

Abstract
Ciliary disorders , which are also referred to as ciliopathies , are a group of hereditary disorders that result from dysfunctional cilia. The latter are cellular organelles that stick up from the apical plasma membrane. Cilia have important roles in signal transduction and facilitate communications between cells and their surroundings. Ciliary disruption can result in a wide variety of clinically and genetically heterogeneous disorders with overlapping phenotypes. Because cilia occur widespread in our bodies many organs and sensory systems can be affected when they are dysfunctional. Ciliary disorders may be isolated or syndromic, and common features are cystic liver and/or kidney disease, blindness, neural tube defects, brain anomalies and intellectual disability, skeletal abnormalities ranging from polydactyly to abnormally short ribs and limbs, ectodermal defects, obesity, situs inversus , infertility, and recurrent respiratory tract infections. In this review, we summarize the features, frequency, morbidity, and mortality of each of the different ciliopathies that occur in pediatrics. The importance of genetics and the occurrence of genotype-phenotype correlations are indicated, and advances in gene identification are discussed. The use of next-generation sequencing by which a gene panel or all genes can be screened in a single experiment is highlighted as this technology significantly lowered costs and time of the mutation detection process in the past. We discuss the challenges of this new technology and briefly touch upon the use of whole-exome sequencing as a diagnostic test for ciliary disorders. Finally, a perspective on the future of genetics in the context of ciliary disorders is provided.
Keywords: ciliopathy, cilia, next-generation sequencing, diagnostics, genotype-phenotype
Cilia
Structure of Cilia
Cilia are cellular organelles with an antenna-like structure that protrude from the cellular membrane of almost all cell types. 1 Some cells have bundles with 200 to 300 cilia, whereas most cells only form a single cilium. 1 Cilia can be divided in two types: motile cilia, which often occur as groups of cilia on the cell surface and are capable of beating in a constant frequency whereby a fluid flow is generated, and primary cilia that occur one per cell and are immotile and involved in the communication of the cell with its surroundings. 2 Besides their difference in capacity to actively move, motile and immotile cilia also differ structurally. Both cilia types erect from a basal body, a centriolar-based structure that is used as a foundation to build the ciliary microtubular skeleton; however, the number of microtubule pairs in motile and immotile cilia is unequal and components that drive active movement are only present on motile cilia ( Fig. 1 ). 1 3 4 Formation of motile and immotile cilia also slightly differs as centriole replication precedes cilia formation in multiciliated cells, whereas this step is not required in monociliated cells. 2 Generally, when a cell starts to assemble a cilium, the centrosome that is normally involved in separating the chromosomes during mitosis is relocated and anchors just below the surface of the apical membrane. 5 During the conversion of a centriole to a basal body, distal and subdistal centriolar appendages transform into transition fibers and basal feet, which act as physical attachments to the plasma membrane ( Fig. 1 ). These elements are also required to establish the orientation of the basal body in the plasma membrane. 6 Distal to the transition fibers resides the ciliary transition zone, which functions as a gate and regulates the entry and exit of proteins into and out of the cilium. 7 8 Vesicles with membrane proteins are targeted here to be integrated into the ciliary membrane, a process that is coordinated by the BBSome, a complex of proteins that are mutated in Bardet-Biedl syndrome (BBS). 9 10 Vesicle delivery to the cilium contributes to the establishment of a highly specialized ciliary membrane that contains various receptors and signaling proteins. 11 Within the cilium proteins are transported from base to tip and back over the axonemal microtubules ( Fig. 1 ). This transport, first described in unicellular algae, is performed by the intraflagellar transport (IFT) machinery. 12 13 IFT is essential for ciliary functionality as protein synthesis does not occur within the cilium. 14
Fig. 1.
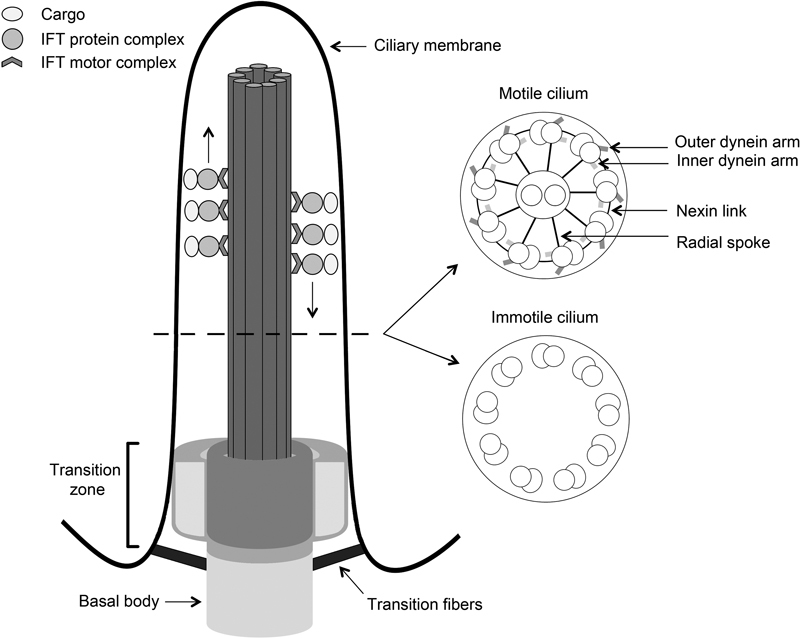
Schematic structure of motile and immotile cilia. Cilia are composed of a basal body and a ciliary axoneme that protrudes from the apical plasma membrane. These two structures are linked via the transition zone (TZ) that consists of three compartments, that is, a base and two compartments filling the space between the microtubules and the axonemal membrane, which are thought to act as a diffusion barrier for regulation of ciliogenesis and signaling. Cilia contain microtubule skeletons that are composed of nine microtubule doublets that organize in a ring. A central microtubule pair is usually also present in motile cilia but not in immotile cilia. Inner and outer dynein arms, radial spokes, and nexin links are also solely present in motile cilia and actively regulate ciliary movement. During this process, dynein heavy chains of one doublet slide against microtubules of a neighboring doublet, thereby orchestrating the beating of the cilium in an ATP-dependent fashion. In both cilium types, the ciliary microtubules also function as rails for intraflagellar transport (IFT) trains that control bidirectional ciliary transport. During this process, IFT-B particles are associated with kinesin-2 motors and regulate transportation of cargo from the ciliary base to the tip, while IFT-A particles and ciliary dynein motors transport cargo in the opposite direction.
Function and Occurrence of Motile and Immotile Cilia
Motile cilia are found in only a few tissues where they transport extracellular fluid over the epithelial surface. For example, they occur on respiratory epithelial cells in the trachea where they are essential for mucus clearance, and on the apical surface of epithelial cells that line the fallopian tubes where they facilitate transportation of the (fertilized) ovum. 1 Cells with a single motile cilium also exist; sperm cells have a specialized motile cilium (flagellum) that provides their motility, and single motile cilia are also found on the apical surface of nodal cells that regulate left-right asymmetry during embryogenesis. 15 Nonmotile cilia are found on almost all quiescent cells and function differently. 1 They generally act as a sensory signaling hub of the cell, and are key players in embryonic development and tissue morphogenesis and homeostasis by regulating Wnt, Hedgehog, and many other signaling pathways. 2 16 17 They initiate or regulate signaling by sensing their environment and ensuring delivery and/or removal of signaling proteins to or from the cilium. 16 Taken together, over the last decades, cilia demonstrated to be essential organelles that regulate a wide array of developmental, cellular, and physiologic processes wherein their motile and/or sensory capacities play a major role.
Ciliopathies
Ciliopathies are disorders that are caused by defects in motile or immotile cilia. As cilia occur on virtually all cells of the human body, their dysfunction can lead to a broad spectrum of features. 1 Common features are renal cystic disease, blindness, neural tube defects, intellectual disability, skeletal abnormalities ranging from polydactyly to abnormally short ribs and limbs, ectodermal defects, obesity, situs inversus , infertility, and respiratory anomalies. These features can occur isolated or may be part of a recognizable syndrome. Here, we describe the different types of ciliopathies, their characteristics, and to what extent they genetically and phenotypically overlap.
Neurodevelopmental Ciliopathies
Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) are rare neurodevelopmental disorders that genetically and phenotypically overlap. JBTS was first described by Marie Joubert in 1969 and is diagnosed when patients have hypotonia, ataxia, developmental delay, and distinctive cerebellar and brain stem malformations that can be recognized as a “molar tooth sign” (MTS; Fig. 2A ) on magnetic resonance imaging (MRI). 18 19 20 JBTS patients also often present with kidney cysts, polydactyly, coloboma, retinal dystrophy, respiratory abnormalities (episodic tachypnea or apnea), oral frenula, and hepatic fibrosis. 21 The health of JBTS patients largely depends on the occurrence and severity of liver and kidney disease that require appropriate management and treatment. 19 22 MKS shows marked clinical overlap with JBTS but is more severe and perinatal lethal. This disorder is characterized by occipital encephalocele, polycystic kidneys, polydactyly, and hepatic fibrosis ( Fig. 2B–D ). 23 MKS is diagnosed when two of these four features are present or when one is present in combination with two other anomalies, including cleft lip/palate, cardiac septal defects, holoprosencephaly, agenesis of corpus callosum, Dandy-Walker malformation, microphthalmia, situs inversus , ambiguous genitalia, and shortening and bowing of long bones. 24 25 The incidence of JBTS ranges from 1:80,000 to 1:100,000 live births, 22 whereas the prevalence of MKS is less frequent at 1:140,000. 26 Exceptions exist as the occurrence of MKS in the Finnish population is as high as 1:9,000 births. 27 28
Fig. 2.
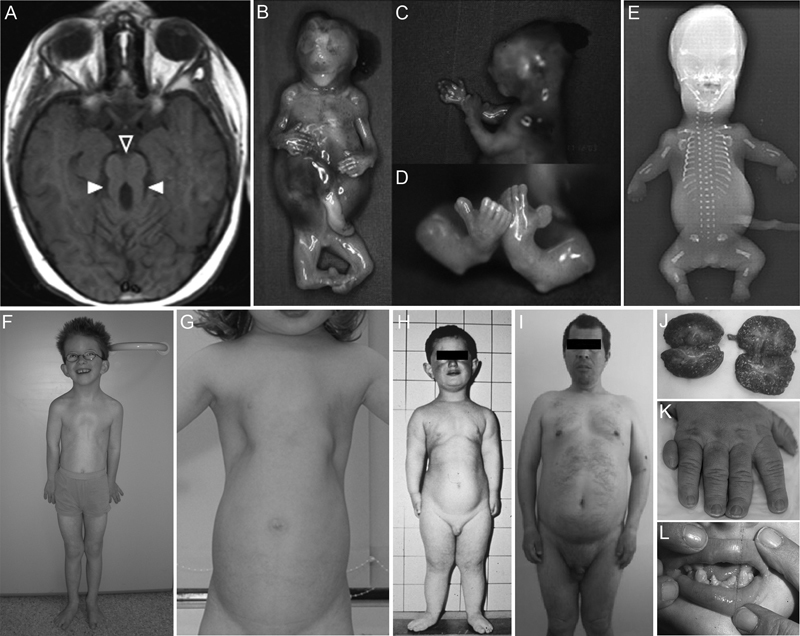
Phenotypic features of ciliopathies. The following features are commonly seen in ciliopathies. ( A ) Distinctive cerebellar and brain stem malformation that is known as the “molar tooth sign” in a JBTS patient. Open and filled arrowheads point out a deepened interpeduncular fossa and elongated superior cerebellar peduncles, respectively. ( B–D ) MKS fetus at the 14th week of gestation showing swollen stomach due to enlarged kidneys (B), occipital encephalocele (C), and polydactyly of hands (B) and feet (D). ( E ) Radiograph of an SRTD fetus displaying a short and narrow thorax, horizontally oriented ribs, short tubular bones with smooth ends, short and ovoid tibiae, and postaxial polysyndactyly. ( F ) The patient with CED showing a narrow thorax, pectus excavatum, and rhizomelic shortening of limbs. ( G ) Narrow thorax in a JATD patient. (H, L) Images of an EVC patient with a long narrow chest and shortness of the limbs (H), and hypodontia, that is, absence of upper and lower conical incisors (L). ( I ) BBS patient with truncal obesity, micropenis, and apathetic facial features. ( J ) Cystic kidneys of a patient with infantile NPHP. ( K ) Postaxial polydactyly of the right hand in a patient with OFDS. Reprinted from Brancati et al. Joubert syndrome and related disorders. Orphanet J Rare Dis 2010 Jul 8;5:20, Copyright (2010), with permission from BioMed Central Ltd. (A); Tallila et al. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet 2008 Jun;82(6):1361–1367, Copyright (2008) with permission from Elsevier (B–D); El Hokayem et al. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. J Med Genet 2012 Apr;49(4):227–233, Copyright (2012) with permission from BMJ Publishing Group Ltd. (E); Gilissen et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet 2010 Sep 10;87(3):418–423, Copyright (2010) with permission from Elsevier (F); Schmidts. Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet 2014 Nov;3(2):46–94, Copyright (2014) Thieme Publishers (G); Baujat and Le Merrer. Ellis-van Creveld syndrome. Orphanet J Rare Dis 2007 Jun; 4;2:27, Copyright (2007) with permission from Central Ltd. (H, L); Sahin et al. Two brothers with Bardet-Biedl syndrome presenting with chronic renal failure. Case Rep Nephrol 2015 Apr 3;2015:764973, Copyright (2015) (I); Oud et al. Early presentation of cystic kidneys in a family with a homozygous INVS mutation. Am J Med Genet A 2014 Jul;164A(7):1627–1634, Copyright (2014) with permission from John Wiley and Sons Inc. (J); Poretti et al. Delineation and diagnostic criteria of oral-facial-digital syndrome type VI. Orphanet J Rare Dis 2012 Jan 11;7:4, Copyright (2012) with permission from BioMed Central Ltd (K).
Both JBTS and MKS are usually autosomal recessive, but a few JBTS cases with X-linked inheritance have been reported. 29 JBTS and MKS overlap genetically; 25% of the 25 known JBTS genes have also been found mutated in MKS ( Table 1 ). 20 30 It remains elusive why certain mutations in a gene lead to JBTS, while other mutations in the same gene cause MKS. In some cases mutation type may determine the phenotypic outcome. For example, missense mutations in CC2D2A cause JBTS, whereas null alleles are associated with MKS. 20 Modifier genes have also been proposed to play a role in phenotypic outcome and could explain intrafamilial phenotypic differences. 20 30 Genotype-phenotype correlations have been reported and can facilitate choices for specific genetic testing. For example, AHI1 mutations often occur in JBTS patients with retinal dystrophy, whereas NPHP1 and RPGRIP1L mutations are most frequent in JBTS with renal dysplasia. Moreover, in 50% of JBTS patients with both retinal and renal phenotypes CEP290 mutations are detected. 19 22 When considering the protein localization, it is evident that most JBTS and MKS proteins reside in the ciliary transition zone ( Fig. 1 ). When they are mutated, the structure of the ciliary transition zone is perturbed causing disruption of its function as a ciliary gate, thereby affecting a variety of signaling pathways including Wnt/β-catenin and Hedgehog signaling. 20 30
Table 1. Genes mutated in ciliopathies.
| Gene | OMIM | Description |
|---|---|---|
| Neurodevelopmental ciliopathies | ||
| AHI1 | 608894 | JBTS |
| ARL13B | 608922 | JBTS |
| ATXN10 | 611150 | JBTS-like |
| B9D1 | 614144 | MKS |
| B9D2 | 611951 | MKS |
| C5orf42 | 614571 | JBTS |
| CC2D2A | 612013 | JBTS, MKS |
| CEP104 | NA | JBTS |
| CEP290 | 610142 | JBTS, SLSN, LCA, MKS, BBS |
| CEP41 | 610523 | JBTS |
| CSPP1 | 611654 | JBTS |
| EXOC4 | 608185 | MKS |
| EXOC8 | 615283 | JBTS |
| HYLS1 | 610693 | MKS-like |
| ICK | 612325 | ECO |
| IFT81 | 605489 | JBTS-like |
| INPP5E | 613037 | JBTS |
| KIAA0586 | 610178 | JBTS, SRTD |
| KIF14 | 611279 | MKS |
| KIF7 | 611254 | JBTS, MKS-like, ACLS |
| MKS1 | 609883 | MKS, BBS |
| NPHP1 | 607100 | JBTS, NPHP, SLSN |
| NPHP3 | 608002 | MKS, NPHP |
| PDE6D | 602676 | JBTS |
| PIBF1 | 607532 | JBTS |
| RPGRIP1L | 610937 | JBTS, MKS |
| TCTN1 | 609863 | JBTS |
| TCTN2 | 613846 | JBTS |
| TCTN3 | 613847 | JBTS, OFDS |
| TMEM107 | 616183 | MKS, OFDS |
| TMEM138 | 614459 | JBTS |
| TMEM216 | 613277 | JBTS, MKS |
| TMEM231 | 614949 | JBTS, MKS |
| TMEM237 | 614423 | JBTS |
| TMEM67 | 609884 | MKS, JBTS, NPHP, BBS |
| TTBK2 | 611695 | JBTS-like |
| ZNF423 | 604557 | JBTS, NPHP |
| Ciliopathies with major skeletal involvement | ||
| C2CD3 | 615944 | OFDS |
| CEP120 | 613446 | SRTD |
| C21ORF2 | 603191 | JATD |
| DDX59 | 615464 | OFDS |
| DYNC2H1 | 603297 | JATD, SRTD |
| EVC | 604831 | EVC, WAD |
| EVC2 | 607261 | EVC, WAD |
| IFT122 | 606045 | CED |
| IFT140 | 614620 | SRTD |
| IFT172 | 607386 | JATD, MZSDS, SRTD, RD |
| IFT43 | 614068 | CED |
| IFT80 | 611177 | SRTD |
| NEK1 | 604588 | SRTD |
| OFD1 | 300170 | OFDS, SGBS, JBTS |
| SCLT1 | 611399 | OFDS |
| TBC1D32 | 615867 | OFDS |
| TTC21B | 612014 | JATD, NPHP |
| WDR19 | 608151 | JATD, CED, NPHP |
| WDR34 | 613363 | SRTD |
| WDR35 | 613602 | CED, SRTD |
| WDR60 | 615462 | SRTD |
| Isolated and syndromic obesity | ||
| ALMS1 | 606844 | ALMS |
| ARL6 | 608845 | BBS, RP |
| BBIP1 | 613605 | BBS |
| BBS1 | 209901 | BBS |
| BBS10 | 610148 | BBS |
| BBS12 | 610683 | BBS |
| BBS2 | 606151 | BBS |
| BBS4 | 600374 | BBS |
| BBS5 | 603650 | BBS |
| BBS7 | 607590 | BBS |
| BBS9 | 607968 | BBS |
| CCDC28B | 610162 | BBS |
| CEP19 | 615586 | MOSPGF |
| IFT27 | 615870 | BBS |
| LZTFL1 | 606568 | BBS |
| MKKS | 604896 | BBS, MKKS |
| TRIM32 | 602290 | BBS |
| TTC8 | 608132 | BBS, RP |
| WDPCP | 613580 | BBS |
| Renal ciliopathies | ||
| ANKS6 | 615370 | NPHP |
| CEP164 | 614848 | NPHP |
| CEP83 | 615847 | NPHP |
| DCDC2 | 605755 | NPHP |
| GLIS2 | 608539 | NPHP |
| INVS | 243305 | NPHP |
| IQCB1 | 609237 | SLSN |
| NEK8 | 609799 | NPHP |
| NPHP4 | 607215 | NPHP, SLSN |
| PKD1 | 601313 | ADPKD |
| PKD2 | 173910 | ADPKD |
| PKHD1 | 606702 | ARPKD |
| SDCCAG8 | 613524 | SLSN |
| VHL | 608537 | VHL |
| XPNPEP3 | 613553 | NPHP-like |
| Primary ciliary dyskinesia | ||
| ARMC4 | 615408 | PCD |
| C21orf59 | 615494 | PCD |
| CCDC103 | 614677 | PCD |
| CCDC114 | 615038 | PCD |
| CCDC151 | 615956 | PCD |
| CCDC39 | 613798 | PCD |
| CCDC40 | 613799 | PCD |
| CCDC65 | 611088 | PCD |
| CCNO | 607752 | PCD |
| CENPF | 600236 | PCD |
| DNAAF1 | 613190 | PCD |
| DNAAF2 | 612517 | PCD |
| DNAAF3 | 614566 | PCD |
| DNAAF5 | 614864 | PCD |
| DNAH11 | 603339 | PCD |
| DNAH5 | 603335 | PCD |
| DNAI1 | 604366 | PCD |
| DNAI2 | 605483 | PCD |
| DNAL1 | 610062 | PCD |
| DRC1 | 615288 | PCD |
| DYX1C1 | 608706 | PCD |
| HYDIN | 610812 | PCD |
| LRRC6 | 614930 | PCD |
| NME8 | 607421 | PCD |
| RSPH1 | 609314 | PCD |
| RSPH4A | 612647 | PCD |
| RSPH9 | 612648 | PCD |
| SPAG1 | 603395 | PCD |
| ZMYND10 | 607070 | PCD |
Abbreviations: ACLS, acrocallosal syndrome; ADKPD, autosomal dominant polycystic kidney disease; ALMS, Alström syndrome; ARKPD, autosomal recessive polycystic kidney disease; BBS, Bardet-Biedl syndrome; CED, cranioectodermal dysplasia syndrome; ECO, endocrine-cerebro-osteodysplasia syndrome; EVC, Ellis-van Creveld syndrome; JATD, Jeune asphyxiating thoracic dysplasia; JBTS, Joubert syndrome; LCA, Leber congenital amaurosis; MKKS, McKusick-Kaufman syndrome; MKS, Meckel-Gruber syndrome; MOSPGF, morbid obesity and spermatogenic failure; NPHP, nephronophthisis; OFDS, oral-facial-digital syndrome; PCD, primary ciliary dyskinesia; RP, retinitis pigmentosa; SGBS, Simpson-Golabi-Behmel syndrome; SLSN, Senior-Løken syndrome; SRTD, short-rib thoracic dysplasia; VHL, von Hippel-Lindau syndrome; WAD, Weyers acrodental dysostosis.
Note: This overview of genes mutated in ciliopathies shows the heterogeneity of certain ciliopathies.
Corresponding OMIM identifiers are shown.
NA: OMIM number is not available.
Ciliopathies with Major Skeletal Involvement
Many ciliopathies are characterized by skeletal abnormalities such as dwarfism, short limbs and ribs, and brachydactyly/polydactyly. Ciliopathies with major skeletal involvement can be divided into four groups: cranioectodermal dysplasia (CED), short-rib thoracic dysplasia (SRTD), Ellis-van Creveld syndrome (EVC), and oral-facial-digital syndrome (OFDS). CED is rare and autosomal recessive. Typical features are sagittal craniosynostosis, narrow thorax with pectus excavatum, rhizomelia, brachydactyly, and ectodermal dysplasia ( Fig. 2F ). 31 Vital organ dysfunction often occurs and causes death in approximately 20% of CED patients before the age of 7. 31 SRTD is clinically and genetically related to CED. This family of disorders includes the following syndromes; short-rib polydactyly syndrome (SRPS), Jeune asphyxiating thoracic dysplasia (JATD), and Mainzer-Saldino syndrome (MZSDS). These disorders are autosomal recessive and frequencies range from 1:100,000 to 1:1,000,000. 32 The most prominent SRTD features are limb shortening and a small rib cage. These features are most pronounced in perinatal lethal SRPS ( Fig. 2E ). Additional SRPS features may include polydactyly, cleft lip/palate, and anomalies of a variety of organs and tissues including the brain, heart, eyes, kidneys, liver, pancreas, intestines, and genitalia. 33 Patients with JATD present with similar, but milder skeletal anomalies compared with SRPS. The main complication in JATD patients is the narrow thorax phenotype that causes severe respiratory insufficiency; this phenotype causes lethality in 20 to 60% of JATD infants ( Fig. 2G ). Besides skeletal abnormalities, JATD patients also occasionally present with nephronophthisis (NPHP), blindness, liver fibrosis, and intellectual disability. 32 Similar as in CED, MZSDS is also featured by a relatively mild narrow thorax phenotype. Other features of MZSDS are shortened limbs, blindness, NPHP, liver fibrosis, and pancreatic abnormalities. 34 EVC is another ciliopathy that is featured by major skeletal anomalies. Besides dwarfism, polydactyly, and ectodermal dysplasia, congenital heart malformations are commonly seen in EVC patients ( Fig. 2H , L ). The latter have a major influence on the prognosis for individual patients. 35 Finally, a variety of OFDSs have been reported; however, many are based on single or few cases that are not molecularly tested. 36 The best known OFD type is OFD1, which is X-linked and characterized by bifid tongue, oral frenula, cleft lip/palate, dental anomalies, syndactyly, polydactyly, polycystic kidney disease, and central nervous system abnormalities ( Fig. 2K ).
Ciliary chondrodysplasias are genetically heterogenous. EVC is caused by mutations in EVC and EVC2 . EVC mutations have also been reported in patients with autosomal dominant Weyers acrodental dysostosis, which presents with a milder phenotype than EVC. 32 Genetic overlap is mostly seen between CED, JATD, SRPS, and MZSDS ( Table 1 ), but it also occurs between OFDS and JBTS ( Table 1 ). 29 36 37 38 Most CED and SRTD mutations have been found in genes encoding IFT and associated motor proteins ( Fig. 1 and Table 1 ). OFDS is also genetically heterogeneous and recent data suggest that a proportion of OFDS proteins reside in centriolar appendages where they regulate ciliogenesis in an antagonistic manner; C2CD3 promotes ciliary growth, whereas OFD1 is a repressor. 39 40
Isolated and Syndromic Obesity
Nonsyndromic obesity is a growing problem in the general population and mainly caused by multifactorial factors. Yet in some cases isolated obesity is a mendelian disorder that results from ciliary anomalies. 41 Previously, ciliary defects had already been described in obesity syndromes such as in BBS and Alström syndrome (ALMS). Both syndromes display an autosomal recessive inheritance; however, oligogenic inheritance has also been described in BBS. 42 43 The prevalence of BBS varies markedly between populations—from 1:160,000 in northern Europe to 1:13,500 births in isolated communities in Kuwait. 44 Cardinal BBS features are rod-cone dystrophy, obesity, polydactyly, intellectual disability, hypogonadism, and renal dysfunction ( Fig. 2I ). Secondary features include cardiovascular abnormalities, craniofacial anomalies, psychomotor delay, type 2 diabetes mellitus (T2DM), and hearing loss. The clinical diagnosis of BBS is based on the presence of at least four primary features or a combination of three primary plus two secondary features. In general, the BBS phenotype evolves during the first decade of life, except for polydactyly that is present at birth; generating an accurate clinical diagnosis may therefore be challenging in infancy and early childhood. 45 BBS has an adverse prognosis, with renal impairment being a frequent and important cause of death. 46 The phenotypically related ALMS is characterized by cone-rod dystrophy, obesity, hearing impairment, cardiomyopathy, T2DM diabetes, and hepatic and renal disease. 47 A clinical diagnosis is based on the aforementioned cardinal features and differs from BBS by the presence of hearing impairment and absence of polydactyly and learning difficulties. 44 While it may be difficult to make a clinical diagnosis of BBS and ALMS in early childhood, molecular testing clearly distinguishes both disorders. To date, only one gene, ALMS1 , is known to be mutated in ALMS ( Table 1 ). 48 In BBS mutations in 19 genes are reported to be causative ( Table 1 ), explaining more than 80% of the patients. 44 The protein encoded by ALMS1 and most of the proteins encoded by BBS-associated genes localize to the base of the primary cilium. ALMS1 is also found in endosomal structures and is suggested to play a role in the recycling endosome pathway. 49 The BBSome, composed of BBS1–2, 4–5, 7–9, and BBIP10, plays an important role in trafficking proteins into and out of the cilium, whereas the chaperonin complex that consists of BBS6 and BBS10–12 is required for assembly of the BBSome. 44 50 51 The remaining BBS proteins have variable functions. Some are thought to act as GTPases or E3 ubiquitin ligases; others have been shown to regulate centriole migration and IFT. 44
Renal Ciliopathies
Polycystic kidneys are commonly seen in ciliopathies. The best-known renal ciliopathies are NPHP, and autosomal dominant and autosomal recessive polycystic kidney disease (ADPKD and ARPKD, respectively). ADPKD is most common with an incidence of approximately 1:1,000 and is characterized by enlarged polycystic kidneys. 52 Liver cysts and abnormalities of the vasculature and heart may also be present. 52 PKD1 and PKD2 mutations explain 85% and 15% of ADPKD patients, respectively. Most individuals with ADPKD have a normal renal function during childhood; however, renal insufficiency progresses during life and results in end-stage renal disease (ESRD) in 50% of affected individuals. 52 In contrast, ARPKD is usually diagnosed in late pregnancy or at birth by the detection of enlarged cystic kidneys by ultrasound. Grossly enlarged kidneys cause pulmonary hypoplasia and thoracic compression leading to respiratory distress and death in approximately 30 to 50% of affected neonates. In the majority of remaining patients ESRD evolves in adulthood. In addition, virtually all ARPKD patients show hepatic involvement, that is, hepatomegaly and liver fibrosis. 53 To date, only mutations in PKHD1 have been reported and these explain at least 80 to 87% of ARPKD patients. 54 PKHD1 encodes the fibrocystin protein that forms a ciliary protein complex together with PKD1 and PKD2. 55 Another monogenetic cause of ESRD in children is NPHP. Three types have been described based on age of onset of ESRD; infantile (< 2 years), juvenile (∼13 years), and adolescent (∼19 years) NPHP. 56 Juvenile NPHP is most common and affects 5 to 10% of the children with ESRD. 57 It is estimated to occur in 1:50,000 births in Canada and 1:900,000 in the United States. 57 Typical histopathologic characteristics are renal cysts, tubular basement membrane disruption, and tubulointerstitial fibrosis. Most features are shared between the different NPHP types; however, tubular basement membrane disruption is usually absent in infantile NPHP. Also, kidneys of patients with infantile NPHP are usually enlarged, whereas normal-sized or smaller kidneys are seen in patients with other forms of NPHP ( Fig. 2J ). 57 NPHP can both be isolated and syndromic. Twenty NPHP genes are known so far and explain roughly one-third of the patients. All NPHP proteins reside in the basal body, transition zone, or axonemal base of primary cilia; however, they have also been found in other cellular sites such as cell junctions and nuclei. 58 59 Mutations in NPHP genes perturb a variety of signaling cascades including Wnt, Hedgehog, Hippo, and DNA damage response signaling, which in turn disrupt renal development and tissue homeostasis. 59
Primary Ciliary Dyskinesia
Most phenotypes associated by dysfunction of motile cilia result in primary ciliary dyskinesia (PCD). Cardinal symptoms are neonatal respiratory abnormalities and airway infections during childhood, which eventually develop into bronchiectasis. Patients also often present with situs inversus , dextrocardia, and infertility. 60 61 Diagnosis of PCD is challenging due to the phenotypic overlap with other respiratory diseases such as asthma, immune deficiencies, and bronchomalacia, and the absence of clear diagnostic criteria further complicates making an accurate diagnosis. 61 The prevalence of PCD, being 1:10,000 to 1:20,000, may therefore be and underestimate. 61 Mutations in 30 genes have been associated with PCD and explain 60 to 70% of the cases. 3 61 The inheritance pattern associated with PCD is usually autosomal recessive; however, X-linked or autosomal dominant inheritance have also been reported in a few cases. 61 Roughly 60% of proteins that are encoded by PCD genes are found in protein complexes and axonemal structures that are typically found in motile cilia such as inner and outer dynein arms, radial spokes, the central microtubule pair, and nexin links ( Fig. 1 ), whereas the remaining proteins are cytoplasmic with a role in (pre)assembly and transportation of dynein components. 3
Why Is Genetic Screening Important?
The phenotypic spectrum of ciliopathies as a whole is very broad, but phenotypic overlap between certain ciliopathies can be marked, such as in JBTS and MKS or BBS and ALMS. 20 A correct diagnosis can thus not always be made solely based on the clinical phenotype but requires molecular testing. Molecular information facilitates making an accurate diagnosis and is therefore essential for proper genetic counseling, prognosis, recurrence risk estimation, and clinical management in the clinic. In the past years, gene panels and exomes have been analyzed by next-generation sequencing (NGS) technologies. This resulted in the detection of numerous pathogenic mutations and dozens of novel disease genes in a nick of time. NGS thus enormously facilitated our ability to make an early diagnosis and to recognize important genotype-phenotype correlations. 62 63 This knowledge contributes to the clinician's ability to correctly inform patients about expected disease progression and allows clinicians to form appropriate interdisciplinary teams leading to optimal clinical care and disease management. For example, early diagnosis is highly beneficial to PCD patients in whom progression to irreversible lung disease continues unless treated early on. 64 Gene and mutation identification also allows for development of personalized therapies such as targeted gene therapy. This is important as no cures are available for ciliopathies to date. Gene therapy successes have been booked in patients with a variety of hereditary disorders, 65 66 67 68 including retinal degeneration. The latter is a common feature in many ciliopathies and is probably treatable in the future. 69 In addition to the retina, researchers also started to investigate possibilities for gene therapy in ciliopathies affecting the respiratory system. 70 71 72 For example, ciliary beating was restored in ex vivo cell cultures from PCD patients harboring mutations in DNAI1 by introducing a healthy copy of this gene via lentiviral transduction. 72
Ciliary Genes and Next-Generation Sequencing
Research
Since 1994, 73 127 ciliopathy genes have been identified ( Table 1 ), initially with linkage studies and positional cloning (1980s–2005), later with single nucleotide polymorphism (SNP) microarrays and candidate gene analysis (2006–2010), and most recently with various NGS methods (2009–2015). 74 75 76 77 78 79 80 81 Targeted gene panels have been sequenced with NGS methods in dozens or even hundreds of subsets of patients for efficient and cheap mutation detection in known genes in a variety of ciliopathies, including, but not limited to, JBTS, BBS, ALMS, NPHP, and PCD. 62 63 82 83 Some gene panels not only targeted known ciliary disease genes but also included candidate genes, for example genes with a predicted ciliary function. These panels have proven to be successful tools to identify novel disease genes by various groups. 81 83 84 85 Currently, we can genetically explain 45 to 90% of JBTS and MKS patients, 63 86 87 55 to 70% of CED and SRTD patients, 32 88 84% of EVC patients, 89 80% of BBS and ALMS patients, 44 21% of NPHP patients, 90 and 65% of PCD patients with panels. 91 The advantage of gene panel testing versus genomic methods such as whole-exome sequencing (WES), whereby all exons of the genome are sequenced, is that panels usually result in higher coverage data at a relatively low cost and with high-throughput. 92 93 In addition, the chance of identification of incidental findings, which are clinically relevant findings that are unrelated to the clinical problem for which screening was initially intended, is extremely low when only known ciliopathy-specific genes are being sequenced 94 ; however, it is difficult to add new genes to existing gene panels. 94 The possibility of detection of incidental findings is an important concern when analyzing WES data, however, WES has proven to be highly successful for the identification of a mutation in a gene that has not previously been implicated in ciliary disease. Furthermore, retrospective analysis of the data is possible. 94 A recent research study reported that WES provides a molecular diagnosis in 44% of ciliopathy patients. 95
Diagnostics
In past and current genome diagnostics for ciliopathies, prescreening methods such as (SNP) microarrays for homozygosity mapping (and copy number detection, CNV) or targeted panels with only known mutations in ciliary disease genes have been used to reduce costs by limiting the list of genes to be tested by gene-specific methods such as Sanger sequencing. More recently, WES started to be implemented as a diagnostic test for ciliopathies and is considered to be cost-effective when a clinician needs to investigate more than two genes to identify the genetic cause of disease, a scenario that often applies when a ciliopathy patient presents in the clinic considering the enormous genetic heterogeneity in these disorders. 95 96 While incidental findings are a concern in WES, these can be easily circumvented in diagnostics by first (or only) analyzing known ciliopathy genes and leaving the remainder of WES data concealed. This approach has proven to be an effective tool for gene and mutation identification with reasonable throughput and acceptable costs. 96 97 Diagnostic WES has an estimated success rate of approximately 30% for ciliopathies in our institute (personal communication, D. Lugtenberg and www.genomediagnosticsnijmegen.nl ). Other groups reported general success rates for rare diseases that vary between 20 and 45% based on various studies that include thousands of patients. 98 99 100 101
Current Next-Generation Sequencing Obstacles
Although NGS sequencing has tremendously improved mutation detection for numerous disorders, ciliopathies included, there are still several hurdles to take with respect to accurate variant detection and prioritization.
Technical Challenges
In WES the coverage, referring to the number of times that a genomic region is read, is not always complete for each ciliopathy gene, which prevents detection of causative variants. One gene that is not sufficiently covered in any standard WES procedure is PKD1 . This is explained by the existence of six PKD1 pseudogenes that share greater than 97% sequence identity with the first 33 exons of PKD1 , which complicates standard alignment algorithms. 102 103 This problem can be resolved by combining NGS with long-range PCR 104 105 or by using custom-designed targeted enrichment libraries 106 and applying mapping algorithms that facilitate specific mapping of reads to the PKD1 gene. 103 Pseudogenes are not the sole cause for the occurrence of low coverage; genomic regions with high GC contents and repetitive sequences are also difficult to read. 107 Another NGS challenge is represented by the occurrence of pathogenic mosaicisms and postzygotic mutations. 108 Such variation is difficult to detect because of the sporadic occurrence of these variants in sequence reads. Stringent filtering criteria may thus prevent us from identifying these mutations in WES data. Also, this type of variation may not be detectable at all when occurring in other tissues than the tested specimen. Other variants that cannot be detected by WES are noncoding mutations, translocations, and imprinting changes. 95 97 Copy number variations (CNVs) can be detected by different algorithms, but detection may be limited to three or more exons depending on the software used. 109
Variant Interpretation
Besides technical improvements, genome analysis would also highly benefit from improved variant understanding. Many WES-detected variants have not been associated with a clinical phenotype before and their clinical significance is therefore unclear. Such variants are referred to as variants of unknown clinical significance (VUS). 110 VUS may be found in genes that have previously been associated with ciliopathies and in genes in which clinical variation has not been described yet. One can distinguish between benign and pathogenic variants based on bioinformatic information, but also by assessing effects of VUS in vitro in ciliated cells derived from patient's shed deciduous teeth, skin biopsy, or urine samples. 111 112 Besides cellular phenotyping, simple organisms with ciliated cells such as Danio rerio (zebrafish), Caenorhabditis elegans (worm), and Drosophila melanogaster (fruit fly) can also be used to model mutational effects. 113 114 115 Although functional testing would allow for a better understanding of VUS, this type of testing is often not available in a clinical setting and is difficult to implement as each variant or gene requires a different assay. Another method that facilitates variant interpretation in both research and clinic is data sharing and public registration of identified variants and associated phenotypes 95 ; if patients with similar ciliopathy features are identified with identical variants in known disease genes or if deleterious variants in the same (previously unreported) gene are found, these replicative findings can render important clues about the nature of a variant. An initiative that promotes data sharing for rare disorders is the “Matchmaker Exchange” database ( http://www.matchmakerexchange.org/ ). When genotypic and phenotypic data are entered on this Web site, it is registered in various connected databases that in turn allows bonding between scientists and clinical staff with matching interests, 95 which will benefit patients and their families in the long run.
Future Perspectives
This review discussed mutation detection for ciliary disorders through gene panels and WES in both research and diagnostics. We did not highlight the power of whole-genome sequencing yet, while this is the technology of the future. The general advantage of WGS compared with WES is that it leads to improved mutation detection in protein-coding DNA. 116 In addition, WGS allows detection of variation in noncoding genomic regions. Although our understanding of variation in noncoding DNA is still poor, future research will allow for improved interpretation of variants in these regions, probably in particular in conserved regulatory elements such as promoters, enhancers, insulators, and other regulators. 117 Other WGS challenges concern data storage and throughput, which rely on the availability of excellent bioinformatic platforms; however, WGS platforms and procedures are improving and fastening as we speak. For example, it was recently shown that it is possible to effectively conduct WGS and data analysis within 26 to 50 hours periods in ill neonates. 118 119 This strategy is also of interest for newborns with a suspected ciliary disorder as clinical features of classic syndromes may be incomplete and difficult to recognize in infancy, while molecular knowledge allows accurate and early diagnosis and prognosis and directs decisions for optimal disease management in these children. Besides implementing NGS for ciliopathies in postnatal diagnostics, there also is a need to conduct such testing prenatally as clinical features of ciliopathies may present as early as the first trimester of pregnancy. 120 In addition, improved prenatal testing also allows for optimal management and counseling for parents who have a risk of giving birth to a child with a ciliopathy. The future implementation of NGS-based methods in prenatal diagnostics will rely on short turnaround times of clinical reports as limited time is available for counseling and decision making. Prenatal diagnostics will further benefit from the possibility to conduct genetic screening on fetal DNA derived from maternal blood as that avoids the need for invasive amniocentesis and chorionic villus sampling. Notably, NGS is already successfully being used for detection of aneuploidies and CNVs in fetal DNA extracts from maternal blood, whereas technological advances may allow for genome-wide single nucleotide variation (SNV) detection in fetal-maternal DNA in the future. 121 122 123
In conclusion, NGS-based screening revolutionized the ciliopathy research and diagnostics in recent years, and this process is still ongoing. While technical and bioinformatic challenges remain, NGS methods are gradually being implemented in clinical laboratories with reasonable throughput, for acceptable costs, and with significant mutation identification success for ciliopathies. In the future, return of molecular reports will accelerate with turnaround times of only one or a few days, and screening of ciliopathy panels, WES, and WGS will probably evolve as part of noninvasive procedures in prenatal genome diagnostics. These developments will further contribute to improved health care for families affected by ciliary disorders.
Acknowledgments
The authors thank Dorien Lugtenberg for critical discussions and review of the manuscript, Christian Gilissen and Stefan Lelieveld for bioinformatic consultation, and Jeroen van Reeuwijk for his contribution to Table 1 . This manuscript was written with funding from the Dutch Kidney Foundation (KOUNCIL consortium project CP11.18 to HHA), the Netherlands Organization for Health Research and Development (ZonMW Veni-91613008 to HHA) and an institutional grant from the Radboud Institute for Molecular Life Sciences (RIMLS) to HHA.
References
- 1.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8(11):880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 2.Satir P, Christensen S T. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 3.Horani A, Ferkol T W, Dutcher S K, Brody S L. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev. 2016;18:18–24. doi: 10.1016/j.prrv.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salathe M. Regulation of mammalian ciliary beating. Annu Rev Physiol. 2007;69:401–422. doi: 10.1146/annurev.physiol.69.040705.141253. [DOI] [PubMed] [Google Scholar]
- 5.Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Dynlacht B D. Assembling a primary cilium. Curr Opin Cell Biol. 2013;25(04):506–511. doi: 10.1016/j.ceb.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szymanska K, Johnson C A.The transition zone: an essential functional compartment of cilia Cilia 201210110. Doi: 10.1186/2046-2530-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang T T, Su J, Wang W Jet al. Super resolution pattern recognition reveals the architectural map of the ciliary transition zone Sci Rep 2015514096. Doi: 10.1038/srep14096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin H, White S R, Shida T et al. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141(07):1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J C, Badano J L, Sibold S et al. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36(05):462–470. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- 11.Malicki J, Avidor-Reiss T. From the cytoplasm into the cilium: bon voyage. Organogenesis. 2014;10(01):138–157. doi: 10.4161/org.29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozminski K G, Johnson K A, Forscher P, Rosenbaum J L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A. 1993;90(12):5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbaum J L, Witman G B. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3(11):813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen L B, Rosenbaum J L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 15.Powles-Glover N. Cilia and ciliopathies: classic examples linking phenotype and genotype—an overview. Reprod Toxicol. 2014;48:98–105. doi: 10.1016/j.reprotox.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Singla V, Reiter J F.The primary cilium as the cell's antenna: signaling at a sensory organelle Science 2006313(5787):629–633. [DOI] [PubMed] [Google Scholar]
- 17.Rao Damerla R, Gabriel G C, Li Y et al. Role of cilia in structural birth defects: insights from ciliopathy mutant mouse models. Birth Defects Res C Embryo Today. 2014;102(02):115–125. doi: 10.1002/bdrc.21067. [DOI] [PubMed] [Google Scholar]
- 18.Joubert M, Eisenring J J, Robb J P, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19(09):813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 19.Romani M, Micalizzi A, Valente E M. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12(09):894–905. doi: 10.1016/S1474-4422(13)70136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szymanska K, Hartill V L, Johnson C A. Unraveling the genetics of Joubert and Meckel-Gruber syndromes. J Pediatr Genet. 2014;3(02):65–78. doi: 10.3233/PGE-14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parisi M A, Doherty D, Chance P F, Glass I A. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15(05):511–521. doi: 10.1038/sj.ejhg.5201648. [DOI] [PubMed] [Google Scholar]
- 22.Brancati F, Dallapiccola B, Valente E M.Joubert syndrome and related disorders Orphanet J Rare Dis 2010520. Doi: 10.1186/1750-1172-5-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salonen R, Paavola P. Meckel syndrome. J Med Genet. 1998;35(06):497–501. doi: 10.1136/jmg.35.6.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logan C V, Abdel-Hamed Z, Johnson C A. Molecular genetics and pathogenic mechanisms for the severe ciliopathies: insights into neurodevelopment and pathogenesis of neural tube defects. Mol Neurobiol. 2011;43(01):12–26. doi: 10.1007/s12035-010-8154-0. [DOI] [PubMed] [Google Scholar]
- 25.Wright C, Healicon R, English C, Burn J. Meckel syndrome: what are the minimum diagnostic criteria? J Med Genet. 1994;31(06):482–485. doi: 10.1136/jmg.31.6.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C P. Meckel syndrome: genetics, perinatal findings, and differential diagnosis. Taiwan J Obstet Gynecol. 2007;46(01):9–14. doi: 10.1016/S1028-4559(08)60100-X. [DOI] [PubMed] [Google Scholar]
- 27.Barisic I, Boban L, Loane M et al. Meckel-Gruber syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur J Hum Genet. 2015;23(06):746–752. doi: 10.1038/ejhg.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salonen R, Norio R. The Meckel syndrome in Finland: epidemiologic and genetic aspects. Am J Med Genet. 1984;18(04):691–698. doi: 10.1002/ajmg.1320180415. [DOI] [PubMed] [Google Scholar]
- 29.Coene K L, Roepman R, Doherty D et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85(04):465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelhamed Z A, Wheway G, Szymanska K et al. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet. 2013;22(07):1358–1372. doi: 10.1093/hmg/dds546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arts H, Knoers N, Pagon R A, Adam M P, Ardinger H H.Cranioectodermal DysplasiaIn:et al. Seattle, WA: Gene Reviews(R)1993 [Google Scholar]
- 32.Schmidts M. Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet. 2014;3(02):46–94. doi: 10.3233/PGE-14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C(03):165–174. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 34.Keppler-Noreuil K M, Adam M P, Welch J, Muilenburg A, Willing M C. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy) Am J Med Genet A. 2011;155A(05):1021–1032. doi: 10.1002/ajmg.a.33892. [DOI] [PubMed] [Google Scholar]
- 35.Baujat G, Le Merrer M.Ellis-van Creveld syndrome Orphanet J Rare Dis 2007227. Doi: 10.1186/1750-1172-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toriello H V. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009;149A(05):1089–1095. doi: 10.1002/ajmg.a.32799. [DOI] [PubMed] [Google Scholar]
- 37.Lopez E, Thauvin-Robinet C, Reversade B et al. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet. 2014;133(03):367–377. doi: 10.1007/s00439-013-1385-1. [DOI] [PubMed] [Google Scholar]
- 38.Srour M, Schwartzentruber J, Hamdan F F et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet. 2012;90(04):693–700. doi: 10.1016/j.ajhg.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thauvin-Robinet C, Lee J S, Lopez E et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet. 2014;46(08):905–911. doi: 10.1038/ng.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye X, Zeng H, Ning G, Reiter J F, Liu A. C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci U S A. 2014;111(06):2164–2169. doi: 10.1073/pnas.1318737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shalata A, Ramirez M C, Desnick R J et al. Morbid obesity resulting from inactivation of the ciliary protein CEP19 in humans and mice. Am J Hum Genet. 2013;93(06):1061–1071. doi: 10.1016/j.ajhg.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katsanis N, Ansley S J, Badano J Let al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder Science 2001293(5538):2256–2259. [DOI] [PubMed] [Google Scholar]
- 43.Beales P L, Badano J L, Ross A J et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003;72(05):1187–1199. doi: 10.1086/375178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forsythe E, Beales P L. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(01):8–13. doi: 10.1038/ejhg.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beales P L, Elcioglu N, Woolf A S, Parker D, Flinter F A. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(06):437–446. [PMC free article] [PubMed] [Google Scholar]
- 46.O'Dea D, Parfrey P S, Harnett J D, Hefferton D, Cramer B C, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis. 1996;27(06):776–783. doi: 10.1016/s0272-6386(96)90513-2. [DOI] [PubMed] [Google Scholar]
- 47.Marshall J D, Bronson R T, Collin G B et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165(06):675–683. doi: 10.1001/archinte.165.6.675. [DOI] [PubMed] [Google Scholar]
- 48.Collin G B, Marshall J D, Ikeda A et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31(01):74–78. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 49.Collin G B, Marshall J D, King B Let al. The Alström syndrome protein, ALMS1, interacts with α-actinin and components of the endosome recycling pathway PLoS One 2012705e37925. Doi: 10.1371/journal.pone.0037925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loktev A V, Zhang Q, Beck J S et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell. 2008;15(06):854–865. doi: 10.1016/j.devcel.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Liew G M, Ye F, Nager A R et al. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev Cell. 2014;31(03):265–278. doi: 10.1016/j.devcel.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dell K M. The spectrum of polycystic kidney disease in children. Adv Chronic Kidney Dis. 2011;18(05):339–347. doi: 10.1053/j.ackd.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30(01):15–30. doi: 10.1007/s00467-013-2706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guay-Woodford L M. Autosomal recessive polycystic kidney disease: the prototype of the hepato-renal fibrocystic diseases. J Pediatr Genet. 2014;3(02):89–101. doi: 10.3233/PGE-14092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ward C J, Hogan M C, Rossetti S et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30(03):259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 56.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(01):23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol. 2009;24(12):2333–2344. doi: 10.1007/s00467-008-0840-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saunier S, Salomon R, Antignac C. Nephronophthisis. Curr Opin Genet Dev. 2005;15(03):324–331. doi: 10.1016/j.gde.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 59.Wolf M T. Nephronophthisis and related syndromes. Curr Opin Pediatr. 2015;27(02):201–211. doi: 10.1097/MOP.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Afzelius B A.A human syndrome caused by immotile cilia Science 1976193(4250):317–319. [DOI] [PubMed] [Google Scholar]
- 61.Paff T, Daniels J MA, Pals G, Haarman E G. Primary ciliary dyskinesia: from diagnosis to molecular mechanisms. J Pediatr Genet. 2014;3(02):115–127. doi: 10.3233/PGE-14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Halbritter J, Porath J D, Diaz K A et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132(08):865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bachmann-Gagescu R, Dempsey J C, Phelps I G et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52(08):514–522. doi: 10.1136/jmedgenet-2015-103087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collins S A, Walker W T, Lucas J S. Genetic testing in the diagnosis of primary ciliary dyskinesia: state-of-the-art and future perspectives. J Clin Med. 2014;3(02):491–503. doi: 10.3390/jcm3020491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maguire A M, High K A, Auricchio Aet al. Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial Lancet 2009374(9701):1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Testa F, Maguire A M, Rossi S et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013;120(06):1283–1291. doi: 10.1016/j.ophtha.2012.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brimble M A, Reiss U M, Nathwani A C, Davidoff A M. New and improved AAVenues: current status of hemophilia B gene therapy. Expert Opin Biol Ther. 2016;16(01):79–92. doi: 10.1517/14712598.2015.1106475. [DOI] [PubMed] [Google Scholar]
- 68.Hawkes N.Gene therapy trial for cystic fibrosis shows modest benefits BMJ 2015351h3608. Doi: 10.1136/bmj.h3608 [DOI] [PubMed] [Google Scholar]
- 69.Estrada-Cuzcano A, Roepman R, Cremers F P, den Hollander A I, Mans D A.Non-syndromic retinal ciliopathies: translating gene discovery into therapy Hum Mol Genet 201221(R1):R111–R124. [DOI] [PubMed] [Google Scholar]
- 70.Simons D L, Boye S L, Hauswirth W W, Wu S M. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet-Biedl syndrome mouse model. Proc Natl Acad Sci U S A. 2011;108(15):6276–6281. doi: 10.1073/pnas.1019222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McIntyre J C, Williams C L, Martens J R. Smelling the roses and seeing the light: gene therapy for ciliopathies. Trends Biotechnol. 2013;31(06):355–363. doi: 10.1016/j.tibtech.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chhin B, Negre D, Merrot Oet al. Ciliary beating recovery in deficient human airway epithelial cells after lentivirus ex vivo gene therapy PLoS Genet 2009503e1000422. Doi: 10.1371/journal.pgen.1000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.The European Polycystic Kidney Disease Consortium.The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16 Cell 19947706881–894. [DOI] [PubMed] [Google Scholar]
- 74.Mykytyn K, Braun T, Carmi R et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet. 2001;28(02):188–191. doi: 10.1038/88925. [DOI] [PubMed] [Google Scholar]
- 75.Mykytyn K, Nishimura D Y, Searby C C et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31(04):435–438. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 76.Gilissen C, Arts H H, Hoischen A et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87(03):418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang L, Szymanska K, Jensen V L et al. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011;89(06):713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Otto E A, Hurd T W, Airik R et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42(10):840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srour M, Hamdan F F, McKnight D et al. Joubert syndrome in French Canadians and identification of mutations in CEP104. Am J Hum Genet. 2015;97(05):744–753. doi: 10.1016/j.ajhg.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Delous M, Baala L, Salomon R et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39(07):875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 81.Hopp K, Heyer C M, Hommerding C J et al. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum Mol Genet. 2011;20(13):2524–2534. doi: 10.1093/hmg/ddr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Redin C, Le Gras S, Mhamdi O et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alström syndromes. J Med Genet. 2012;49(08):502–512. doi: 10.1136/jmedgenet-2012-100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Onoufriadis A, Shoemark A, Schmidts M et al. Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects. Hum Mol Genet. 2014;23(13):3362–3374. doi: 10.1093/hmg/ddu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Failler M, Gee H Y, Krug P et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet. 2014;94(06):905–914. doi: 10.1016/j.ajhg.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmidts M, Frank V, Eisenberger T et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum Mutat. 2013;34(05):714–724. doi: 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Knopp C, Rudnik-Schöneborn S, Eggermann T et al. Syndromic ciliopathies: from single gene to multi gene analysis by SNP arrays and next generation sequencing. Mol Cell Probes. 2015;29(05):299–307. doi: 10.1016/j.mcp.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 87.Lambacher N J, Bruel A L, van Dam T J et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol. 2016;18(01):122–131. doi: 10.1038/ncb3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin A E, Traum A Z, Sahai I et al. Sensenbrenner syndrome (cranioectodermal dysplasia): clinical and molecular analyses of 39 patients including two new patients. Am J Med Genet A. 2013;161A(11):2762–2776. doi: 10.1002/ajmg.a.36265. [DOI] [PubMed] [Google Scholar]
- 89.D'Asdia M C, Torrente I, Consoli F et al. Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur J Med Genet. 2013;56(02):80–87. doi: 10.1016/j.ejmg.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 90.Schueler M, Halbritter J, Phelps I G et al. Large-scale targeted sequencing comparison highlights extreme genetic heterogeneity in nephronophthisis-related ciliopathies. J Med Genet. 2016;53(03):208–214. doi: 10.1136/jmedgenet-2015-103304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Djakow J, Kramná L, Dušátková L et al. An effective combination of sanger and next generation sequencing in diagnostics of primary ciliary dyskinesia. Pediatr Pulmonol. 2016;51(05):498–509. doi: 10.1002/ppul.23261. [DOI] [PubMed] [Google Scholar]
- 92.Saudi Mendeliome G; Saudi Mendeliome Group.Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases Genome Biol 201516134. Doi: 10.1186/s13059-015-0693-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Koning T J, Jongbloed J D, Sikkema-Raddatz B, Sinke R J. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: the opportunities and challenges. Expert Rev Mol Diagn. 2015;15(01):61–70. doi: 10.1586/14737159.2015.976555. [DOI] [PubMed] [Google Scholar]
- 94.Renkema K Y, Stokman M F, Giles R H, Knoers N V. Next-generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol. 2014;10(08):433–444. doi: 10.1038/nrneph.2014.95. [DOI] [PubMed] [Google Scholar]
- 95.Sawyer S L, Hartley T, Dyment D A et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet. 2016;89(03):275–284. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neveling K, Feenstra I, Gilissen C et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34(12):1721–1726. doi: 10.1002/humu.22450. [DOI] [PubMed] [Google Scholar]
- 97.Valencia C A, Husami A, Holle Jet al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center's experience Front Pediatr 2015367. Doi: 10.3389/fped.2015.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soden S E, Saunders C J, Willig L Ket al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders Sci Transl Med 20146265265ra168. Doi: 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang Y, Muzny D M, Reid J G et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Y, Muzny D M, Xia F et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee H, Deignan J L, Dorrani N et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li F, Cao Y, Han L et al. Gene expression signature: an R package for discovering functional connections using gene expression signatures. OMICS. 2013;17(02):116–118. doi: 10.1089/omi.2012.0087. [DOI] [PubMed] [Google Scholar]
- 103.Eisenberger T, Decker C, Hiersche Met al. An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease PLoS One 20151002e0116680. Doi: 10.1371/journal.pone.0116680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tan A Y, Michaeel A, Liu G et al. Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J Mol Diagn. 2014;16(02):216–228. doi: 10.1016/j.jmoldx.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rossetti S, Hopp K, Sikkink R A et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. 2012;23(05):915–933. doi: 10.1681/ASN.2011101032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trujillano D, Bullich G, Ossowski S et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol Genet Genomic Med. 2014;2(05):412–421. doi: 10.1002/mgg3.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kirby A, Gnirke A, Jaffe D B et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45(03):299–303. doi: 10.1038/ng.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Acuna-Hidalgo R, Bo T, Kwint M P et al. Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am J Hum Genet. 2015;97(01):67–74. doi: 10.1016/j.ajhg.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.de Ligt J, Boone P M, Pfundt R et al. Platform comparison of detecting copy number variants with microarrays and whole-exome sequencing. Genom Data. 2014;2:144–146. doi: 10.1016/j.gdata.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang J, Liao J, Zhang Jet al. ClinLabGeneticist: a tool for clinical management of genetic variants from whole exome sequencing in clinical genetic laboratories Genome Med 201570177. Doi: 10.1186/s13073-015-0207-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ajzenberg H, Slaats G G, Stokman M Fet al. Non-invasive sources of cells with primary cilia from pediatric and adult patients Cilia 201548. Doi: 10.1186/s13630-015-0017-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Arts H H, Bongers E M, Mans D A et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48(06):390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 113.Davis E E, Zhang Q, Liu Q et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43(03):189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roberson E C, Dowdle W E, Ozanturk A et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. 2015;209(01):129–142. doi: 10.1083/jcb.201411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Diggle C P, Moore D J, Mali Get al. HEATR2 plays a conserved role in assembly of the ciliary motile apparatus PLoS Genet 20141009e1004577. Doi: 0.1371/journal.pgen.1004577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gilissen C, Hehir-Kwa J Y, Thung D Tet al. Genome sequencing identifies major causes of severe intellectual disability Nature 2014511(7509):344–347. [DOI] [PubMed] [Google Scholar]
- 117.Kundaje A, Meuleman W, Ernst Jet al. Integrative analysis of 111 reference human epigenomes Nature 2015518(7539):317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller N A, Farrow E G, Gibson Met al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases Genome Med 2015701100. Doi: 10.1186/s13073-015-0221-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saunders C J, Miller N A, Soden S Eet al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units Sci Transl Med 20124154154ra135. Doi: 10.1126/scitranslmed.3004041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones D, Fiozzo F, Waters B, McKnight D, Brown S. First-trimester diagnosis of Meckel-Gruber syndrome by fetal ultrasound with molecular identification of CC2D2A mutations by next-generation sequencing. Ultrasound Obstet Gynecol. 2014;44(06):719–721. doi: 10.1002/uog.13381. [DOI] [PubMed] [Google Scholar]
- 121.Stokowski R, Wang E, White K et al. Clinical performance of non-invasive prenatal testing (NIPT) using targeted cell-free DNA analysis in maternal plasma with microarrays or next generation sequencing (NGS) is consistent across multiple controlled clinical studies. Prenat Diagn. 2015;35(12):1243–1246. doi: 10.1002/pd.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Thung D T, Beulen L, Hehir-Kwa J, Faas B H. Implementation of whole genome massively parallel sequencing for noninvasive prenatal testing in laboratories. Expert Rev Mol Diagn. 2015;15(01):111–124. doi: 10.1586/14737159.2015.973857. [DOI] [PubMed] [Google Scholar]
- 123.Amor D J. Future of whole genome sequencing. J Paediatr Child Health. 2015;51(03):251–254. doi: 10.1111/jpc.12634. [DOI] [PubMed] [Google Scholar]