

Abstract
Formation of a fully functional artery proceeds through a multistep process. Here we show that Notch3 is required to generate functional arteries in mice by regulating arterial differentiation and maturation of vascular smooth muscle cells (vSMC). In adult Notch3–/– mice distal arteries exhibit structural defects and arterial myogenic responses are defective. The postnatal maturation stage of vSMC is deficient in Notch3–/– mice. We further show that Notch3 is required for arterial specification of vSMC but not of endothelial cells. Our data reveal Notch3 to be the first cell-autonomous regulator of arterial differentiation and maturation of vSMC.
Keywords: Notch3, artery, smooth muscle cell, differentiation
Full elaboration of the vascular system proceeds through highly coordinated and tightly regulated processes. During the early stages of vascular development, endothelial precursors differentiate and proliferate in situ to form a primary tubular network in a process termed vasculogenesis. This primary capillary plexus is subsequently remodeled through the process of angiogenesis. The ensuing stabilization and maturation of the vessel depend on the recruitment of mural cells and their differentiation into vascular smooth muscle cells (vSMC) or pericytes (Carmeliet 2003). Blood vessels are specified as artery or vein before circulation commences (Lawson and Weinstein 2002). Arteries are also specified into different calibers and types of vessels to perform different functions. The major arteries of the trunk are elastic, conducting arteries, whereas the distal arteries are muscular, resistance arteries. In mature arteries, vSMC exist in a quiescent contractile state, but vSMC can change their phenotype and differentiated state when responding to changing environmental cues. To achieve homeostasis, factors in the arterial wall must instruct vSMC to maintain this quiescent differentiated phenotype (Owens 1998).
The Notch signaling pathway is an evolutionarily conserved, intercellular signaling mechanism that plays a central role in the development of most vertebrate organs (Artavanis-Tsakonas et al. 1999). Notch pathway components have been recently added to the growing list of signaling molecules that control vascular development and homeostasis (Lawson and Weinstein 2002; Shawber and Kitajewski 2004). Notch family genes encode evolutionarily conserved transmembrane receptors. In mammals, four Notch family receptors have been described. Notch activation is triggered by interactions with ligands of the Delta and Serrate/Jagged families and results in the induction of the expression of Hairy and Enhancer of Split-related basic helix–loop–helix transcriptions factors (Artavanis-Tsakonas et al. 1999; Iso et al. 2003). Targeted mutagenesis in mice disrupting Notch1/Notch4, Jagged1, or Hey1/Hey2 results in embryonic lethality with severe defects in angiogenic vascular remodeling (Xue et al. 1999; Krebs et al. 2000; Fischer et al. 2004). Recent work indicates that Notch signaling may regulate arterial differentiation of endothelial cells (Lawson et al. 2001; Fischer et al. 2004). On the other hand, highly stereotyped mutations in human Notch3 cause CADASIL, a hereditary vascular dementia, suggesting a role for Notch3 in vessel homeostasis (Joutel et al. 1996). CADASIL is a late-onset disorder, and neurological symptoms arise from a slowly developing systemic vasculopathy, characterized ultimately by degeneration of vSMC (Ruchoux et al. 1995). Yet, the function of Notch3, which is expressed predominantly in blood vessels in late embryos and in adults, is poorly understood (Joutel et al. 2000). Notch3 function is not required for viability and fertility in mice (Krebs et al. 2003). In this study we investigated the in vivo function of Notch3 in the adult blood vessel.
Results and Discussion
Adult Notch3–/– mice exhibited marked arterial defects. Mutant arteries were enlarged and exhibited a less festooned elastica lamina (Fig. 1A–J). Staining with smooth muscle cell-specific markers, including α-smooth muscle actin (α-SMA) and smooth muscle myosin heavy chain (SMMHC) showed no evidence of vSMC deficiency in mutant arteries, but revealed that the vSMC coat was thinner than in wild-type arteries (Fig. 1C–F,I,J). The caudal artery within the tail exhibited a thinner, disorganized tunica media composed of discontinuous layers of noncohesive smooth muscle cells, especially within the outermost layers (Fig. 1I,J). High resolution optic microscopy and ultrastructural analysis of mutant arteries indicated that vSMC were present but had marked alterations in shape and size. Mutant vSMC were thinner and often extended thin elongated cytoplasmic processes, which frequently overlapped (Fig. 1G,H,K,L). Endothelial cells of mutant arteries appeared morphologically normal. Arterial defects were observed in all the organs analyzed from Notch3–/– mice. In contrast, major elastic arteries of the trunk in mutant mice appeared indistinguishable from those of control mice (data not shown). Arterial vessels from Notch3+/– heterozygous mice appeared similar to those of wild-type mice (data not shown). Despite these arterial defects, there was no indication of parenchyma damage. In particular, there was no evidence of brain pathology, even in older mice up to 12 mo of age (data not shown).
Figure 1.

Arterial defects in adult Notch3–/– mice. (A–D) Cerebral arteries from wild-type and Notch3–/– mice. Toluidine blue staining of semithin sections (A,B) shows that the Notch3–/– artery is enlarged; SMMHC staining (C,D) shows the thinner profile of Notch3–/– vSMC. (E–H) Tail lateral arteries. αSMA staining (E,F) shows that the Notch3–/– artery is enlarged and has a thinner vSMC coat; electron micrographs (G,H) show the marked alteration in the shape and size of vSMC in Notch3–/– mice. Arrowheads point to the thin elongated cell processes from distinct vSMC that overlap. (I–L) Caudal arteries. αSMA staining (I,J) shows the disorganized tunica media in Notch3–/– artery with noncohesive vSMC in the outermost layers (arrows); toluidine blue staining of semithin sections (K,L) shows the thinner and irregular profiles of mutant vSMC. Bars: A,B,I,J, 85 μm; C–F,K,L, 34 μm; G, 8 μm; H, 14 μm.
We next determined the impact of Notch3 deficiency on the functional properties of the arteries. Defects of vSMC in resistance arteries may affect systemic blood pressure. However, resting arterial blood pressure did not differ between control and mutant mice, suggesting a compensatory role of other factors involved in its regulation, such as cardiac output, autonomic innervation, or circulating hormones (Fig. 2A). Constrictive response to pressure elevation is a highly characteristic function of arteries, particularly distal arteries. This behavior, which primarily involves a vascular myogenic mechanism, is particularly well developed in cerebral arteries. It underlies autoregulation of cerebral blood flow (CBF), the maintenance of constant blood flow during variations in systemic blood pressure, and cerebrovascular resistance (CVR) (Davis and Hill 1999). In response to angiotensin II or phenylephrine injections, Notch3–/– mice showed normal arterial blood pressure elevations but strongly impaired CBF reactivity and CVR (Fig. 2B,C; data not shown). The slight increase in CBF elicited by acute induced hypertension in control mice was exacerbated in Notch3–/– mice (Fig. 2C, middle panel). Consistently, in wild-type mice CVR exhibited a dose-dependent increase in response to injections of angiotensin II or phenylephrine, while in Notch3–/– mice acute induced hypertension did not elicit any change in CVR (Fig. 2C, upper panel; data not shown). Thus, the results indicate that Notch3–/– mice exhibit strongly defective arterial myogenic responses. Taken together these observations indicate that Notch3 function is required in adult mice for the structural and functional integrity of arteries, particularly smaller-diameter arteries.
Figure 2.

Impaired cerebral blood flow reactivity in Notch3–/– mice. (A) Arterial blood pressure measurements in conscious wild-type (open bars, n = 8) and Notch3–/– mice (filled bars, n = 8; 5–7 mo old). Mean values ± SEM of systolic, diastolic, and mean arterial pressure (MABP) do not differ between wild-type and mutant mice. (B) Representative blood pressure (lower) and CBF (upper) tracings from wild-type (left) and mutant (right) mice in response to angiotensin II injection. (C) The effects on MABP (in millimeter Hg change, lower panel), on CBF (in percent change, middle panel), and CVR (in percent change, upper panel) were measured in response to three distinct doses of angiotensin II (0.15, 0.37, and 0.74 μg/kg) in wild-type mice (open bars, n = 6) and mutant mice (filled bars, n = 6). Angiotensin II infusion induces similar blood pressure response in wild-type and mutant mice but strongly defective CBF and CVR responses in Notch3–/– mice. Bars represent the mean ± SEM. Statistical analysis was carried out using the unpaired Student's t-test. (*** P < 0.001).
Arterial defects in adult Notch3–/– mice could result from a defect in vessel maturation or a perturbation in vessel homeostasis. To distinguish between these two possibilities, we searched for the earliest detectable defects in Notch3–/– mice. We focused on the tail vasculature because of its highly stereotyped pattern (Supplementary Fig. 1). At birth, arterial vessels in Notch3–/– neonates were indistinguishable from those of control littermates. However, at this stage both wild-type and mutant vessels were immature. At birth, both wild-type and mutant arteries located in the lateral bundles, hereafter designated lateral arteries, were surrounded by a single layer of αSMA- and SMMHC-positive cells (Fig. 3A,B; data not shown). The tunica media of both wild-type and mutant caudal arteries was composed of a loose meshwork of mural cells with αSMA and SMMHC expression predominating within the two innermost cell layers (Supplementary Fig. 2A,B; data not shown). These presumptive vSMC, in lateral and caudal arteries, had a polygonal or rounded shape with little cytoplasmic content and were poorly oriented around the lumen (Fig. 3D,E; Supplementary Fig. 2C,D). Subsequently, vSMC underwent coordinated changes in their shape and orientation, lending the arterial vessels their final appearance by postnatal day 28 (P28). This process was strongly impaired in Notch3–/– mice.
Figure 3.

Impaired postnatal maturation of the lateral arteries from the tail in Notch3–/– mice. (A–C) α-SMA staining of P0.5 arteries from wild-type (A) and Notch3–/– mice (B) and vein from wild-type mice (C) showing that arteries and veins are surrounded by α-SMA-positive mural cells. (D–L) Toluidine blue staining of semithin sections at P0.5 (D–F), P7 (G–I), and P28 (J–L) from wild-type arteries (D,G,J), Notch3–/– arteries (E,H,K), and wild-type veins (F,I,L). At P0.5 wild-type and Notch3–/– arteries and wild-type vein appear similar. At later stages, vSMC in wild-type arteries harmoniously increase in length and thickness and become circumferentially oriented around the lumen, while vSMC in Notch3–/– arteries exhibit thin, irregular, and overlapping cytoplasmic processes (arrows), and form abnormal clusters of poorly oriented cells (black arrowheads). Note the similar aspect of vSMC from veins to those from Notch3–/– arteries at the same age. (M–O) Electron micrographs at P28 of wild-type artery (M), Notch3–/– artery (N), and wild-type vein (O), showing the irregular shape of vSMC with thin cytoplasmic expansions as well as the marked reduction of dense plaques (white arrowheads) in the mutant artery and the wild-type vein (ec, endothelial cells). Bars: A–L, 17 μm; M, 7.5 μm; N, 10 μm; O, 5 μm.
In wild-type mice, vSMC harmoniously increased in length and thickness and reached a circumferential orientation around the lumen. By P28 wild-type lateral arteries exhibited a single, regular layer of cohesive and continuous cells closely applied to the elastica lamina (Fig. 3J). In the wild-type caudal artery, morphological changes of vSMC occurred with a luminal to abluminal gradient. These changes were coincident with the progressive juxtaposition of the vSMC layers toward the elastica lamina and with an increase in the levels of αSMA and SMMHC. At P28 and beyond, the wild-type caudal artery harbored four cohesive, continuous layers of circumferentially elongated vSMC (Supplementary Fig. 2G). In contrast, in Notch3–/– mice vSMC did not appropriately remodel their shape and did not reach their proper place and orientation in the tunica media and around the lumen. In mutant lateral arteries, vSMC extended abnormal thin elongated cytoplasmic processes, which frequently overlapped. Additionally, mutant vSMC formed abnormal clusters of cells that were not properly oriented around the lumen (Fig. 3H,K). The Notch3–/– mutant caudal artery exhibited, by P7, abnormal aggregates of αSMA-, SMMHC-, and desmin-positive cells, that did not subsequently apply to the inner layers and did not acquire the proper orientation (Supplementary Fig. 2F,H; data not shown). In addition, several cytoskeletal components characteristic of mature arterial smooth muscle cells, including dense plaques and dense bodies, were markedly reduced in number in Notch3–/– mice (Fig. 3M,N; Supplementary Fig. 2I,J). These results indicate that the postnatal maturation of vSMC that lends arterial vessels their final shape is deficient in Notch3–/– mice.
We next examined whether altered cell proliferation or survival contributed to the failure of arterial maturation in Notch3–/– mice. No differences were observed in neonatal mutant (17.5% ± 2.5%) or control (17.4% ± 3.4%) mice in the percentage of presumptive smooth muscle cells in the arterial vessels that stained with the mitotic marker KI67. Moreover, we did not find apoptotic, TUNEL-positive cells either in mutant or control arteries (data not shown). These data indicate that alterations in cell proliferation and cell death did not contribute to the arterial maturation defects of Notch3–/– mice.
Remarkably, during all the postnatal maturation stages examined, vSMC in mutant arterial vessels exhibited several features that were highly reminiscent of those of vSMC in wild-type veins, suggesting that in Notch3–/– mice, vSMC exhibited a venous instead of an arterial pattern of maturation. At birth, vSMC of veins appeared similar to those of wild-type and mutant arteries (Fig. 3A–C). Subsequently, venous vSMC did not undergo the extensive morphological changes and cell re-orientation that wild-type arterial vSMC did. Instead, venous vSMC, like vSMC of mutant arterial vessels, extended thin elongated cytoplasmic processes that frequently overlapped and formed clusters of poorly oriented cells around the lumen (Fig. 3, cf. H and I, K and L, N and O).
This observation raised the question whether Notch3 was required for arterial differentiation of vSMC. We examined expression of smoothelin, one of the rare smooth muscle markers that is expressed predominantly in arterial rather than venous vSMC (van der Loop et al. 1997). Smoothelin expression was markedly reduced in mutant arteries, suggesting an impaired arterial identity of mutant vSMC (Fig. 4C,D). Alternatively, the diminished expression of smoothelin may also simply reflect an impaired terminal differentiation of mutant vSMC, since smoothelin is a late-stage vSMC differentiation marker. To directly assess vSMC arterial differentiation, we introduced an arterial smooth muscle-specific reporter into the Notch3-deficient background. The SM22α gene is normally transcribed in arterial, venous, and visceral smooth muscle cells (Li et al. 1996). Previous work has identified arterial-specific regulatory elements within the SM22α promoter. A 2.1-kb genomic fragment of the SM22α gene was shown to drive expression of a lacZ reporter in arterial but not venous or visceral smooth muscle cells in transgenic mice (Moessler et al. 1996). We examined β-galactosidase expression in the vessels of freshly dissected tails. In control mice heterozygous for the SM22α–LacZ transgene, β-galactosidase expression was detected in arterial vSMC but was absent from both venous and visceral SMC; β-galactosidase expression was detectable in arterial vessels of the tail by late embryonic stages (Fig. 4E,G; data not shown). Notch3–/– mice heterozygous for the SM22α–LacZ transgene showed strongly reduced β-galactosidase expression in arterial vSMC by late embryogenesis and beyond (Fig. 4F,H; data not shown). Similarly, in the brain β-galactosidase expression was markedly reduced in mutant pial arteries (Supplementary Fig. 3A,C), which exhibited morphological alterations similar to those observed in mutant tail arteries. However, β-galactosidase expression in the aorta, which appeared morphologically normal, was retained (Supplementary Fig. 3B,D). Expression of endogenous SM22α protein was maintained in both venous and visceral smooth muscle cells in Notch3-deficient mice, indicating that the other nonarterial regulatory elements of the SM22α gene were transcriptionally active. Importantly, normal levels of SM22α protein expression were maintained in vSMC of the mutant arteries, indicating that in these cells the endogenous SM22α gene was transcribed from the nonarterial promoter elements (Fig. 4I,J). These results indicate that, in the absence of Notch3 function, arterial differentiation of vSMC was impaired.
Figure 4.
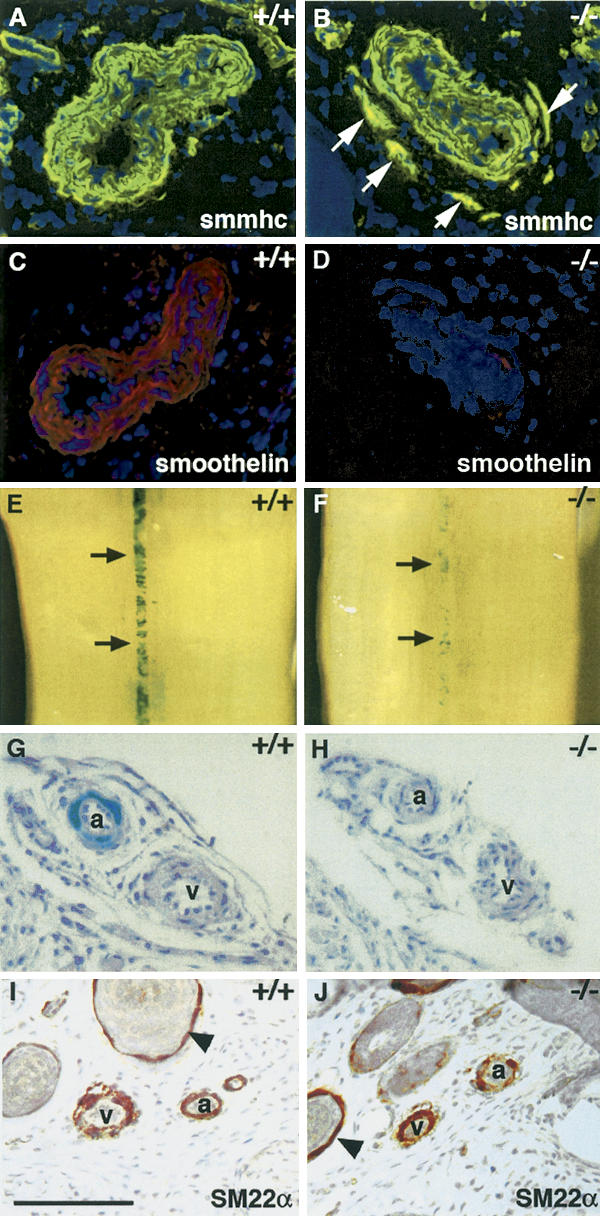
Expression of arterial markers of smooth muscle cells is impaired in Notch3–/– mice. SMMHC (A,B) and smoothelin (C,D) staining of wild-type and Notch3–/– caudal arteries. Notch3–/– arteries exhibit normal SMMHC expression levels but markedly reduced smoothelin expression levels. White arrows point to the ectopic vSMC stained with SMMHC in the mutant artery. (E–H) β-Gal staining of tails from Notch3+/+ and Notch3–/– mice heterozygous for the SM22α–LacZ transgene. Macroscopic view of caudal artery (E,F, black arrows) and microscopic view through artery (a) and vein (v) in a lateral bundle (G,H) showing that β-gal staining is restricted to arterial vSMC in control mice and is markedly reduced in mutant arteries. (I,J) SM22α staining of Notch3+/+ and Notch3–/– tail sections. SM22α is expressed in arterial, venous, and visceral (arrowheads) smooth muscle cells in both control and Notch3–/– mice. Mice shown in panels A–D were P28 and those in panels E–J were P7. Bars: A–D,G–J, 85 μm; E,F, 560 μm.
Recent studies in zebrafish and mouse have established a critical role for Notch signaling in arterial specification of endothelial cells during embryogenesis (Lawson et al. 2001; Lawson and Weinstein 2002; Fischer et al. 2004). Additionally, avian studies indicated that components of the vessel wall might control endothelial cell identity (Moyon et al. 2001). We examined expression of ephrinB2 and Connexin 40, which are well-established arterial-specific endothelial markers (Wang et al. 1998; Mukouyama et al. 2002). Notch3–/– mice exhibited expression of ephrinB2 and Connexin 40 within endothelial cells of arterial vessels at normal levels, indicating that arterial identity of endothelial cells was retained (Supplementary Fig. 4A–D). Moreover, in situ hybridization analysis showed that expression of Notch3 was detected primarily in the presumptive arterial smooth muscle cells and was absent in endothelial cells, indicating that Notch3 was acting autonomously in vSMC (Supplementary Fig. 4E,F). Taken together the results suggest that Notch3 acts cell-autonomously to specifically regulate the arterial differentiation of vSMC without affecting arterial identity of endothelial cells.
We next investigated effects downstream from Notch3 function. Using in situ hybridization and quantitative RT–PCR, expression of immediate downstream targets of Notch signaling (including Hes1, Hey1, Hey2, and HeyL, but not Hes5) was detected in vessels. However, these genes exhibited distinct expression levels and profiles with respect to endothelial versus smooth muscle cells and arteries versus veins. We did not find any differences between Notch3–/– mice and control littermates in the transcript levels or expression pattern of these genes in vessels (Supplementary Fig. 5). Thus, in striking contrast to Notch-dependent arterial differentiation of endothelial cells, in which Hey1 and Hey2 were found to be down-regulated (Fischer et al. 2004), effects downstream from Notch3 in vSMC do not seem to involve previously identified Notch target genes.
How does Notch3 expression regulate postnatal arterial maturation of vSMC? This maturation process may be controlled by regulation of the actin cytoskeleton, since cytoskeletal dynamics and cell shape control are highly interdependent. To test whether Notch3 activation could affect actin cytoskeletal dynamics, we infected vSMC in vitro with a recombinant adenovirus expressing a constitutively active Notch3 receptor construct. Results of these in vitro studies suggest a direct effect of Notch3 signaling on actin cytoskeletal dynamics. Expression of the constitutively active Notch3ΔE in cultured vSMC resulted in cell shape changes, an increase of actin stress fibers, and steady-state levels of polymerized actin (Fig. 5A–E). Additionally, Notch3ΔE expressing vSMC had higher resistance to actin-depolymerizing drugs (Fig. 5F,G; data not shown). The postnatal arterial maturation of vSMC does not correlate with the temporal expression of the Notch ligands Jagged1 and Delta4 (data not shown). Instead, it parallels the increase in arterial blood pressure, which occurs during the first month after birth (Tiemann et al. 2003). Thus, our current mechanistic hypothesis is that Notch3 may act as a sensor or transducer that affords vSMC the ability to rearrange the actin cytoskeleton in response to mechanical stretching of the vessel wall by the intraluminal blood pressure.
Figure 5.

Activated Notch3 modulates cell shape and actin dynamics in cultured vSMC. (A,B) Cells expressing an activated Notch3 construct (N3ΔE) have a markedly different morphological appearance compared to GFP-expressing cells, being larger and having a less elongated shape. (C,D) N3ΔE cells exhibit a more robust actin stress fibers network compared to GFP cells. (E) The F:G actin ratio is increased in N3ΔE-expressing cells. *P < 0.05 compared with GFP cells. Results are mean ± SEM of three independent experiments performed in triplicate. (F,G) GFP cells are latrunculin B sensitive, with marked actin cytoskeleton disassembly, while N3ΔE cells have higher latrunculin B resistance. Bars: A,B, 135 μm; C,D,F,G, 69 μm.
In summary, we describe here a dynamic process that is essential for the elaboration of fully functional arteries. This process commences during late embryogenesis with arterial differentiation of vSMC, continues postnatally with the subsequent maturation of vSMC, and ends around P28 when the artery acquires its final shape. We identify Notch3 to be the first key player of this process, by regulating cell-autonomously the arterial differentiation and maturation of vSMC. Our findings extend the current concept of arterial/venous specification, initially defined at the level of endothelial cells, to vSMC and provide new molecular and cellular insights into this process. First, the results indicate that arteries are genetically distinct from veins at both the level of vSMC and endothelial cells, and suggest that the pathways controlling arterial identity of these two vascular cell types are regulated independently. Second, while it has been shown that arterial specification of endothelial cells is required during early embryonic vascular development to maintain boundaries between arteries and veins, this study indicates that arterial differentiation of vSMC is required to ensure integrity of the structure and function of adult arterial vessels.
Finally, the finding of vascular defects in Notch3-deficient mice provides the groundwork for future study of the in vivo effects of CADASIL mutations on Notch3 receptor activity and function.
Materials and methods
Mice
Notch3 mutant mice and SM22α–lacZ transgenic mice have been described previously (Moessler et al. 1996; Krebs et al. 2003). Experimental procedures involving mice were approved by the relevant French legal authority (Préfecture de Paris, authorization numbers 75–572 and 75–071).
Histology, immunohistochemistry, and X-gal histochemistry
For histological analyses, tissues were fixed in formaldehyde and paraffin-embedded or fixed in CARSON and embedded in Epon E812, then sectioned and stained using standard protocols. For electron microscopy, tissues were processed as described (Ruchoux et al. 2003). Immunohistochemistry was performed on acetone-fixed cryostat sections or paraffin sections using the following primary antibodies: desmin (clone D33; DAKO), SM22α (gift from M. Gimona, Department of Genetics and Cell Biology, Austrian Academy of Sciences, Salzburg, Austria), SMMHC (Biomedical Technologies Inc.), α-SMA (clone 1A4; DAKO), smoothelin (gift from G. Van Eys, Department of Genetics and Cell Biology, University of Maastricht, Maastricht, The Netherlands), and connexin 40 (gift from M. Théveniau-Ruissy, CNRS-UMR 6545, Institut de Biologie du Developpement de Marseille, Marseille, France). We carried out LacZ staining on freshly dissected tissues as described (Moessler et al. 1996).
Cell proliferation and apoptosis assay
We used antibody to KI-67 (clone MM1; Novocastra Laboratories Ltd) to detect mitotic cells. Apoptotic cell death was analyzed by TUNEL staining of paraffin sections using digoxigenin-labeled dNTP and a TRITC-conjugated anti-digoxigenin antibody (Intergen).
In situ hybridization
We carried out in situ hybridization of Hes1, Hes5 (from V. Taylor, Department of Molecular Embryology, Max Planck Institute of Immunobiology, Freiburg, Germany), Hey1, Hey2, and HeyL (from M. Gessler, Theodor-Boveri-Institut fur Biowissenschaften, Universitat Wurzburg, Wurzburg, Germany), Jagged1 (from T. Mitsiadis, CNRS-UMR 5665 CNRS/ENS de Lyon, Lyon, France), Delta4, Notch3, and ephrin B2 using 35S-labeled cRNA probes, as described (Joutel et al. 2000).
RNA analysis
Total RNA was extracted from snap-frozen caudal arteries dissected from P15 mice, treated with DNase I using the SV total RNA isolation system (Promega) and converted into cDNA using the MMLV (Invitrogen). Quantitative PCR was performed using the SYBR Green PCR master mix (Bio-Rad) on an MyiQ Single-Color Real-Time PCR detection system (Bio-Rad). Each sample was amplified in triplicate using primers specific for Hes1, Hes5, Hey1, Hey2, HeyL, and GAPDH. Expression levels were normalized for GAPDH expression.
Cerebral blood flow reactivity
We used laser-Doppler flowmetry to study cerebral blood flow (CBF) in mice (Bonvento et al. 1994). Surgical preparation was carried out on anesthetized mice with inhaled halothane. Tygon catheters were inserted into the femoral artery for heart rate and blood pressure recording, and into the femoral vein for drug infusions. The head was then placed in a Kopf stereotaxic frame. The left and right parietal bones (∼2.5 × 2.5 mm2 area) were thinned to translucency with a dental drill for positioning of the two laser-Doppler flowmeter probes (Moor Instrument MBF3-Dual) to monitor CBF. Mice were allowed to recover from anesthesia and held under minimal restraint. Body temperature was monitored with a rectal probe and maintained between 37.0°C and 37.7°C. Heart rate, blood pressure, and CBF were continuously monitored using a 4-Multichannel recorder (Gould RS 3400). CBF reactivity was examined in response to acute hypertension produced by intravenous administration of angiotensin II (0.15–0.74 μg/kg) or phenylephrine (11–37 μg/kg; Sigma). Cerebrovascular resistance (CVR) was calculated as the ratio of mean arterial blood pressure to concomitant CBF. Changes in CBF and CVR were expressed as percent change relative to the resting level.
Morphological analysis of cultured vSMC
Porcine aorta SMC were prepared and cultured as described (Berrou and Bryckaert 2001). A Notch3ΔE cDNA (corresponding to amino acids 1617–2321 of human Notch3) was used to generate a recombinant adenovirus expressing a constitutively activated Notch3 as described (Chartier et al. 1996). An adenovirus expressing the Green Fluorescent Protein (GFP) was used as the control. Cells were infected with the appropriate virus using 25 TCID50 per cell. Filamentous (F) actin was detected using TRITC-labeled phalloidin and monomeric globular G actin using Alexa Fluor 594-labeled DNase I. The F:G ratio was expressed as the ratio of both fluorescence measured in cells plated in triplicate wells, using a plate reader (Fluoroscan Ascent FL; Labsystems).
Acknowledgments
We thank J.M. Gasc for advice with in situ hybridization; A. Fournier for adenoviruses preparation; G. Van Eys, M. Gessler, M. Gimona, T. Mitsiadis, V. Taylor, and M. Théveniau-Ruissy for reagents; and C. Babinet for discussions. This work was supported by grants from GIS Maladies Rares, CADASIL Foundation of America, Ministère de la Recherche (ACI no. 169), and Fédération pour la Recherche sur le Cerveau to A.J.; and by grants from the NIH (NS36437) and the March of Dimes Foundation to T.G. P.F. is a recipient from Fondation pour la Recherche Médicale and Fédération des Maladies Orphelines.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.308904.
References
- Artavanis-Tsakonas S., Rand, M.D., and Lake, R.J. 1999. Notch signaling: Cell fate control and signal integration in development. Science 284: 770-776. [DOI] [PubMed] [Google Scholar]
- Berrou E. and Bryckaert, M. 2001. Platelet-derived growth factor inhibits smooth muscle cell adhesion to fibronectin by ERK-dependent and ERK-independent pathways. J. Biol. Chem. 276: 39303-39309. [DOI] [PubMed] [Google Scholar]
- Bonvento G., Charbonne, R., Correze, J.L., Borredon, J., Seylaz, J., and Lacombe, P. 1994. Is α-chloralose plus halothane induction a suitable anesthetic regimen for cerebrovascular research? Brain Res. 665: 213-221. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. 2003. Angiogenesis in health and disease. Nat. Med. 9: 653-660. [DOI] [PubMed] [Google Scholar]
- Chartier C., Degryse, E., Gantzer, M., Dieterle, A., Pavirani, A., and Mehtali, M. 1996. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 70: 4805-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.J. and Hill, M.A. 1999. Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79: 387-423. [DOI] [PubMed] [Google Scholar]
- Fischer A., Schumacher, N., Maier, M., Sendtner, M., and Gessler, M. 2004. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes & Dev. 18: 901-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T., Kedes, L., and Hamamori, Y. 2003. HES and HERP families: Multiple effectors of the Notch signaling pathway. J. Cell Physiol. 194: 237-255. [DOI] [PubMed] [Google Scholar]
- Joutel A., Corpechot, C., Ducros, A., Vahedi, K., Chabriat, H., Mouton, P., Alamowitch, S., Domenga, V., Cecillion, M., Marechal, E., et al. 1996. Notch3 mutations in CADASIL, an hereditary adult-onset condition causing stroke and dementia. Nature 383: 707-710. [DOI] [PubMed] [Google Scholar]
- Joutel A., Andreux, F., Gaulis, S., Domenga, V., Cecillon, M., Battail, N., Piga, N., Chapon, F., Godfrain, C., and Tournier-Lasserve, E. 2000. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 105: 597-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs L.T., Xue, Y., Norton, C.R., Shutter, J.R., Maguire, M., Sundberg, J.P., Gallahan, D., Closson, V., Kitajewski, J., Callahan, R., et al. 2000. Notch signaling is essential for vascular morphogenesis in mice. Genes & Dev. 14: 1343-1352. [PMC free article] [PubMed] [Google Scholar]
- Krebs L.T., Xue, Y., Norton, C.R., Sundberg, J.P., Beatus, P., Lendahl, U., Joutel, A., and Gridley, T. 2003. Characterization of Notch3-deficient mice: Normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis 37: 139-143. [DOI] [PubMed] [Google Scholar]
- Lawson N.D. and Weinstein, B.M. 2002. Arteries and veins: Making a difference with zebrafish. Nat. Rev. Genet. 3: 674-682. [DOI] [PubMed] [Google Scholar]
- Lawson N.D., Scheer, N., Pham, V.N., Kim, C.H., Chitnis, A.B., Campos-Ortega, J.A., and Weinstein, B.M. 2001. Notch signaling is required for arterial–venous differentiation during embryonic vascular development. Development 128: 3675-3683. [DOI] [PubMed] [Google Scholar]
- Li L., Miano, J.M., Cserjesi, P., and Olson, E.N. 1996. SM22 α, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ. Res. 78: 188-195. [DOI] [PubMed] [Google Scholar]
- Moessler H., Mericskay, M., Li, Z., Nagl, S., Paulin, D., and Small, J.V. 1996. The SM22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development 122: 2415-2425. [DOI] [PubMed] [Google Scholar]
- Moyon D., Pardanaud, L., Yuan, L., Breant, C., and Eichmann, A. 2001. Plasticity of endothelial cells during arterial–venous differentiation in the avian embryo. Development 128: 3359-3370. [DOI] [PubMed] [Google Scholar]
- Mukouyama Y.S., Shin, D., Britsch, S., Taniguchi, M., and Anderson, D.J. 2002. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell 109: 693-705. [DOI] [PubMed] [Google Scholar]
- Owens G.K. 1998. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol. Scand. 164: 623-635. [DOI] [PubMed] [Google Scholar]
- Ruchoux M.M., Guerouaou, D., Vandenhaute, B., Pruvo, J.P., Vermersch, P., and Leys, D. 1995. Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. (Berl) 89: 500-512. [DOI] [PubMed] [Google Scholar]
- Ruchoux M.M., Domenga, V., Brulin, P., Maciazek, J., Limol, S., Tournier-Lasserve, E., and Joutel, A. 2003. Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am. J. Pathol. 162: 329-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawber C.J. and Kitajewski, J. 2004. Notch function in the vasculature: Insights from zebrafish, mouse and man. Bioessays 26: 225-234. [DOI] [PubMed] [Google Scholar]
- Tiemann K., Weyer, D., Djoufack, P.C., Ghanem, A., Lewalter, T., Dreiner, U., Meyer, R., Grohe, C., and Fink, K.B. 2003. Increasing myocardial contraction and blood pressure in C57BL/6 mice during early postnatal development. Am. J. Physiol. Heart Circ. Physiol. 284: H464-H474. [DOI] [PubMed] [Google Scholar]
- van der Loop F.T., Gabbiani, G., Kohnen, G., Ramaekers, F.C., and van Eys, G.J. 1997. Differentiation of smooth muscle cells in human blood vessels as defined by smoothelin, a novel marker for the contractile phenotype. Arterioscler. Thromb. Vasc. Biol. 17: 665-671. [DOI] [PubMed] [Google Scholar]
- Wang H.U., Chen, Z.F., and Anderson, D.J. 1998. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93: 741-753. [DOI] [PubMed] [Google Scholar]
- Xue Y., Gao, X., Lindsell, C.E., Norton, C.R., Chang, B., Hicks, C., Gendron-Maguire, M., Rand, E.B., Weinmaster, G., and Gridley, T. 1999. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8: 723-730. [DOI] [PubMed] [Google Scholar]