

Abstract
Research in the last decades has widely investigated the anti-oxidant properties of natural products as a therapeutic approach for the prevention and the treatment of oxidative-stress related disorders. In this context, several studies were aimed to evaluate the therapeutic potential of phytocannabinoids, the bioactive compounds of Cannabis sativa. Here, we examined the anti-oxidant ability of Cannabigerol (CBG), a non-psychotropic cannabinoid, still little known, into counteracting the hydrogen peroxide (H2O2)-induced oxidative stress in murine RAW264.7 macrophages. In addition, we tested selective receptor antagonists for cannabinoid receptors and specifically CB1R (SR141716A) and CB2R (AM630) in order to investigate through which CBG may exert its action. Taken together, our in vitro results showed that CBG is able to counteract oxidative stress by activation of CB2 receptors. CB2 antagonist pre-treatment indeed blocked the protective effects of CBG in H2O2 stimulated macrophages, while CB1R was not involved. Specifically, CBG exhibited a potent action in inhibiting oxidative stress, by down-regulation of the main oxidative markers (iNOS, nitrotyrosine and PARP-1), by preventing IκB-α phosphorylation and translocation of the nuclear factor-κB (NF-κB) and also via the modulation of MAP kinases pathway. On the other hand, CBG was found to increase anti-oxidant defense of cells by modulating superoxide dismutase-1 (SOD-1) expression and thus inhibiting cell death (results focused on balance between Bax and Bcl-2). Based on its anti-oxidant activities, CBG may hold great promise as an anti-oxidant agent and therefore used in clinical practice as a new approach in oxidative-stress related disorders.
Key words: RAW264.7 macrophages, immunohistochemistry, cannabigerol, oxidative stress, apoptosis
Introduction
The oxidative stress is induced by an imbalanced redox states, involving either excessive generation of reactive oxygen species (ROS) or dysfunction of the antioxidant system.1 It is widely recognized that oxidative stress is the major cause of development of many chronic diseases, such as aging, cancer, cardiovascular and neurodegenerative diseases.2-4 Several line of evidence have shown that the excessive production of ROS from metabolism or exogenous intake, can lead to damage of biomolecules, including lipids, proteins and nucleic acids, thereby affecting normal physiological functions.5 The brain is one of the organs especially vulnerable to the effects of ROS because of its high oxygen demand and its abundance of peroxidation-susceptible lipid cells.2
Over the last years, there has been an increasing attention to antioxidants from natural compounds without undesirable side effects and toxicity. In this context, phytocannabinoids the bioactive compounds of Cannabis sativa, that do not produce psychotropic effects, such as cannabidiol (CBD), cannabigerol (CBG), Δ9 tetrahydrocannabivarin (Δ9THCV) and cannabidivarin (CBDV) were considered of special interest as novel therapeutic agents especially in the treatment of diseases involving both inflammation and oxidative stress.6 Generally, cannabinoids exert many of their beneficial effects by binding cannabinoid (CB) receptors. To date, two types of receptors, both belonging to the family of G-protein coupled receptors (GPCRs), have been identified. CB1 receptors are expressed mainly on neurons and glial cells in various parts of the brain, CB2 receptors are found predominantly in the cells of immune system7. Moreover, there is increasing evidence supporting the existence of additional cannabinoid receptors (no-CB1 and no-CB2) in both central and peripheral system, identified in CB1 and CB2-knockout mice.8 To date, there are few studies about the pharmacological actions of CBG. The small body of in vitro evidence suggests that CBG exhibits a pharmacological profile relatively similar to cannabidiol (CBD) regarding its CB receptors activities,6,9 however its affinity for CB receptors has not been fully clarified. Although, CBG was proven to exert anti-inflammatory and anti-oxidant effects, the molecular mechanism of action is not yet known. Borrelli and co-authors10 demonstrated both of these properties in an experimental model of inflammatory bowel disease. CBG was found to reduce nitric oxide production and consequently to protect macrophages against oxidative stress by binding CB2 receptors.10 In addition, it was demonstrated that CBG was able to inhibit COX-2 enzymes in human colon adenocarcinoma cell line HT29.11 CBG was found also to suppress lipopolysaccharide-induced (LPS) release of pro-inflammatory cytokines and prostaglandin E2 in primary microglial cells.12 A recent study of 2015, suggests CBG as a potential candidate for pharmacological therapies in a experimental model of Huntington’s disease. Specifically, authors demonstrated that CBG by activating the peroxisome proliferator-activated receptor gamma (PPARγ) in striatal cells was able to alleviate symptomatology and neuroinflammation.13 Moreover, by using a murine model of Huntington’s disease, it was demonstrated that the treatment with VCE-003, a CBG quinone derivative, inhibited the upregulation of pro-inflammatory markers and improved antioxidant defenses in the brain through PPARγ activation.14
In the present study, we investigated whether treatment with purified CBG may counteract oxidative stress induced by hydrogen peroxide (H2O2) in a murine macrophage cells line. Specifically, we have chosen RAW 264.7 macrophages since it was widely demonstrated that following toxicant exposure, they are able to trigger mechanisms associated with over-production of pro-inflammatory mediators as well as over-production of ROS.15-17 Therefore, this cell line provides an excellent model for screening anti-oxidant drugs and consequent evaluation of inhibitors of the signaling pathways that lead to induction of oxidative mediators. In addition, in order to understand whether the inhibition of oxidative stress by CBG treatment could be mediated by CB receptors, we have pre-treated RAW 264.7 macrophages with selective receptor antagonists for cannabinoid receptors and specifically CB1R (SR141716A) and CB2R (AM630) before CBG administration.
Materials and Methods
Plant material
Cannabis sativa var. Carma was derived from a greenhouse cultivation at CREA-CIN, Rovigo (Italy), where a voucher specimen is kept. The plant material was collected in November 2010 and was supplied by Dr. Gianpaolo Grassi (CREA, Rovigo, Italy). The manipulation of the plant was done in accordance with its legal status (Authorization SP/101 of the “Ministero della Salute”, Rome, Italy).
General experimental procedures
1H NMR spectra were measured on JEOL ECP 300-300 MHz spectrometer. Chemical shifts were referenced to the residual solvent signal (CDCl3: δH 7.26). Reverse phase (RP) C-18 (POLYGOPREP 60-30 C18) was used for the removal of waxes and pigments. Silica gel 60 (70-230 mesh) was used for gravity column chromatography. CBG purification was monitored by TLC on Merck 60 F254 (0.25 mm) plates, which were visualized by UV inspection and/or spraying with 5% H2SO4 in ethanol and heating.
Extraction and isolation of CBG
One kg of dried, powdered flowered aerial parts were heated at 120°C for 2.5 h to decarboxylate precannabinoids and then extracted exhaustively with acetone (2x9 L) in a shaker. Removal of the solvent left a black resinous residue (74 g, 7.4%), which was dissolved in MeOH (30 mL/g of extract) and filtered through RP C-18 silica gel for the removal of waxes and pigments. The evaporation of methanol afforded 36 g of a dark-green extract that was further purified by gravity column chromatography on silica gel (75 g, petroleum ether-EtOAc, 8:2, as eluent) to afford 5 g of a yellow oil then crystallized with petroleum ether to give 3 g of pure CBG (0.3%). Pure CBG was stored at -8°C.
Cell culture conditions and drug treatment
The murine macrophage cell line RAW 264.7 was acquired from the “Centro substrati cellulari, Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia” (Italy). Macrophage cells were cultured in monolayer using RPMI-1640 medium (Carlo Erba, Cornaredo, Italy) containing 10% fetal bovine serum (FBS) (Sigma-Aldrich Co. Ltd., St. Louis, MO, USA). The cells were grown in logarithmic phase at 37°C in a moisturized atmosphere of 5% CO2 and 95% air. Experiments were performed with cells not exceeding 25 passages. In order to assess the potential cytotoxic effects of CBG, macrophage cells were grown in RPMI-1640 medium containing 10% of FBS until 70-80% confluence (4×105 cells/cm2) followed by 24 h of incubation at 37°C with CBG at following concentration range (μM): 2,5, 5, 10, 15, 20, 25 and 30. Moreover in order to evaluate the effect of H2O2 time-dependent, cells were stimulated with H2O2 (500 μM; Sigma-Aldrich) for 3, 6 and 9 h by adding directly H2O2 (500 μM; Sigma-Aldrich) into cell culture medium. Then, cells were either fixed or harvested for further analyses. For drug treatment, cells were grown until 70-80% confluence (4×105 cells/cm2) followed by 2 h pre-treatment with CBG at 10 μM dose and then cells were stimulated with H2O2 (500 μM) for 6 h by adding directly H2O2 into CBG-treated cell culture medium. For antagonists study, before CBG administration, cells were incubated for 2 h with different combinations of SR141716A (CB1 antagonist; 1 μM, Tocris Bioscience, Bristol, UK) or AM630 (CB2 antagonist; 100 nM, Tocris Bioscience) dissolved in 0.1% DMSO. Untreated cells (CTR) without H2O2 stimulation and RAW 264.7 treated with 0.1% DMSO (vehicle of CBG) were also included as controls. After H2O2 stimulation, the cells were either fixed or harvested for further analyses. All the experiments were made in triplicates and repeated for three independent times.
Eosin and hematoxylin staining
Morphological changes in the cells were assessed by eosin and hematoxylin staining (E&H) applying a standard protocol. Briefly, cells on coverslips (10 mm; Thermo Scientific, Darmstadt, Germany) were fixed with 4% paraformaldehyde (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 15 min at room temperature. After washing with 1X PBS, cells were stained with hematoxylin Harris (Bio-Optica, Milan, Italy) for 1 min, rinsed with tap water, stained with eosin (Bio-Optica) for 5 min, and rinsed with distilled water. Then, coverslips were dehydrated with ethanol series (Carlo Erba Reagents, Valde-Reuil, France; 50%, 70%, 80%, 96%, and 100%) and xylene (J.T. Baker, Deventer, The Netherlands). Coverslips were mounted on microscope slides using mounting medium (Eukitt, Carlo Erba Reagents) and were allowed to dry. Microscopy was performed using light microscope (Leica DM 2000 combined with Leica ICC50 HD camera). All images are representative of three independent experiments.
Immunocytochemistry
Cells on coverslips (10 mm; Thermo Scientific) were fixed with 4% paraformaldehyde at room temperature for 15 min followed by phosphate buffered saline (PBS, pH 7.5) washes. Then, cells were incubated with 3% hydrogen peroxide (H2O2) at room temperature for 15 min to suppress the endogenous peroxidase activity. Following three washes with PBS, cells were blocked with horse serum +0.1% Triton X-100 for 20 min followed by incubation for overnight at 4°C with primary antibodies against examined proteins:
anti-JNK(1:100 Santa Cruz Biotechnology);
anti-IκB-α (1:100 Cell Signaling Technology, Danvers, MA, USA);
anti-NF-kB (1:100 Cell Signaling Technology);
anti-Nitrotyrosine (1:250 Millipore, Billerica, MA, USA);
anti-PARP-1 (1:100 Santa Cruz Biotechnology);
anti-Bax (1:100 Santa Cruz Biotechnology);
anti-Bcl-2 (1:100 Santa Cruz Biotechnology).
After PBS wash, cells were incubated with biotinylated secondary antibody (1:200, Vector Laboratories, Burlingame, CA) and streptavidin AB Complex-HRP (ABC-kit from Dako, Glostrup, Denmark). The immunostaining was developed with the DAB peroxidase substrate kit (Vector Laboratories) (brown color; positive staining) and counterstaining with nuclear fast red (Vector Laboratories) (pink background; negative staining).
The immunocytochemical assays were repeated three times and each experimental group was plated in duplicate. Totally, were assessed 6 coverslips for each antibody. In order to calculate the percentage of positive cell stained, the images were captured by using a light microscopy (LEICA DM 2000 combined with LEICA ICC50 HD camera) with an objective of 40X and subjected to densitometric analysis by using the software LEICA Application Suite ver. 4.2.0. Quantitative analysis was performed on 6 coverslips by covering about 90% of total area.
Protein extraction and western blot analysis
Cells were harvested following 6 h of incubation with H2O2. After washing with ice-cold PBS, the cells were lysed using buffer A [320 mM sucrose, 10 mM, 1 mM EGTA, 2 mM EDTA, 5 mM NaN3, 50 mM NaF, β-mercaptoethanol, and protease/ phosphatase inhibitor cocktail (Roche Molecular Systems, Inc., Branchburg, NJ, USA)] in ice for 15 min, followed by centrifugation at 1000 g for 10 min at 4°C. The supernatant was served as cytosolic extract. The pellet was further lysed using buffer B [150 mM NaCl, 10 mM Tris-HCl (pH 7.4), 1 mM EGTA, 1 mM EDTA, Triton x-100, and protease/phosphatase inhibitor cocktail (Roche)] in ice for 15 min, followed by centrifugation at 15,000 g for 30 min at 4°C. The supernatant was collected and used as nuclear extract. Protein concentrations were calculated using the Bradford assay (Bio-Rad, Hercules, CA, USA). Twenty micro-grams of proteins were heated for 5 min at 95°C, resolved by 8% or 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a PVDF membrane (Immobilon-P, Millipore). Membranes were blocked in 5% skim milk in PBS for 1 h at room temperature followed by overnight incubation at 4°C with particular primary antibodies. The following primary antibodies were used: iNOS (1:500; Cell Signaling Technology) and SOD-1 (1:1000; Abcam, Cambridge, UK). Then, membranes were washed in PBS 1X and incubated with HRP-conjugated anti-mouse or rabbit IgG secondary antibody (1:2000; Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. To ascertain that blots were loaded with equal amounts of protein lysates, they were also incubated with antibody for GAPDH HRP Conjugated (1:1000; Cell Signaling Technology). The relative expression of protein bands was visualized using an enhanced chemiluminescence system (Luminata Western HRP Substrates, Millipore) and protein bands were acquired and quantified with ChemiDoc™ MP System (Bio-Rad) and a computer program (ImageJ software) respectively. All blots are representative of three independent experiments.
Statistical data analysis
Statistical analysis of immunocytochemistry was performed using GraphPad Prism version 6.0 computer software program (GraphPad Software, La Jolla, CA, USA). The data were statistically analyzed by one-way ANOVA test and Bonferroni post-hoc test for multiple comparisons. A P-value less than or equal to 0.05 was considered statistically significant. Results are reported as the mean ± SEM of N experiments.
Results
Morphological assessment in H2O2 stimulated RAW 264.7 treated with CBG
To investigate the cytotoxicity of CBG we treated the normal macrophage cells with CBG for 24 h at the following concentrations (μM): 2,5, 5, 10, 15, 20, 25 and 30. We found that treatment with CBG at the concentrations range from 2,5 to 10 μM was not cytotoxic for the cells and did not cause cell death. Otherwise, starting from 15 until to 30 μM concentrations, CBG induced cell death in a dose dependent manner (Figure 1). Therefore, we have chosen 10 μM as optimal dose. Moreover, RAW 264.7 incubated with H2O2 (500 μM) showed only less cell death after 3 h (Figure 2A). A severe cell death was noticed both at 6 h that at 9 h (Figure 2 B,C). Since no differences in cell death were found, we decided to incubate the cells with H2O2 (500 μM) for 6 h to obtain a better management of the entire experiment. By eosin and hematoxylin staining we found that the DMSO, vehicle of CBG, was not cytotoxic for macrophage cells treated with it (Figure 3B). DMSO-treated cells displayed indeed normal morphology, indicated by the presence of ruffles as CTR cells (Figure 3A). On the contrary, morphological changes, including presence of surface smooth and shrinkage, were observed in H2O2 stimulated RAW 264.7 macrophages (Figure 3C). Treatment with CBG instead attenuated these H2O2 triggered morphological features (Figure 3D). In addition, by looking to the possible involvement of cannabinoid receptors CB1 and CB2, our data demonstrated that CBG acts via CB2R. More in detail, pre-treatment with AM630 (CB2 antagonist) blocked the protective effect of CBG in H2O2-stimulated macrophages (Figure 3E), while SR141716A (CB1 antagonist) did not show inhibition towards the protective role of CBG (Figure 3F).
Figure 1.

Cytotoxicity of CBG. Eosin/hematoxylin (E&H) staining of untreated RAW 264.7 cells (CTR) (A), incubated with DMSO (B), with CBG 2,5 µM (C), 5 µM (D), 10 µM (E), 15 µM (F), 20 µM (G), 25 µM (H) and 30 µM (I).
Figure 2.
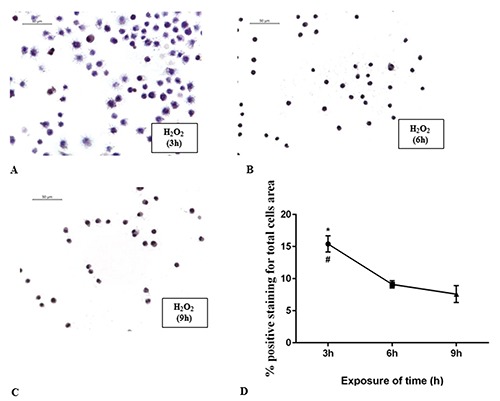
Curve of cytotoxicity of H2O2. RAW 264.7 cells incubated with H2O2 (500 µM) for 3 h (A), 6 h (B) and 9 h (C). The graph (D) shows the percentage of positive staining for total cells area. *P<0.0321 3 h vs 6 h; #P=0.0174 3 h vs 9 h.
Figure 3.
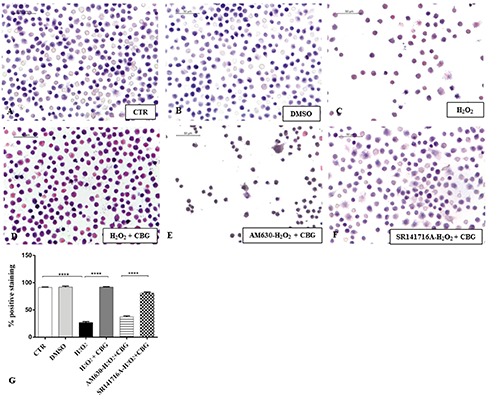
Eosin/hematoxylin (E/H) staining. Eosin/hematoxylin (E/H) staining of untreated RAW 264.7 cells (CTR) (A), incubated with DMSO (B), H2O2 (C), H2O2 + CBG (D), AM630+ H2O2 + CBG (E) and SR141716A + H2O2 + CBG (F). The percentage of positive staining was showed in graph (G), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A.
Effects of CBG treatment on MAPK signal-transduction pathway
By immunocytochemical analysis, we investigated the expression of JNK, a mitogen-activated protein kinase (MAPK) that plays a key role in relaying extracellular signals from the cell membrane to the nucleus via a cascade of phosphorylation events.18 As known, MAPKs can be activated also by the ROS production.19 Our results confirmed a positive cytoplasmic immunolocalization for JNK in macrophages stimulated with H2O2 (Figure 4C). On the contrary, cells treated with CBG showed negative cytoplasmic staining for JNK (Figure 4D), as observed in CTR cells (Figure 4A) and DMSO cells (Figure 4B). In addition, our results suggest that JNK modulation involved solely CB2R activation. Pre-treatment with CB2 antagonist (AM630) indeed blocked the protective effect of CBG in H2O2 stimulated macrophages as proven by positive immunolocalization for JNK (Figure 4E). Contrariwise, administration of CB1 antagonist (SR141716A) exhibited negative staining for JNK (Figure 4F, see densitometric analysis in Figure 4G), suggesting in this way that CB1R was not involved.
Figure 4.
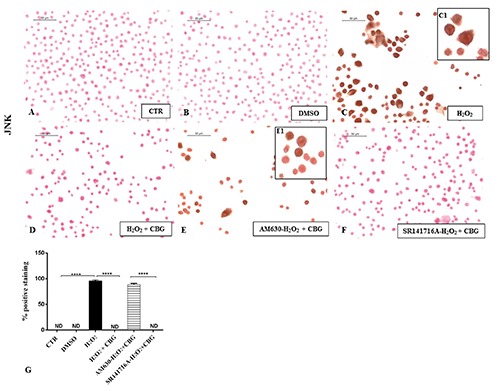
Immunocytochemical analysis for JNK in untreated RAW 264.7 cells (CTR) (A), incubated with DMSO (B), H2O2 (C, 40x; C1, 100x), H2O2 + CBG (D), AM630+ H2O2 + CBG (E, 40x; E1, 100x) and SR141716A + H2O2 + CBG (F). Densitometric analysis for JNK (G), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A; ND not detectable.
Effects of CBG treatment on IκB-α degradation and NF-κB activation in H2O2 stimulated RAW 264.7 treated with CBG
NF-kB is a dimeric transcription factor present in the cytoplasm of cells in an inactive form due to its association with a class of inhibitory proteins called IκBs. Following different stimulus effects, like oxidative stress, IκB undergoes phosphorylation and subsequently degraded, allowing NF-κB to translocate into the nucleus and induce gene expression.20 As expected, a positive cytoplasmic staining for IκB-α and a in parallel a negative nuclear staining for NF-κB was observed in CTR cells (Figure 5 A,G) as well as in DMSO group (Figure 5 B,H). Upon stimulation with H2O2, cells did not stain for IκB-α in cytoplasm (Figure 5C), but showed a marked nuclear positive staining for NF-κB (Figure 5I). CBG administration significantly increased the degree of cytoplasmic positive staining for IκB-α (Figure 5D) and reduced nuclear positive staining for NF-κB in H2O2-stimulated cells (Figure 5J). Moreover, the protection of CBG can be attributed only to CB2R. The pre-treatment with AM630 (Figure 5 E, K) indeed was found to block the efficacy of CBG, restoring a condition similar to that observed in cell incubated with H2O2. Pre-treatment with SR141716A (Figure 5 F,L, see densitometric analysis in Figure 5 M,N) on the contrary did not altered the efficacy of the CBG treatment, suggesting that CB1R did not affect this signaling.
Figure 5.
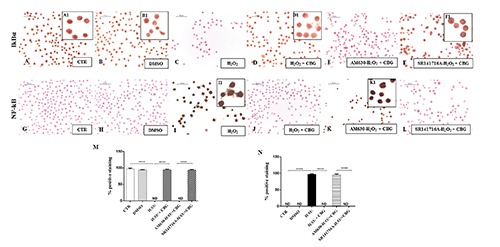
Immunocytochemical analysis for IκB-α in untreated RAW 264.7 cells (CTR) (A, 40x; A1, 100x), incubated with DMSO (B, 40x; B1, 100x), H2O2 (C), H2O2 + CBG (D, 40x; D1, 100x), AM630+ H2O2 + CBG (E) and SR141716A + H2O2 + CBG (F, 40x; F1, 100x). Immunocytochemical analysis for NF-κB in untreated RAW 264.7 cells (CTR) (G), incubated with DMSO (H), H2O2 (I, 40x; I1, 100x), H2O2 + CBG (J), AM630+ H2O2 + CBG (K, 40x; K1, 100x) and SR141716A + H2O2 + CBG (L). Densitometric analysis for IκB-α (M), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A. Densitometric analysis for NF-κB (N), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A.
CBG inhibits the production of oxidative stress markers in H2O2 stimulated RAW 264.7
We assessed the anti-oxidative effects of CBG by evaluating the expression of some oxidative damage markers in the mouse RAW 264.7 cells stimulated with H2O2. Immunocytochemistry results showed negative staining for nitrotyrosine in CTR cells (Figure 6A) as well as in DMSO cells (Figure 6B). Conversely, a dense positive staining for nitrotyrosine in H2O2-stimulated cells was found (Figure 6C). CBG administration significantly inhibited the expression of nitrotyrosine (Figure 6D). Positive staining for macrophages pre-treated with AM630 (Figure 6E) and in concert a negative staining for SR141716A pretreated ones (Figure 6F, see densitometric analysis in Figure 6G) in the same conditions, demonstrated the involvement only of CB2R and not of CB1R in the protective effects exhibited by CBG. In addition, Western blot data showed enhanced expression of another oxidative stress marker - iNOS - in H2O2-stimulated cells, while CBG treatment significantly reduced its expression (Figure 7H).
Figure 6.
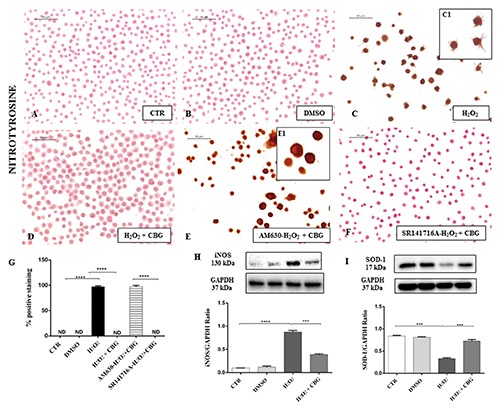
Immunocytochemical analysis for nitrotyrosine in untreated RAW 264.7 cells (CTR) (A), incubated with DMSO (B), H2O2 (C, 40x; C1, 100x), H2O2 + CBG (D), AM630+ H2O2 + CBG (E, 40x; E1, 100x) and SR141716A + H2O2 + CBG (F). Densitometric analysis for nitrotyrosine (G), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A. Western blot analysis for iNOS (H), ****P<0.0001 CTR vs H2O2; ***P=0.002 H2O2 vs H2O2 + CBG. Western blot analysis for SOD-1 (I), ***P=0.0001 CTR vs H2O2; ***P=0.003 H2O2 vs H2O2 + CBG.
Figure 7.
Immunocytochemical analysis for PARP-1 in untreated RAW 264.7 cells (CTR) (A), incubated with DMSO (B), H2O2 (C, 40x; C1, 100x), H2O2 + CBG (D), AM630+ H2O2 + CBG (E, 40x; E1, 100x) and SR141716A + H2O2 + CBG (F). Densitometric analysis for PARP-1 (G), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A.
As a result of an increased production of radical species, cells have developed enzymatic cellular defense systems in the effort to detoxify these molecules and regenerate balanced redox homeostasis.21 Therefore, we investigated the expression of SOD-1, chosen as anti-oxidant defense marker. Western blot analysis showed a basal expression of SOD-1 in CTR and DMSO cell groups. H2O2 stimulated RAW 264.7 showed a reduced level expression of SOD-1, increased instead by administration of CBG (Figure 6I). Moreover, among the first cellular responses to DNA damage is the production of Poly (ADP-ribose) polymerase (PARP), a marker of oxidative stress synthesized in large quantities by PARP enzymes, whose activity increases when cells are subjected to damage.22 By immunocytochemical evaluation we assessed the presence of nuclear PARP-1 activation, showing negative nuclear staining for PARP-1 in CTR cells and DMSO cells (Figure 7 A,B, respectively). Conversely, a dense nuclear positive staining for PARP-1 observed in H2O2-stimulated cells (Figure 7C), was totally deleted by CBG administration (Figure 7D). Anti-oxidant effects exhibited by CBG are due to involvement of CB2R and not of CB1R. Immunocytochemical results obtained from cells pre-treated with AM630 (Figure 7E) indeed were compatible with what has been observed in cell incubated only with H2O, suggesting that use of CB2R antagonist blocked the activity of CBG. Pre-treatment with SR141716A (Figure 7F, see densitometric analysis in Figure 7G) on the contrary did not affect the anti-oxidant effects of CBG, thus we can assess that CB1R was not involved in this cellular signaling.
CBG regulates apoptosis pathway in H2O2 stimulated RAW 264.7
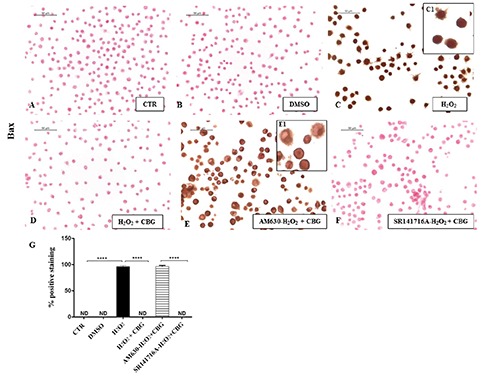
Finally, as the close link between ROS and apoptosis is well known, we evaluated whether CBG could potentially exert apoptosis regulatory functions in RAW cells stimulated with H2O2. By immunocytochemical analysis, we noticed a completely negative staining for Bax and a in parallel a notable positive staining for Bcl-2 in CTR cells (Figures 8A and 9A) as well as in DMSO ones (Figures 8B and 9B). On the contrary, upon H2O2 stimulation, macrophage cells showed positive staining for Bax and negative staining for Bcl-2 (Figures 8C and 9C). Moreover, CBG exhibited a significant ability in protecting the unbalance between Bax/Bcl-2 (Figures 8D and 9D). Also in this case, by pre-treating cells with the cannabinoid receptor antagonists, we found that Bax/Bcl2 modulation is only mediated by the CB2R, not by CB1R (Figures 8 F and 9F, see densitometric analysis in Figures 8 and 9G). The use of CB2R antagonist (AM630) blocked the protective effects of CBG as proven by positive immunolocalization for Bax (Figure 8E) and negative staining for Bcl2 (Figure 9E). Administration of CB1 antagonist, in contrast, exhibited negative staining for Bax (Figure 8F) with consequent positive staining for Bcl2 (Figure 9F, see densitometric analysis in Figures 8G and 9G).
Figure 8.
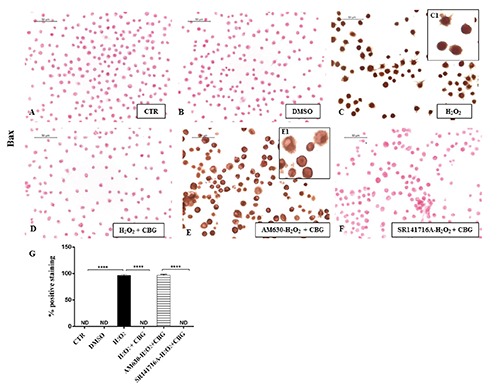
Immunocytochemical analysis for Bax in untreated RAW 264.7 cells (CTR) (A), incubated with DMSO (B), H2O2 (C, 40x; C1, 100x), H2O2 + CBG (D), AM630+ H2O2 + CBG (E, 40x; E1, 100x) and SR141716A + H2O2 + CBG (F). Densitometric analysis for Bax (G). ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A.
Figure 9.
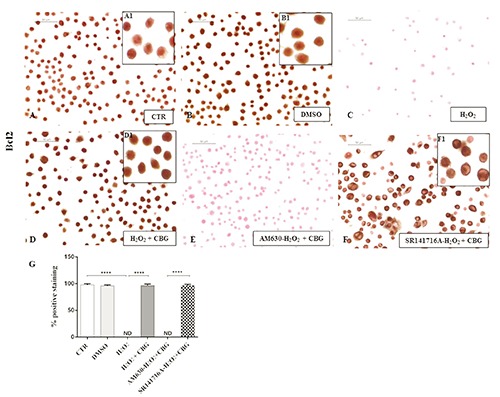
Immunocytochemical analysis for Bcl2 in untreated RAW 264.7 cells (CTR) (A, 40x; A1, 100x), incubated with DMSO (B, 40x; B1, 100x), H2O2 (C), H2O2 + CBG (D, 40x; D1, 100x), AM630+ H2O2 + CBG (E) and SR141716A + H2O2 + CBG (F, 40x, F1, 100x). Densitometric analysis for Bcl2 (G), ****P<0.0001 CTR vs H2O2; ****P<0.001 H2O2 vs H2O2 + CBG; ****P<0.001 AM630 vs SR141716A.
Discussion
Oxidative stress was first characterized by Sies23 as “a disturbance in the pro-oxidant to anti-oxidant balance in favor of the oxidant species, leading to potential damage”. Oxidative stress is a crucial mechanism of the aging that can cause direct injury to the central nervous system. Free radicals affect indeed both the structure and function of neural cells, playing a key role in to a wide range of neurodegenerative diseases, such as Parkinson’s disease and Alzheimer’s disease.24 There is currently a debate whether oxidative stress is a cause or a consequence of these disorders, mostly due to a lack of understanding of the mechanisms by which reactive oxygen species (ROS) act in both normal physiological and disease conditions.25 Indeed, high concentrations of ROS can lead to impaired physiological functions through cellular damage of DNA, proteins, phospholipids, and other macromolecules.26 On the other hand, low doses of ROS are essential to cell signaling and regulation.27
In the last decades, a growing interest was aimed to identifying pharmacological inhibitors of oxidative stress mediators as a potential therapeutic strategy for preventing diseases correlated with oxidative stress. In this context, cannabinoids, the bioactive compounds of Cannabis sativa, exhibited a potent action in inhibiting oxidative and nitrosative stress, modulating the expression of inducibile nitric oxide synthase (iNOS) and reducing the production of ROS.28,29 In agreement with these findings, we found that CBG, a non-psychotropic phytocannabinoid yet little known, was able to counteract oxidative stress in mouse macrophage cell line RAW264.7 stimulated with H2O2 by activation of CB2 receptors.
Upon stimulation, macrophage cells are involved in host defense and also they are able to increase the production of reactive oxygen and nitrogen species dramatically for a short period of time.15 Our data showed that the stimulation of macrophages with H2O2 induced cell morphological changes, such as appearance of surface smooth and shrinkage, reduced by CBG administration. It is well known that cannabinoids can exert multiple pharmacological effects via CB1/CB2 receptors. As regards CB1R, it is not directly implied in the mechanism by which cannabinoids are able to protect cells from oxidative stress in vitro.30 Conversely, as proven by Borrelli et al.,10 CB2R is involved in counteracting oxidative stress. Here, by pre-treating macrophages with selective receptor antagonists for CB1R (SR141716A) and CB2R (AM630), we clearly confirmed that CBG acts via CB2R. CB2 antagonist pre-treatment indeed blocked the protective effect of CBG in H2O2 stimulated macrophages, while CB1R was not involved. This result is not surprising considering that the CB1 receptors are localized mainly in the central nervous system,31 while CB2 receptors are expressed predominantly in cells of the immune system and hematopoietic.32
To date, the molecular mechanism by which ROS cause oxidative damage is poorly characterized. Several line of evidence showed that H2O2 can induce or mediate the activation of the mitogen-activated protein kinases (MAPK).33-36 The MAPKs, a family of ubiquitous proline-directed, protein-serine/threonine kinases, including extracellular signal-regulated kinases 1 and 2 (ERK1/2), p38, and c-Jun amino-terminal kinase (JNK), play an essential role in sequential transduction of biological signals from the cell membrane to the nucleus.37 In agreement with these findings, we reported direct evidence showing that stimulation with H2O2 induced increased expression of JNK in H2O2-stimulated macrophages, whereas CBG administration by activating CB2R reduced significantly its expression.
Moreover, it is widely accepted that MAP kinases can participate in the regulation of NF-κB transcriptional activity, by mediating downstream phosphorylation of IκB-α and subsequent activation of NF-κB.38 NF-κB is generally inactive in cytoplasm due to the inhibition of IκB-α. In response to a wide range of stimuli, including ROS production, IκB-α is phosphorylated by the enzyme IκB-α kinase, so that NF-κB is free to translocate into the nucleus and to promote the expression of a wide range of mediators involved not only in oxidative stress pathway, but also in inflammation and apoptosis molecular mechanisms. By immunocytochemical analysis we observed a negative immunolocalization for IκB-α and conversely dense positive immunolocalization for NF-κB in H2O2-stimulated macrophages. In attempting to coordinate the responses of macrophages against oxidative stimuli, CBG led to up-regulation of IκB-α and consequently down-regulation of NF-κB. CB2-dependent effects of CBG could be involved in upstream events that could affect intracellular oxidative pathways. Furthermore, upon H2O2 stimulation, macrophages exhibit excessive accumulation of both nitric oxide (NO) and superoxide anion (O2-), whose interaction results in formation of toxic peroxynitrite (ONOO–). Our results showed increased expression of two principal oxidative markers - iNOS and nitrotyrosine - in H2O2-stimulated macrophages, inhibited instead by treatment with CBG. Our results are in accordance with the literature which reported that cannabidiol, another non-psychotropic cannabinoid, by blocking upstream MAP kinase and the transcription factor NF-κB activation, can suppress iNOS expression and NO production.28 Specifically, it was demonstrated that inhibition of MAP kinase pathway was due to CB2R activation.39-41 Coherently with these data, we have found that CBG counteracts oxidative stress by inhibiting some key oxidative mediators via a CB2R dependent mechanism. Moreover, as a result of an increased production of radical species, cells have developed enzymatic cellular defense systems, such as SOD, catalase, glutathione peroxidase, in the effort to detoxify these molecules and regenerate balanced redox homeostasis.
Here, we investigated the expression of SOD-1, that regulates oxidative response genes involved in resistance to oxidative stress and DNA damage repair.42 Our data showed that stimulation with H2O2 in RAW264.7 cells caused a decrease of SOD-1 expression, restored by CBG administration. In this cellular cascade, another critical event is poly(ADP-ribose) polymerase-1 (PARP-1) activation following DNA damage.22 Continuous or excessive activation of PARP resulted in a substantial depletion of ATP, leading to cellular dysfunction and cell death.43 Here, we demonstrated that treatment with CBG inhibited PARP-1 activation in CB2R dependent manner. Moreover, radical species production is implicated in the progression of oxidative stress-related apoptosis and cell death.44 Finally, we evaluated the role of CBG in counteracting cell death by looking to the main apoptosis-regulatory genes, such Bax and Bcl-2. Immunocytochemistry evaluation showed positive staining for Bax in H2O2-stimulated macrophages, inhibited by treatment with CBG. In parallel, it was found a negative staining of Bcl-2 in H2O2-stimulated macrophages, and conversely a positive staining in CBG-treated ones. We have also established that the anti-apoptotic effect of CBG are mediated by the CB2R. These results are corroborated by other studies which have showing that CB2R activation correlated with reduced apoptosis.45 Taken together, our results suggest CBG as a new relevant helpful approach to use in clinical practice for oxidative stress-related disorders. CBG may effectively scavenge free radicals, increase anti-oxidant activity of cells, by modulating multiple signaling pathways such as MAPK kinases and NF-kB translocation and lastly inhibiting cell death. Moreover, by looking to specific antagonists of the CB1 and CB2 receptors, we found that during cellular processes CBG administered in H2O2-stimulated macrophages, compared with control cells, exerts negative feedback towards the expression of proteins investigated by immunocytochemistry analyses. The increase in positive signal in the H2O2-stimulated macrophages treated with AM630, antagonist of CB2R, is due to an important positive feedback effect in cellular processes.
Acknowledgments
The authors would like to thank Prof. Appendino Giovanni (University of Eastern Piedmont "Amedeo Avogadro", Novara, Italy), for his technical assistance and precious contribution during manuscript drafting.
Funding Statement
Funding: this study was supported by current research funds 2016 of IRCCS "Centro Neurolesi Bonino-Pulejo", Messina, Italy.
References
- 1.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 2002;82:47-95. [DOI] [PubMed] [Google Scholar]
- 2.Kim GH, Kim JE, Rhie SJ, Yoon S. The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol 2015;24:325-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pham-Huy LA, He H, Pham-Huy C. Free radicals, antioxidants in disease and health. Int J Biomed Sci 2008;4:89-96. [PMC free article] [PubMed] [Google Scholar]
- 4.Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, Me LL. Oxidative stress and cancer: an overview. Ageing Res Rev 2013;12:376-90. [DOI] [PubMed] [Google Scholar]
- 5.Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 2004;10:1611-26. [DOI] [PubMed] [Google Scholar]
- 6.Hill AJ, Williams CM, Whalley BJ, Stephens GJ. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol Ther 2012;133:79-97. [DOI] [PubMed] [Google Scholar]
- 7.Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev 2010;62:588-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckley NE. The peripheral cannabinoid receptor knockout mice: an update. Br J Pharmacol 2008;153:309-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izzo AA, Borrelli F, Capasso R., Di Marzo V, Mechoulam R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci 2009;30:515-27 [DOI] [PubMed] [Google Scholar]
- 10.Borrelli F, Fasolino I, Romano B, Capasso R, Maiello F, Coppola D, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol 2013;85:1306-16. [DOI] [PubMed] [Google Scholar]
- 11.Ruhaak LR, Felth J, Karlsson PC, Rafter JJ, Verpoorte R, Bohlin L. Evaluation of the cyclooxygenase inhibiting effects of six major cannabinoids isolated from Cannabis sativa. Biol Pharm Bull 2011;34:774-8. [DOI] [PubMed] [Google Scholar]
- 12.Granja AG, Carrillo-Salinas F, Pagani A, Gómez-Cañas M, Negri R, Navarrete C, et al. A cannabigerol quinone alleviates neuroinflammation in a chronic model of multiple sclerosis. J Neuroimmune Pharmacol 2012;7:1002-16. [DOI] [PubMed] [Google Scholar]
- 13.Valdeolivas S, Navarrete C, Cantarero I, Bellido ML, Munoz E, Sagredo O. Neuroprotective properties of cannabigerol in Huntington’s disease: studies in R6/2 mice and 3-nitropropionate-lesioned mice. Neurotherapeutics 2015;12:185-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Díaz-Alonso J, Paraíso-Luna J, Navarrete C, Del Río C, Cantarero I, Palomares B, et al. VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Sci Rep 2016;6:29789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gieche J, Mehlhase J, Licht A, Zacke T, Sitte N, Grune T. Protein oxidation and proteolysis in RAW264.7 macrophages: effects of PMA activation. Biochim Biophys Acta 2001;1538:321-8. [DOI] [PubMed] [Google Scholar]
- 16.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014;20:1126-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol 2011;51:267-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev 2008;60:261-310. [DOI] [PubMed] [Google Scholar]
- 19.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct 2011;2011:792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makarov SS. NF-kappaB as a therapeutic target in chronic inflammation: recent advances. Mol Med Today 2000;6:441-8. [DOI] [PubMed] [Google Scholar]
- 21.B Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J 2012;5:9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong WM, Cortes U, Wang ZQ. Poly(ADP-ribose) polymerase: a guardian angel protecting the genome and suppressing tumorigenesis. Biochim Biophys Acta 2001;1552:27-37. [DOI] [PubMed] [Google Scholar]
- 23.Sies H. Oxidative stress: from basic research to clinical application. Am J Med 1991;91:S31-8. [DOI] [PubMed] [Google Scholar]
- 24.Jiang T, Sun Q, Chen S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog Neurobiol 2016; 147:1-19. [DOI] [PubMed] [Google Scholar]
- 25.Pitzschke A, Hirt H. Mitogen-activated protein kinases and reactive oxygen species signaling in plants. Plant Physiol 2006;141:351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 2011;7:504-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med 2007;42:926-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esposito G, De Filippis D, Maiuri MC, De Stefano D, Carnuccio R, Iuvone T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci Lett 2006;399:91-5. [DOI] [PubMed] [Google Scholar]
- 29.Booz GW. Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radic Biol Med 2011;51: 1054-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C. Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem 2002;80: 448-56. [DOI] [PubMed] [Google Scholar]
- 31.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990;346:561-4. [DOI] [PubMed] [Google Scholar]
- 32.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993;365:61-5. [DOI] [PubMed] [Google Scholar]
- 33.Konishi H, Tanaka M, Takemura Y, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci USA 1997;94:11233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal 2006;8:1775-89. [DOI] [PubMed] [Google Scholar]
- 35.Ruffels J, Griffin M, Dickenson JM. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: role of ERK1/2 in H2O2-induced cell death. Eur J Pharmacol 2004;483:163-73. [DOI] [PubMed] [Google Scholar]
- 36.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. BioFactors 2003;17:287-96. [DOI] [PubMed] [Google Scholar]
- 37.Brown MD, Sacks DB. Protein scaffolds in MAP kinase signalling. Cell Signal 2009;21:462-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schulze-Osthoff K, Ferrari D, Riehemann K, Wesselborg S. Regulation of NF-kappa B activation by MAP kinase cascades. Immunobiology 1997;198:35-49. [DOI] [PubMed] [Google Scholar]
- 39.Stefano GB, Liu Y, Goligorsky MS. Cannabinoid receptors are coupled to nitric oxide release in invertebrate immunocytes, microglia, and human monocytes. J Biol Chem 1996;27: 19238-42. [DOI] [PubMed] [Google Scholar]
- 40.Ross RA, Brockie HC, Pertwee RG. Inhibition of nitric oxide production in RAW264.7 macrophages by cannabinoids and palmitoylethanolamide. Eur J Pharmacol 2000;401:121-30. [DOI] [PubMed] [Google Scholar]
- 41.Merighi S, Gessi S, Varani K, et al. Cannabinoid CB(2) receptors modulate ERK-1/2 kinase signalling and NO release in microglial cells stimulated with bacterial lipopolysaccharide. Br J Pharmacol 2012;165:1773-88. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XFS. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun 2014;5:3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci 2002;23:122-9. [DOI] [PubMed] [Google Scholar]
- 44.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem 1996;271:4138-42. [DOI] [PubMed] [Google Scholar]
- 45.Fujii M, Sherchan P, Soejima Y, Hasegawa Y, Flores J, Doycheva D, et al. Cannabinoid receptor type 2 agonist attenuates apoptosis by activation of phosphorylated CREB-Bcl-2 pathway after subarachnoid hemorrhage in rats. Exp Neurol 2014;261:396-403. [DOI] [PMC free article] [PubMed] [Google Scholar]