-α gene sequences showed 99.8 to 100% identity.
Phylogenetic analyses based on distinct genes do not always result in consistent phylogenies for F. oxysporum, probably due to hybridizations and/or horizontal transfer events (46, 47). In this EF1-α gene phylogeny, clade A1 to clade A4 were associated with known VCGs of FOL. This suggests that this method can be used to quickly determine the clonal lineage (hence VCG) for isolates without resorting to VCG analysis. Identification of a new clonal lineage (clade A5) for FOL suggests that hitherto-unidentified distinct lineages still exist in nature. Overall, these results are consistent with the findings of previous studies in which FOL was shown to be polyphyletic, meaning that pathogenicity to tomato has evolved multiple times independently (36, 47). The polyphyletic nature of FOL can be explained by horizontal chromosome transfer (HCT). This might be true at least for FOL, where a small chromosome, designated chromosome 14, has been shown to transfer between different isolates of F. oxysporum (46). The effector genes in FOL that facilitate virulence on tomato are located on chromosome 14 and are most likely absent from other formae speciales (42, 45). The conservation of these genes with 100% identity in FOL isolates belonging to different clades suggests that pathogenicity toward cultivated tomato has spread relatively recently to different clades through horizontal chromosome transfer. Moreover, the occurrence of all three races from diverse geographical locations in the same clonal lineage implies that new races emerge independently at different locations, either from local populations or at a center of origin followed by long-distance dispersal. Figure 7 represents a model to explain how pathogenicity to tomato cultivars has evolved in different clonal lines of FOL. We consider VCG0030 to be the ancestral lineage because of its widespread occurrence and genotype diversity (see below). As depicted in Fig. 7, factors facilitating virulence on tomato may have evolved initially within VCG0030, and then a chromosome carrying these virulence factors (e.g., chromosome 14) might have been transferred horizontally to other clonal lines (VCGs) (Fig. 7). Due to selection pressure from host resistance, races have evolved within each clonal line independently by loss of function of avirulence genes. VCG0033 contains only race 3 isolates. These isolates may have evolved from hitherto-unidentified race 1 or race 2 isolates in the same lineage. An alternative explanation is that the race 3 isolates emerged by the acquisition of chromosome 14 through horizontal transfer from an already-existing race 3 isolate in a different lineage, as shown in Fig. 7.
FIG 7.

A model for gain of pathogenicity toward tomato in different clonal lineages (VCGs) of FOL. Each box indicates a genotype; the AVR1 genomic region of each isolate is represented schematically within the box, along with sequence types for Avr2 and Avr3. Boxes are colored according to race: green, race 1; blue, race 2; pink, race 3. Colored circles within the boxes indicate geographical locations. We propose that different VCGs obtained their various genotypes through horizontal chromosome transfer (HCT) and independent losses of the AVR1 locus, possibly after homologous recombination between flanking Helitron transposons.
Pathways for the evolution of races and avirulence genotypes within VCG0030.
The present study provides evidence for the independent evolution of different FOL races within clonal lineages (VCGs). We have shown that FOL race 2 evolved from race 1 isolates by deletion of a genomic region containing AVR1, apparently through homologous recombination between two identical Helitron transposons flanking the locus. Within a given race, isolates can be divided into various types based on the event that led to AVR1 deletion and the sequences of the AVR2 and AVR3 alleles in their genome (Table 1). From these data, we propose a model of the minimal number of events leading to the evolution of races from an ancestral race 1 type in the major lineage, VCG0030 (Fig. 8). Five independent pathways (A to E, indicated in red within the boxes describing molecular events) are hypothesized, each one with one or multiple evolutionary events that led to the evolution of different types and races in a stepwise manner. In the model, the common ancestor for all races and types is a FOL race 1 without HelB but with AVR1, AVR2, and AVR3E present as well as HelA and HelC upstream and downstream of AVR1, respectively. We believe this FOL race 1 type (framed red in Fig. 8) to be the common ancestor for several reasons. First, this FOL race 1 type occurs in all five global regions from which the isolates used in our studies originate: North America, Europe, Australia, Japan, and North Africa/Israel. Furthermore, as discussed above, it is very unlikely that a race 2 (or race 3) isolate represents an ancestral isolate without AVR1 and that at some point in time this isolate acquired a genomic fragment containing AVR1. All race 1 isolates scored positive for primer pairs B, C, H, and I, indicative for the presence of both HelA and HelC. As discussed before as well, we assume that HelB was inserted into the AVR1 locus upstream of HelC rather than that HelB was excised from the locus. However, conclusive evidence for this is not available. Race 1 isolates all contain AVR1 and AVR2, and in all cases analyzed (approximately 50% of the isolates) these appeared to be wild type. All FOL isolates contain AVR3, either the AVR3E or the AVR3K allele. Since isolates with the AVR3K allele are more virulent than isolates with the AVR3E allele, we assume the common ancestor to contain the AVR3E allele.
FIG 8.

Proposed events underlying evolution of FOL races and AVR genotypes within VCG0030. As in Fig. 7, each box indicates a genotype; the AVR1 genomic region of each isolate is represented schematically within the box, along with sequence types for Avr2 and Avr3. Boxes are colored according to race: green, race 1; blue, race 2; pink, race 3. Colored circles within the boxes indicate geographical locations. Molecular events that led to emergence of new races and genotypes are described in open boxes. We inferred five potential parsimonious evolutionary pathways that lead from the ancestral genotype (thick black box) to the various genotypes observed. These pathways are named A to E, and individual steps are indicated with a number (e.g., A1 is the first step in pathway A). Pathways and steps are indicated in red within the open boxes describing molecular events.
In pathway A, race 2 evolved from the ancestral race 1 by deletion of a 102-kb genomic region carrying the AVR1 locus, due to a homologous recombination event between HelA and HelC (indicated as event A1 in Fig. 8). Subsequently, a single point mutation in the AVR3 gene (event A2 in Fig. 8) led to the emergence of race 2 with the AVR3K allele. Race 3 then evolved from this race 2 in two independent events (A3 and A4 in Fig. 8). In each event, a point mutation in the AVR2 gene caused the loss of its avirulence function. Since some race 1 isolates also carry the AVR3K allele, these isolates presumably evolved from the ancestral race 1 type independently (E1 in Fig. 8).
In pathway B, first HelB was inserted 12.9 kb downstream of AVR1 (B1 in Fig. 8). Subsequently, race 2 evolved by deletion of a 31-kb genomic region containing AVR1 (B2 in Fig. 8), due to homologous recombination between HelA and HelB. A single nucleotide change in AVR3 then led to the emergence of race 2 with the AVR3K allele (B3 in Fig. 8). An alternative pathway for evolution of race 2 with the AVR3K allele is as follows. First, a single nucleotide change in AVR3 of a race 1 isolate with HelB led to the emergence of race 1 with the AVR3K allele (B5 in Fig. 8). Thereafter, a homologous recombination event caused the deletion of the 31-kb genomic region containing the AVR1 locus, resulting in race 2 with the AVR3K allele (B6 in Fig. 8). However, because of the widespread distribution of race 2 with the AVR3E allele, we believe that the first pathway (B1 to B2 to B3 in Fig. 8) is the most plausible explanation for evolution of race 2 with the AVR3K allele. In the type that evolved upon event B2, a copy of transposable element Hop3 was inserted into the 5′ end of HelAB (B4 in Fig. 8). To explain evolution of race 3 within pathway B, four independent events are proposed (B7, -8, -9, and -10 in Fig. 8), all related to the loss of the avirulence function of AVR2.
Pathway C is proposed for the evolution of the Kochi-1 isolate (FOL081), a race 3 isolate from Japan. FOL race 2 isolates FOL018 and FOL015 (VCG0031 race 2 isolate FOL076) could have evolved from the ancestral race 1 isolate by independent deletion events (D1 in Fig. 8). Since neither HelA nor HelB was involved in the deletion of the AVR1 locus in FOL018, FOL015, and FOL076, these isolates may also have evolved from another, hitherto-unknown ancestral race 1 type without these Helitron transposons.
In the proposed model, a number of events occur multiple times independently. For example, in four independent events (A2, B3, B5, and E1), the AVR3E allele changed into the AVR3K allele. Since the AVR3K allele renders isolates more virulent than the AVR3E allele, an opposite change (AVR3K into AVR3E) is not likely to be selected for and is hence not proposed in the model. Remarkably, no other mutations in AVR3 have been observed. Another example is the emergence of race 3 isolates within pathway B in four independent mutational events to explain evolution of race 3 from race 2 by mutations in AVR2: a deletion of a triplet (B9) or the whole gene (B8) or an amino acid change at position 45 (R→H; B10) or position 46 (R→P; B7). Also, the deletion of the AVR1 locus between HelA and HelB could have occurred twice (B2 and B6).
The results from the current study confirm the polyphyletic nature of Fusarium oxysporum f. sp. lycopersici. In each genetically distinct lineage, different races of FOL showing different avirulence genotypes have evolved from preexisting races by the loss of function of avirulence genes. However, it is still unknown how the ancestral FOL race originated. Further studies on the pathogenicity determinants on chromosome 14 are required to enhance our understanding of how FOL gained pathogenicity on tomato.
MATERIALS AND METHODS
Fungal isolates.
The fungal isolates used in this study are listed in Table 1. Most of the isolates originate from the United States, Japan, Europe, Australia, and North Africa. For most of these isolates, pathogenicity, vegetative compatibility, and genetic diversity have been assessed in previous studies. All isolates were cultured on Czapek Dox agar (CDA) (Oxoid) and incubated in darkness at 25°C.
Fungal genomic DNA isolation, PCR analysis, and sequencing.
Fungal genomic DNA (gDNA) was extracted using the following method. A patch of mycelium was scraped from the margin of a colony and suspended in 400 μl Tris-EDTA buffer (10 mM Tris [pH 8], 1 mM EDTA [pH 8]) together with 300 μl phenol-chloroform (1:1) and approximately 300 μl glass beads (about 400 μm). The mycelium was mechanically disrupted by vortexing for 2 min. The supernatant (150 μl) was collected after centrifugation (5 min) at maximum speed and mixed with an equal volume of chloroform. The supernatant (100 μl) again was collected after vortexing and centrifugation (5 min) and stored at −20°C for further use. One microliter of genomic DNA was used for PCR experiments. The primers used in this study are listed in Table S1 in the supplemental material. The amplified products were resolved electrophoretically in a 1% agarose gel. PCR products were sequenced and analyzed using SeqBuilder (DNAStar, Madison, WI).
BAC library screening, DNA isolation, and restriction analysis.
A BAC library of race 1 isolate FOL004 was screened with AVR1-specific primers (numbers 1091 and 1033 [Table S1]) (38). Putative BAC clones were verified by a second PCR using DNA isolated from these clones as the template. BAC DNA was isolated with the Qiagen Miniprep kit (Qiagen, Basingstoke, United Kingdom) according to the manufacturer's instructions. To estimate the insert sizes, 5-μl aliquots of extracted BAC DNA were digested with 5 U of either NotI, SwaI, or both for 3 h at 37°C. The digestion products were resolved by contour-clamped homogeneous electric field (CHEF)-gel analysis (CHEF-DRIII system; Bio-Rad) in a 1% wide-range resolute agarose gel in 0.5× Tris-borate-EDTA (TBE) buffer. Electrophoresis was carried out for 18 h at 4°C with an initial switch time of 5 s and a final switch time of 15 s in a voltage gradient of 6 V/cm. Insert sizes were compared to those of the CHEF DNA size standard lambda ladder (Bio-Rad), CHEF DNA size standard 8- to 48-kb ladder (Bio-Rad), and GeneRuler 1-kb DNA ladder (Fermentas).
Sequencing, annotation, and analysis.
The BAC clone sequencing and assembly were carried out by BaseClear (Leiden, The Netherlands). Open reading frames of the insert sequence were predicted using the gene-finding program Fgenesh (Softberry, Inc., Mount Kisco, NY). The predicted ORFs were subjected to BLAST searches against the Fusarium comparative database (www.broadinstitute.org) and NCBI nonredundant databases (http://www.ncbi.nlm.nih.gov/). The genome of FOL029 was searched for copies of FOXG_14241, FOXG_14242, and FOXG_14244 using local Megablast with default settings; FOXG_14241 and FOXG_14244 returned no hits at all, and FOXG_14242 returned only partial hits (spanning up to one-third of the gene). We manually inspected an alignment of this gene with the contig in FOL029 where we found the best hit and concluded that this contig does not contain a copy or close paralog of FOXG_14242. Protein domains were predicted by InterProScan analysis (http://www.ebi.ac.uk/). The repetitive elements were identified by BLAST against the RepeatMasker library (Open 3.2.9) (http://www.repeatmasker.org/), Fusarium comparative database, and NCBI nonredundant database. The transposases/retrotransposases were classified by BLAST against the Repbase (http://www.girinst.org/repbase/) and manual inspection using the system proposed by Wicker and colleagues (39). Comparison between the insert sequence and the FOL reference genome was carried out using Mauve genome alignment software (http://darlinglab.org/mauve/mauve.html).
Phylogenetic analysis.
For phylogenetic analysis, DNA sequences of the EF1-α gene were either retrieved from GenBank or determined in this study. Multiple-sequence alignment was performed using ClustalW on the EBI/EMBL site (http://www.ebi.ac.uk/Tools/clustalw) and then manually adjusted with UniPro ugene software to remove all gap sequences and ambiguously aligned regions. There were a total of 565 positions in the final data set. Phylogenetic analyses were conducted in MEGA 5 software. The evolutionary history was inferred by using the maximum-likelihood method based on the Kimura 2-parameter model. The tree with the highest log likelihood (−1,069.4772) is shown (Fig. 5). The percentage of trees in which the associated taxa clustered together is shown next to the branches. The initial tree(s) for the heuristic search was obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum-composite-likelihood (MCL) approach and then selecting the topology with the superior log-likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites (5 categories [+G, parameter = 0.4697]). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The sequence of Fusarium commune (NRRL28387) (GenBank accession number AF246832.1) was used as an outgroup (44).
Accession number(s).
The sequence of the full 9G3 insert has been deposited in GenBank under accession number KP213325.1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Roger Shivas (Plant Pathology Herbarium, Queensland, Australia), Makoto Kawase (NIAS GenBank, National Institute of Agrobiological Sciences, Japan), Bart Lievens (Scientia Terrae Research Institute, Belgium), Corby H. Kistler (U.S. Department of Agriculture, USA; University of Minnesota, USA), Tsutomu Arie (Graduate School of Agriculture, Tokyo University of Agriculture and Technology [TUAT], Fuchu, Japan), and Susan Barker (School of Plant Biology, The University of Western Australia) for providing fungal isolates.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02548-16.
REFERENCES
- 1.Flor HH. 1971. Current status of the gene-for-gene concept. Annu Rev Phytopathol 9:275–296. doi: 10.1146/annurev.py.09.090171.001423. [DOI] [Google Scholar]
- 2.Dangl JL, Jones JD. 2001. Plant pathogens and integrated defence responses to infection. Nature 411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- 3.Morel JB, Dangl JL. 1997. The hypersensitive response and the induction of cell death in plants. Cell Death Differ 4:671–683. doi: 10.1038/sj.cdd.4400309. [DOI] [PubMed] [Google Scholar]
- 4.Farman ML, Eto Y, Nakao T, Tosa Y, Nakayashiki H, Mayama S, Leong SA. 2002. Analysis of the structure of the AVR1-CO39 avirulence locus in virulent rice-infecting isolates of Magnaporthe grisea. Mol Plant Microbe Interact 15:6–16. doi: 10.1094/MPMI.2002.15.1.6. [DOI] [PubMed] [Google Scholar]
- 5.Orbach MJ, Farrall L, Sweigard JA, Chumley FG, Valent B. 2000. A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi-ta. Plant Cell 12:2019–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joosten MH, Cozijnsen TJ, De Wit PJ. 1994. Host resistance to a fungal tomato pathogen lost by a single base-pair change in an avirulence gene. Nature 367:384–386. doi: 10.1038/367384a0. [DOI] [PubMed] [Google Scholar]
- 7.Van den Ackerveken GF, Van Kan JA, De Wit PJ. 1992. Molecular analysis of the avirulence gene avr9 of the fungal tomato pathogen Cladosporium fulvum fully supports the gene-for-gene hypothesis. Plant J 2:359–366. [DOI] [PubMed] [Google Scholar]
- 8.Kanzaki H, Yoshida K, Saitoh H, Fujisaki K, Hirabuchi A, Alaux L, Fournier E, Tharreau D, Terauchi R. 2012. Arms race co-evolution of Magnaporthe oryzae AVR-Pik and rice Pik genes driven by their physical interactions. Plant J 72:894–907. doi: 10.1111/j.1365-313X.2012.05110.x. [DOI] [PubMed] [Google Scholar]
- 9.Na R, Yu D, Qutob D, Zhao J, Gijzen M. 2013. Deletion of the Phytophthora sojae avirulence gene Avr1d causes gain of virulence on Rps1d. Mol Plant Microbe Interact 26:969–976. doi: 10.1094/MPMI-02-13-0036-R. [DOI] [PubMed] [Google Scholar]
- 10.Zhou E, Jia Y, Singh P, Correll JC, Lee FN. 2007. Instability of the Magnaporthe oryzae avirulence gene AVR-Pita alters virulence. Fungal Genet Biol 44:1024–1034. doi: 10.1016/j.fgb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong GM, Armstrong JK. 1978. Formae speciales and races of Fusarium oxysporum causing wilts of cucurbitaceae. Phytopathology 68:19–28. doi: 10.1094/Phyto-68-19. [DOI] [Google Scholar]
- 12.Michielse CB, Rep M. 2009. Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol 10:311–324. doi: 10.1111/j.1364-3703.2009.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohn GW, Tucker CM. 1939. Immunity to Fusarium wilt in the tomato. Science 89:603–604. doi: 10.1126/science.89.2322.603. [DOI] [PubMed] [Google Scholar]
- 14.Alexander LJ, Tucker CM. 1945. Physiologic specialization in the tomato wilt fungus Fusarium oxysporum f. sp. lycopersici. J Agric Res 70:303–313. [Google Scholar]
- 15.Grattidge R, Obrien RG. 1982. Occurrence of a 3rd race of Fusarium-wilt of tomatoes in Queensland. Plant Dis 66:165–166. doi: 10.1094/PD-66-165. [DOI] [Google Scholar]
- 16.Huang CC, Lindhout P. 1997. Screening for resistance in wild Lycopersicon species to Fusarium oxysporum f sp lycopersici race 1 and race 2. Euphytica 93:145–153. doi: 10.1023/A:1002943805229. [DOI] [Google Scholar]
- 17.Scott JW, Jones JP. 1989. Monogenic resistance in tomato to Fusarium-oxysporum f-sp lycopersici race-3. Euphytica 40:49–53. [Google Scholar]
- 18.Rep M, van der Does HC, Meijer M, van Wijk R, Houterman PM, Dekker HL, de Koster CG, Cornelissen BJ. 2004. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol Microbiol 53:1373–1383. doi: 10.1111/j.1365-2958.2004.04177.x. [DOI] [PubMed] [Google Scholar]
- 19.Houterman PM, Cornelissen BJ, Rep M. 2008. Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog 4:e1000061. doi: 10.1371/journal.ppat.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houterman PM, Ma L, van Ooijen G, de Vroomen MJ, Cornelissen BJ, Takken FL, Rep M. 2009. The effector protein Avr2 of the xylem-colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J 58:970–978. doi: 10.1111/j.1365-313X.2009.03838.x. [DOI] [PubMed] [Google Scholar]
- 21.Lievens B, Houterman PM, Rep M. 2009. Effector gene screening allows unambiguous identification of Fusarium oxysporum f. sp. lycopersici races and discrimination from other formae speciales. FEMS Microbiol Lett 300:201–215. doi: 10.1111/j.1574-6968.2009.01783.x. [DOI] [PubMed] [Google Scholar]
- 22.Inami K, Yoshioka-Akiyama C, Morita Y, Yamasaki M, Teraoka T, Arie T. 2012. A genetic mechanism for emergence of races in Fusarium oxysporum f. sp. lycopersici: inactivation of avirulence gene AVR1 by transposon insertion. PLoS One 7:e44101. doi: 10.1371/journal.pone.0044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kashiwa T, Suzuki T, Sato A, Akai K, Teraoka T, Komatsu K, Arie T. 2016. A new biotype of Fusarium oxysporum f. sp. lycopersici race 2 emerged by a transposon-driven mutation of avirulence gene AVR1. FEMS Microbiol Lett doi: 10.1093/femsle/fnw132. [DOI] [PubMed] [Google Scholar]
- 24.Rep M, Meijer M, Houterman PM, van der Does HC, Cornelissen BJ. 2005. Fusarium oxysporum evades I-3-mediated resistance without altering the matching avirulence gene. Mol Plant Microbe Interact 18:15–23. doi: 10.1094/MPMI-18-0015. [DOI] [PubMed] [Google Scholar]
- 25.de Jong CF, Takken FL, Cai X, de Wit PJ, Joosten MH. 2002. Attenuation of Cf-mediated defense responses at elevated temperatures correlates with a decrease in elicitor-binding sites. Mol Plant Microbe Interact 15:1040–1049. doi: 10.1094/MPMI.2002.15.10.1040. [DOI] [PubMed] [Google Scholar]
- 26.Gordon TR, Martyn RD. 1997. The evolutionary biology of Fusarium oxysporum. Annu Rev Phytopathol 35:111–128. doi: 10.1146/annurev.phyto.35.1.111. [DOI] [PubMed] [Google Scholar]
- 27.Katan T. 1999. Current status of vegetative compatibility groups in Fusarium oxysporum. Phytoparasitica 27:51–64. doi: 10.1007/BF02980727. [DOI] [Google Scholar]
- 28.Elias KS, Zamir D, Lichtmanpleban T, Katan T. 1993. Population-structure of Fusarium-oxysporum f. sp. lycopersici—restriction-fragment-length-polymorphisms provide genetic-evidence that vegetative compatibility group is an indicator of evolutionary origin. Mol Plant Microbe Interact 6:565–572. doi: 10.1094/MPMI-6-565. [DOI] [Google Scholar]
- 29.Cai G, Gale LR, Schneider RW, Kistler HC, Davis RM, Elias KS, Miyao EM. 2003. Origin of race 3 of Fusarium oxysporum f. sp. lycopersici at a single site in California. Phytopathology 93:1014–1022. doi: 10.1094/PHYTO.2003.93.8.1014. [DOI] [PubMed] [Google Scholar]
- 30.Elias KS, Schneider RW. 1991. Vegetative compatibility groups in Fusarium-oxysporum f-sp lycopersici. Phytopathology 81:159–162. doi: 10.1094/Phyto-81-159. [DOI] [Google Scholar]
- 31.Mes JJ, Weststeijn EA, Herlaar F, Lambalk JJM, Wijbrandi J, Haring MA, Cornelissen BJC. 1999. Biological and molecular characterization of Fusarium oxysporum f sp lycopersici divides race 1 isolates into separate virulence groups. Phytopathology 89:156–160. doi: 10.1094/PHYTO.1999.89.2.156. [DOI] [PubMed] [Google Scholar]
- 32.Elias KS, Schneider RW. 1992. Genetic diversity within and among races and vegetative compatibility groups of Fusarium-oxysporum f-sp lycopersici as determined by isozyme analysis. Phytopathology 82:1421–1427. doi: 10.1094/Phyto-82-1421. [DOI] [Google Scholar]
- 33.O'Donnell K, Kistler HC, Cigelnik E, Ploetz RC. 1998. Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc Natl Acad Sci U S A 95:2044–2049. doi: 10.1073/pnas.95.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baayen RP, O'Donnell K, Bonants PJ, Cigelnik E, Kroon LP, Roebroeck EJ, Waalwijk C. 2000. Gene genealogies and AFLP analyses in the Fusarium oxysporum complex identify monophyletic and nonmonophyletic formae speciales causing wilt and rot disease. Phytopathology 90:891–900. doi: 10.1094/PHYTO.2000.90.8.891. [DOI] [PubMed] [Google Scholar]
- 35.Mbofung GY, Hong SG, Pryor BM. 2007. Phylogeny of Fusarium oxysporum f. sp. lactucae inferred from mitochondrial small subunit, elongation factor 1-alpha, and nuclear ribosomal intergenic spacer sequence data. Phytopathology 97:87–98. doi: 10.1094/PHYTO-97-0087. [DOI] [PubMed] [Google Scholar]
- 36.Kawabe M, Kobayashi Y, Okada G, Yamaguchi I, Teraoka T, Arie T. 2005. Three evolutionary lineages of tomato wilt pathogen, Fusarium oxysporum f. sp. lycopersici, based on sequences of IGS, MAT1, and pg1, are each composed of isolates of a single mating type and a single or closely related vegetative compatibility group. J Gen Plant Pathol 71:263–272. doi: 10.1007/s10327-005-0203-6. [DOI] [Google Scholar]
- 37.Takken F, Rep M. 2010. The arms race between tomato and Fusarium oxysporum. Mol Plant Pathol 11:309–314. doi: 10.1111/j.1364-3703.2009.00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teunissen HA, Rep M, Houterman PM, Cornelissen BJ, Haring MA. 2003. Construction of a mitotic linkage map of Fusarium oxysporum based on Foxy-AFLPs. Mol Genet Genomics 269:215–226. [DOI] [PubMed] [Google Scholar]
- 39.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 40.Kapitonov VV, Jurka J. 2001. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A 98:8714–8719. doi: 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapitonov VV, Jurka J. 2007. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet 23:521–529. doi: 10.1016/j.tig.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt SM, Houterman PM, Schreiver I, Ma L, Amyotte S, Chellappan B, Boeren S, Takken FL, Rep M. 2013. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics 14:119. doi: 10.1186/1471-2164-14-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiflett AM, Enkerli J, Covert SF. 2002. Nht2, a copia LTR retrotransposon from a conditionally dispensable chromosome in Nectria haematococca. Curr Genet 41:99–106. doi: 10.1007/s00294-002-0287-x. [DOI] [PubMed] [Google Scholar]
- 44.Skovgaard K, Rosendahl S, O'Donnell K, Nirenberg HI. 2003. Fusarium commune is a new species identified by morphological and molecular phylogenetic data. Mycologia 95:630–636. doi: 10.2307/3761939. [DOI] [PubMed] [Google Scholar]
- 45.van Dam P, Fokkens L, Schmidt SM, Linmans JH, Kistler HC, Ma LJ, Rep M. 2016. Effector profiles distinguish formae speciales of Fusarium oxysporum. Environ Microbiol 18:4087–4102. doi: 10.1111/1462-2920.13445. [DOI] [PubMed] [Google Scholar]
- 46.Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman PM, Kang S, Shim WB, Woloshuk C, Xie X, Xu JR, Antoniw J, Baker SE, Bluhm BH, Breakspear A, Brown DW, Butchko RA, Chapman S, Coulson R, Coutinho PM, Danchin EG, Diener A, Gale LR, Gardiner DM, Goff S, Hammond-Kosack KE, Hilburn K, Hua-Van A, Jonkers W, Kazan K, Kodira CD, Koehrsen M, Kumar L, Lee YH, Li L, Manners JM, Miranda-Saavedra D, Mukherjee M, Park G, Park J, Park SY, Proctor RH, Regev A, Ruiz-Roldan MC, Sain D, et al. 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Donnell K, Gueidan C, Sink S, Johnston PR, Crous PW, Glenn A, Riley R, Zitomer NC, Colyer P, Waalwijk C, Lee T, Moretti A, Kang S, Kim HS, Geiser DM, Juba JH, Baayen RP, Cromey MG, Bithell S, Sutton DA, Skovgaard K, Ploetz R, Corby Kistler H, Elliott M, Davis M, Sarver BA. 2009. A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genet Biol 46:936–948. doi: 10.1016/j.fgb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 48.Chellapan BV, van Dam P, Rep M, Cornelissen BJC, Fokkens L. 2016. Non-canonical helitrons in Fusarium oxysporum. Mobile DNA 7:27. doi: 10.1186/s13100-016-0083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.