

Abstract
Cisplatin is an important antitumor agent, but its clinical utility is often limited by multifactorial mechanism of resistance. Loss of tumor suppressor p53 function is a major mechanism, affected by either mutation in the DNA binding domain or dysregulation by overexpression of p53 inhibitors MDM2 and MDM4 that destabilize p53 by increasing its proteosomal degradation. In the present study, cisplatin-resistant 2780CP/Cl-16 ovarian tumor cells expressed a heterozygous, temperature-sensitive p53V172F mutation, which reduced p53 half-life by 2- to 3-fold compared to homozygous wild-type p53 in parental A2780 cells. Although reduced p53 stability in 2780CP/Cl-16 cells was associated with moderate cellular overexpression of MDM2 or MDM4 (<1.5-fold), their binding to p53 was substantially enhanced (5- to 8-fold). The analogous cisplatin-resistant 2780CP/Cl-24 cells, which express loss of p53 heterozygosity, retained the p53V172F mutation and high p53-MDM4 binding, but demonstrated lower p53-bound MDM2 that was associated with reduced p53 ubiquitination and enhanced p53 stability. The inference that p53 was unstable as a hetromeric p53wt/p53V172F complex was confirmed in 2780CP/Cl-24 cells transfected with wild-type (wt) p53 or multimer-inhibiting p53L344P mutant, and further supported by normalization of p53 stability in both resistant cell lines grown at the permissive temperature of 32.5°C. Surprisingly, in 2780CP/Cl-16 and 2780CP/Cl-24 models, cisplatin-induced transactivity of p53 was attenuated at 37°C, and this correlated with cisplatin resistance. However, downregulation of MDM2 or MDM4 by siRNA in either resistant cell line induced p53 and restored p21 transactivation at 37°C, as did cisplatin-induced DNA damage at 32.5°C that coincided with reduced p53-MDM4 binding and cisplatin resistance. These results demonstrate that cisplatin-mediated p53V172F mutation regulates p53 stability at the normothermic temperature, but it is the increased recruitment of MDM4 by the homomeric or heteromeric mutant-p53V172F complex that inhibits p53-dependent transactivation. This represents a novel cellular mechanism of p53 inhibition and, thereby, induction of cisplatin resistance.
Keywords: p53, MDM2, MDM4, Cisplatin resistance, Stability, V172F mutation
INTRODUCTION
Cisplatin is a clinically effective antitumor drug, demonstrating substantial activity against a number of cancers. It is a DNA damaging agent, and its dependence on the p53 tumor suppressor for potent activity is well acknowledged.1 This is consistent with the critical role of p53 as a tetrameric transcription factor in cell cycle arrest, senescence and apoptosis. In an unstressed environment, however, p53 is prevented from transactivating target genes by its interaction with MDM2 and the closely-related MDM4 proteins.2 Both MDM2 and MDM4 can bind independently to p53 through their N-termini, but can also bind to each other to form heterodimers through the C-terminal RING domain. The interaction between MDM2 and MDM4 is essential for E3 ligase activity of MDM2 and, thereby, inducing proteosomal degradation of p53 and maintaining its levels below a critical threshold for activity.3,4 Moreover, the qualitative nature of negative regulation of p53 is dependent on MDM2 and MDM4 levels, with relatively greater levels of MDM4 inhibiting MDM2-mediated p53 degradation and/or competing with MDM2 for the p53 binding site to inhibit its transcription activity.3,5 DNA damage signals, however, can inhibit interaction between p53 and MDM2 or MDM4, and accelerate MDM4 degradation by MDM2, to stabilize and transcriptionally activate p53.6–8
The critical function of p53 in facilitating antitumor drug response can be severely compromised by its mutation in the DNA-binding domain, and this is generally associated with poor clinical outcome.1 Significantly, p53 can also be inhibited at the wild-type level through cellular overexpression of MDM2 or MDM4 that also results in loss of cytotoxic response to several antitumor agents, including cisplatin.1,2,9–11 However, in a number of drug resistant cancers harboring wild-type p53, overexpression of these negative regulators is not observed.1 Cisplatin-resistance is of critical importance and has been widely studied in models derived from cisplatin-sensitive, wild-type p53 A2780 human ovarian tumor cells; in one such derived cisplatin-resistant model, 2780CP, we have similarly reported that p53-dependent transactivation of p21 with cisplatin was severely attenuated even though expression of MDM2 was not elevated.12 On the other hand, 2780CP cells and analogous, independently-derived cisplatin-resistant A2780/CP70 cells harbor a temperature-sensitive V172F mutation in the DNA binding domain of human p53 in one allele and wild-type p53 in the other,13,14 and it is plausible that such a mutation may be responsible for attenuating cisplatin response, but this has not been investigated previously. Importantly, an identical p53 mutation has been found in human cervical KB-CP20 tumor cells derived from exposing wild-type p53 KB-3-1 cells to cisplatin,15 and this suggests that the V172F mutation may select for cisplatin resistance. Therefore, examination of this p53 mutation to better understand the mechanism of cisplatin resistance has assumed greater significance. Interestingly, in one reported study with A2780/CP70 cells, the half-life of basal p53 was reduced three-fold as compared to p53 in parental A2780 cells.16 In the present study, therefore, we have examined whether p53 destabilization, the V172F mutation or another mechanism is responsible for attenuation of p53 function and contributes to cisplatin resistance. Our results demonstrate that mutant p53V172F is potentially functional, but the heterozygous p53 mutation enhances binding of the p53 heteromer to MDM2 and MDM4 that in turn increases ubiquitination and destabilization of p53. However, increased binding of MDM4 to p53, rather than increased degradation of the tumor suppressor, is the mechanism responsible for inhibiting p53-dependent transactivation function and inducing cisplatin resistance.
RESULTS
Heterozygous p53 gene status enhances p53 destabilization
The increased degradation rate of p53 in cisplatin-resistant A2780/CP70 ovarian tumor cells in comparison to parental A2780 cells has been noted by Yazlovitskaya et al.16 In order to assess whether the analogous cisplatin-resistant 2780CP/Cl-16 cell line developed independently by us also demonstrates this characteristic, cells were incubated at 37°C for various times with the protein synthesis inhibitor cylcoheximde, cell lysates immunoblotted with the DO-1 antibody and the p53 band quantified and normalized to β-actin. The immunoblot and the corresponding p53 vs. time plot in Figure 1A demonstrate that p53 in 2780CP/Cl-16 cells also degrades more rapidly than in A2780 cells. That this was unrelated to the use of the DO-1 antibody was confirmed with the alternative DO-7 antibody (Figure 1B). Based on the combined results with DO-1 and DO-7 antibodies, the half-life of p53 in resistant cells was determined to be 2- to 3-fold shorter than in A2780 cells. Subsequent studies were all conducted using the DO-7 antibody.
Figure 1.
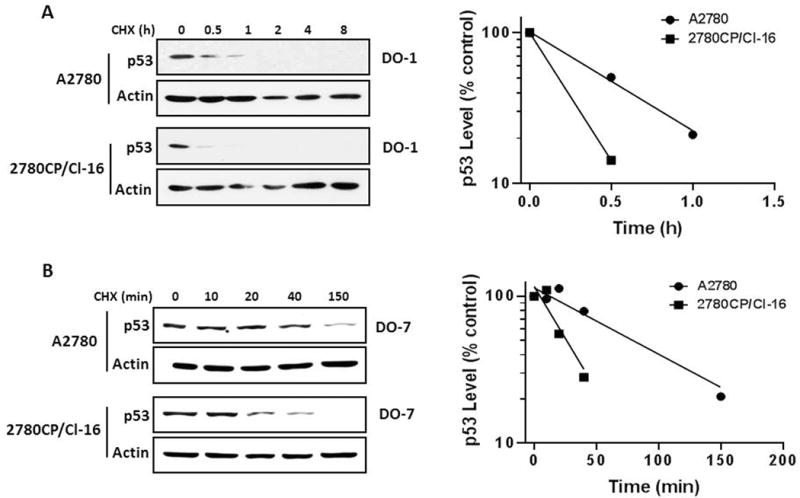
The half-life of p53 is shorter in 2780CP/Cl-16 than in parental A2780 cells. Tumor cells grown at 37°C were treated with 4 μM cychlohexmide (CHX) and then collected at the indicated times. Cell lysates were resolved by SDS-PAGE and p53 levels examined by Western blot using the DO-1 (A) or DO-7 (B) antibody. The p53 and β-actin signals in the immunoblots were quantified by densitometry, the ratios of p53/β-actin were normalized to time zero and plotted against time.
Relative to parental A2780 cells, which are homozygous for wild-type p53, A2780/CP70 cells have a heterozygous missense V172F mutation in p53,14 and an identical p53 status and mutation are also present in 2780CP cells,13 from which the 2780CP/Cl-16 clone was isolated. To examine if the shorter half-life of p53 in 2780CP/Cl-16 may be due to the presence of a mixed population of wild-type and mutant p53 in resistant cells, we included 2780CP/Cl-24 cells that arose spontaneously from 2780CP cells and were characterized as expressing only mutant p53V172F, having lost the wild-type p53 allele through loss of heterozygosity (see Supplementary Figure S1). At 37°C, p53 in 2780CP/Cl-24 cells expressed substantially greater stability than in A2780 or 2780CP/Cl-16 cells (Figure 2A), and this suggested that co-presence of wild-type and mutant p53 in 2780CP/Cl-16 cells effectively destabilizes the protein. This was validated by undertaking studies at 32.5°C since the V172F mutation is reported to be temperature-sensitive13 and confirmed in the present investigation (Supplementary Table S1). At this permissive temperature for wild-type p53, the stability of p53 was similar in all three cell lines (Figure 2A).
Figure 2.
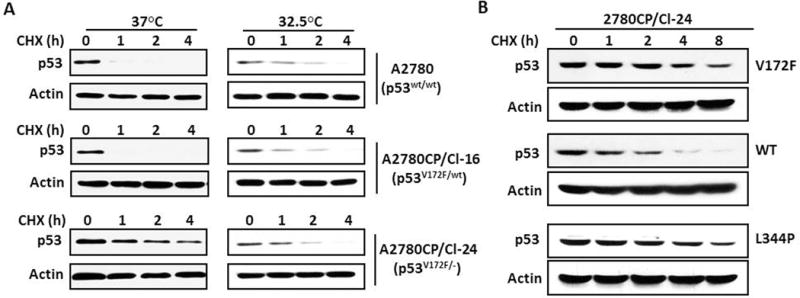
Increased degradation of p53 in 2780CP/Cl-16 cells is due to heterozygous p53 mutation. (A) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells cultured at 37°C or 32.5°C were treated with 4 μM cychlohexmide (CHX) and then collected at the indicated times. Cell lysates were resolved by SDS-PAGE and p53 levels examined by Western blot using the DO-7 p53 antibody. The p53 status of cell lines is indicated in parenthesis. (B) 2780CP/Cl-24 cells grown at 37°C were transfected with wild type (WT) or mutant (V172F or L344P) p53 and the stability of p53 assessed by immunoblot, as in (A).
Since p53 can exist as both dimers and tetramers,17 we examined whether formation of the heteromeric complex between wild-type and mutant p53 is required for the greater instability of p53 in 2780CP/Cl-16 cells at 37°C. For this study, we transfected 2780CP/Cl-24 cells with a p53 construct harboring an L344P mutation that inhibits dimerization and tetramerization of p53,18 and used wild-type and V172F p53 constructs as controls. The results in Figure 2B confirm the relatively greater stability of p53 in 2780CP/Cl-24 cells following ectopic expression of mutant V172F-p53, and this is consistent with expectations, as it is identical to endogenous mutant p53. In contrast, but as anticipated, expression of exogenous wild-type p53 induced destabilization; however, in comparison, the expression of L344P mutant invoked greater p53 protein stability than was observed with wild-type p53. Thus, formation of a complex between wild-type and mutant p53 in 2780CP/Cl-16 is necessary for the observed greater degradation of p53 in these cells.
MDM2 and MDM4 cooperatively regulate p53 degradation
Degradation of p53 mediated by the proteosomal pathway usually involves ubiquitination,4 but ubiquitin-independent pathways are also known.19 Therefore, we examined p53 ubiquitination 6 h after exposure to the proteosomal inhibitor MG132 to reconcile with differential degradation of the p53 protein in the three cell lines at 37°C. With Ponceau S staining establishing equal protein loading, the results in Figure 3A clearly show that p53 ubiquitination was greatest in the 2780CP/Cl-16 cell line and lowest in 2780CP/Cl-24 cells, which provided the anticipated inverse correlation with relative p53 stability in the three tumor models. These observations were reproducible even after 1 h exposure to MG132 (Supplementary Figure S2).
Figure 3.
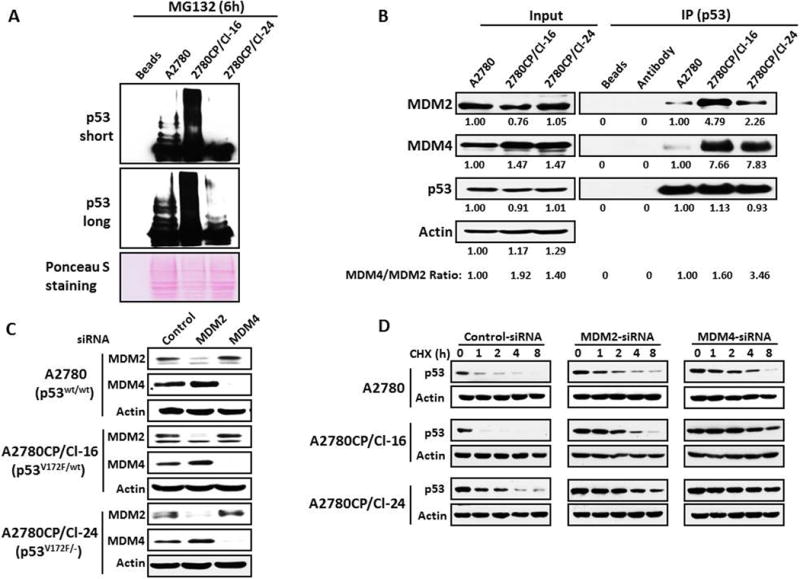
Greater instability of p53 in 2780CP/Cl-16 cells is due to enhanced ubiquitination regulated by MDM2 and MDM4. (A) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells transfected with His-ubiquitin were used to monitor p53 ubiquitination by immunoblot at 6 hours after the addition of MG132 to cell cultures grown at 37°C. The immunoblots were developed using either a short or long exposure condition. Ponceau S staining was used as loading control. (B) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells treated at 37°C with MG132 for 6 hours were collected, and cell lysates immunoprecipitated with the p53 antibody (FL-393), and the proteins resolved by SDS-PAGE and immunoblotted with the indicated antibodies. Whole cell lysates were used as input. The signals in the immunoblots were quantified by densitometry, and the ratios of MDM4/MDM2 were normalized to the ratio in A2780 cells. (C) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells, with the indicated p53 status, were transfected at 37°C with MDM2 or MDM4 siRNA, and the reduction in MDM2 and MDM4 assessed 48 h later by Western blot. (D) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells transfected with MDM2 or MDM4 siRNA at 37°C were exposed 48 hours later to 4 μM cychlohexmide (CHX) to assess p53 stability by Western blot, as described in Figures 1 and 2.
Since ubiquitination of p53 is dependent on the expression of MDM2 and MDM4, cellular levels of these two regulators were also determined to support observations on ubiquitination in Figure 3A. Quantitatively, levels of either MDM2 or MDM4 in the resistant cell lines grown at 37°C were not grossly different from those in A2780 cells (<1.5-fold) to account for differences in p53 ubiquitination, and particularly between 2780CP/Cl-16 and 2780CP/Cl-24 cells (Figure 3B, “input” blots). However, degradation of p53 is reported to be specifically dependent on the ratio of MDM4:MDM2, with a ratio <2 associated with increased p53 degradation and a ratio >2 with increased stabilization.20 Densitometric quantitation of bands normalized to A2780 cells indicated that the ratio in both resistant cell lines were <2, with 2780CP/Cl-16 in fact displaying a higher ratio than 2780CP/Cl-24 cells (1.9 vs. 1.4), and this trend is not consistent with the relatively greater p53 degradation in 2780CP/Cl-16 cells. Therefore, levels of MDM2 and MDM4 and their ratio were next assessed in immunoprecipitates (IP) of p53 from cells exposed to MG132 for 6 h. The results shown in Figure 3B (“IP” blots) indicate that MDM2 bound to p53 was 2- to 5-fold greater in 2780CP/Cl-16 and 2780CP/Cl-24 than in parental A2780 cells, whereas MDM4 binding was about 8-fold greater in the two resistant cell lines. The resultant MDM4:MDM2 ratio of 1.6 in 2780CP/Cl-16 cells vs. 3.5 in 2780CP/Cl-24 cells was highly consistent with the relative stability in the two cell lines. The lower ratio in A2780 than in 2780CP/Cl-16 cells may appear inconsistent with relatively greater p53 stability in parental cells, but the ratio in A2780 cells is likely below a critical threshold for optimal MDM4-dependent ligase activity of MDM2, as has been reported by others20 and observed in the present study in the form of reduced p53 ubiquitination in A2780 cells when compared to 2780CP/Cl-16 cells (see Figure 3A). Thus, the stability of p53 is dependent on relative levels of MDM2 and MDM4 bound to p53.
If degradation of p53 is dependent on an optimal ratio of MDM4 to MDM2, then reduction of either MDM2 or MDM4 in A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells will substantially increase or decrease the MDM4:MDM2 ratio, respectively, and should stabilize p53. Targeting MDM2 or MDM4 in these three models with siRNA at 37°C robustly reduced levels of the protein product (Figure 3C). As regards p53 stability, control siRNA had no effect, with 2780CP/Cl-16 cells again demonstrating the least p53 stability (Figure 3D). However, p53 in these cell lines was stabilized by either MDM2 or MDM4 knockdown, and supported the conclusion that p53 stability is increased by either a low or high MDM4:MDM2 ratio.
Differential p53 stability in resistant cells does not correlate with cytotoxic response to cisplatin
Ectopic overexpression of MDM4 to increase MDM4:MDM2 ratio and p53 stability is reported to induce apoptosis.21 In our studies, however, 2780CP/Cl-24 cells with the higher basal stability of p53 were as resistant to spontaneous apoptosis as were 2780CP/Cl-16 and A2780 cells (data not shown). To investigate whether increased degradation in 2780CP/Cl-16 and greater stability in 2780CP/Cl-24 of p53 cells translated into differential p53-dependent response following DNA damage, experiments were conducted at 37°C to assess the effect of 1–5 μM cisplatin on activating caspase-3, a marker for apoptosis. In parental A2780 cells, cisplatin induced active caspase-3 in a dose-dependent manner (Figure 4A). However, little or no increase in levels of active caspase-3 was observed in 2780CP/Cl-16 and 2780CP/Cl-24 cells, and this strongly indicated that the greater stability of p53 in 2780CP/Cl-24 cells does not result in increased sensitivity to cisplatin.
Figure 4.
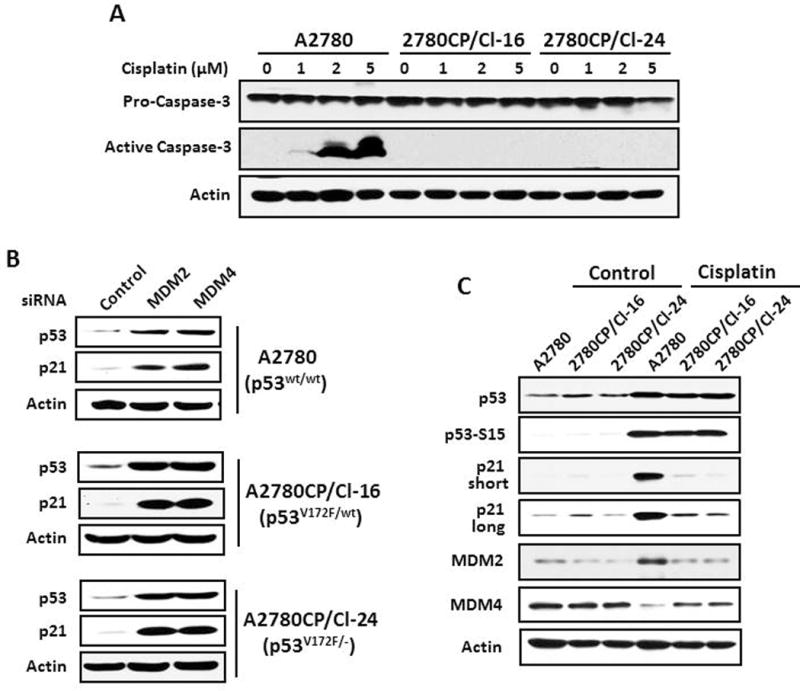
Greater instability of p53 in 2780CP/Cl-16 cells is not associated with cisplatin resistance. (A) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells were exposed at 37°C to increasing concentrations of cisplatin, and cells harvested 48 hours later for assessment of inactive pro-caspase-3 and active cleaved caspase-3 by Western blot. (B) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells, with the indicated p53 status, were transfected at 37°C with MDM2 or MDM4 siRNA as described in Figure 3, and p53 and p21 were examined by Western blot. (C) Tumor cells grown at 37°C were exposed to vehicle or 1 μM (A2780) or 2 μM (2780CP/Cl-16 and 2780CP/Cl-24) cisplatin for 24 hours, and then collected for Western blot analysis using the indicated antibodies. The immunoblots for p21 were developed using either a short or long exposure condition.
Activation of caspase-3 by cisplatin involves p53 induction and p53-dependent transcriptional activation.22 It was possible, therefore, that mutant p53V172F in resistant cells was either unable to be induced or was devoid of transactivation function. Since siRNA targeted against MDM2 or MDM4 reduced p53 degradation (Figure 3D), we examined the effect of this reduced degradation on p53 levels and its transactivation potential for the p21 promoter. The results in Figure 4B clearly demonstrate that knockdown of either MDM2 or MDM4 robustly increased p53 and p21 in all three cell lines grown at 37°C; thus, V172F mutation does not impede p53 induction or its transcriptional function. Based on these findings, we explored the possibility that cisplatin against 2780CP/Cl-16 and 2780CP/Cl-24 cells is unable to activate p53 by failing to regulate MDM2 and/or MDM4 levels. This was tested at the normothermic temperature using a cisplatin concentration of 1 μM in A2780 cells and 2 μM against resistant cells to compensate for a 2-fold reduction in drug uptake and formation of DNA adducts in resistant cells.13 Cisplatin induced p53 to similar degrees in all three cell lines, and this was associated with comparable Serine-15 phosphorylation of p53 (Figure 4C). As expected, p21 and MDM2 as transcriptional targets of p53 were substantially induced in A2780 cells, with MDM4 concomitantly decreased. In contrast, these changes were relatively mild in the two resistant cell lines. Since MDM2-dependent reduction in MDM4 levels is essential for p53-dependent transcriptional activation,6,20,23 the failure of cisplatin to substantially downregulate MDM4 likely prevents p53-dependent transactivation of target genes.
MDM4 inhibits transactivation function of p53 in 2780CP/Cl-16 and 2780CP/Cl-24
The attenuated ability of cisplatin to induce p53-dependent transactivation in the two 2780CP cell lines may also be due to persistent high levels of MDM4 bound to p53, as observed in Figure 3B. At the same time, the permissive temperature of 32.5°C restored p53 stability in resistant cells (Figure 2A), and this provided the rationale to test whether the p53-bound MDM4 is modulated at this lower temperature and restores p53-dependent transactivation response to cisplatin. Initial characterization demonstrated that A2780 cells grown at 32.5°C expressed similar total protein level of p53 or MDM2 when compared to cells grown at 37°C, with MDM4 levels reduced moderately (Figure 5A). In both resistant cell lines grown at 32.5°C, however, MDM2 levels were increased, with concomitant decreases in MDM4. Interestingly, p53 protein levels remained largely unchanged. Similarly, the difference in levels of p53 immunoprecipitated from cells was minimal in comparing the two temperatures (Figure 5B). In contrast, MDM2 levels bound to p53 were moderately increased, with MDM4 concomitantly decreased in the three cell lines grown at 32.5°C. However, MDM4 reduction was the least in 2780CP/Cl-16 cells even though bound MDM2 was the most. To examine how these changes in MDM2 and MDM4 levels in the three cell lines affected cisplatin-induced p53 function, cells grown at 32.5°C were exposed to cisplatin and lysates examined 24 and 48 h later for proteins of interest by immunoblots. At this temperature, induction of p53 and MDM2 were observed in A2780 cells, whereas in 2780CP/Cl-16 and 2780CP/Cl-24 the increases were negligible to mild. On the other hand, a clear reduction in MDM4 was observed at 48 h in the three cell lines, although the reduction in 2780CP/Cl-16 cells was not as extensive (Figure 5C). Nevertheless, concomitant increases in p21 were observed in all tumor models, with lower increases noted again in 2780CP/Cl-16 cells. This indicates that the extent of MDM4 binding to p53 modulates DNA damage-inducible, p53-dependent transcriptional activation.
Figure 5.
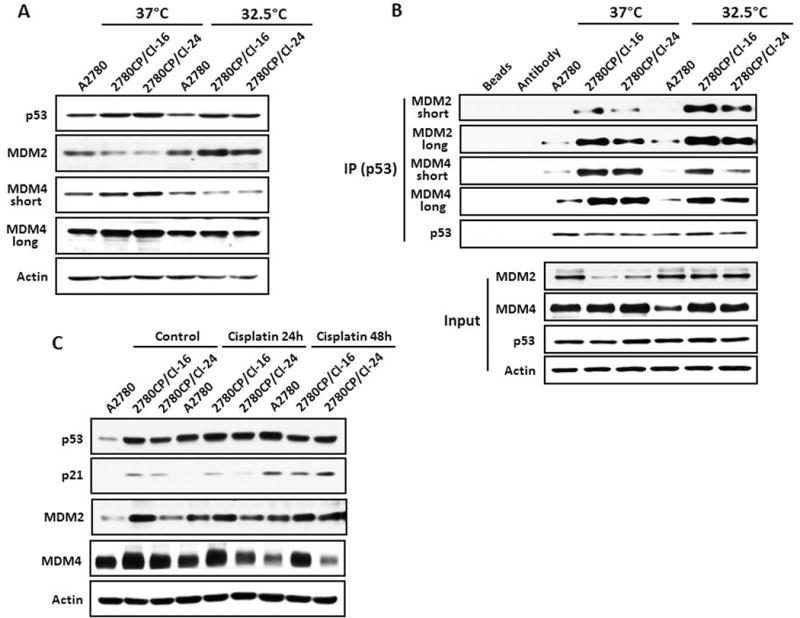
The interaction between p53 and MDM4 decreases in resistant tumor cells grown at 32.5°C to restore cisplatin-induced p53 function. (A) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells cultured at 37°C and 32.5°C, and cell lysates then prepared and subjected to Western blot using the indicated antibodies. (B) A2780, 2780CP/Cl-16 and 2780CP/Cl-24 cells cultured at 37 °C and 32.5 °C exposed to MG132 for 6 hours were collected, and cell lysates immunoprecipitated with the p53 antibody (FL-393) and the proteins immunoblotted with the indicated antibodies, as described in Figure 3. The immunoblots for MDM2 and MDM4 were developed using either a short or long exposure condition. Whole cell lysates were used as input. (C) Tumor cells grown at 32.5°C were exposed to vehicle or 1 μM (A2780) or 2 μM (2780CP/Cl-16 and 2780CP/Cl-24) cisplatin for 24 or 48 hours, and then collected for Western blot analysis using the indicated antibodies.
Cytotoxicity of cisplatin under conditions of reduced MDM4 binding to p53
To strengthen the conclusion that the permissive temperature of 32.5°C restores wild-type p53 status in resistant 2780CP/Cl-16 and 2780CP/Cl-24 cells, decreases MDM4 binding to p53 and enables p21 transactivation following cisplatin exposure, we undertook cytotoxic studies to demonstrate that under these conditions, resistance to cisplatin in resistant cells may be reduced. Studies at 37°C gave typical sigmoidal dose-response curves for cisplatin that clearly show the curves for the two resistant cell lines are shifted substantially to the right of the A2780 curve (Figure 6A). At 32.5°C, the shift in the curves for resistant cells was significantly less (Figure 6B). It is noteworthy that at this lower temperature, the thermodynamic effect caused an ~8-fold increase in the IC50 value for cisplatin in A2780 cells (from 0.25 ± 0.02 to 1.9 ± 0.6 μM) (Figure 6C). However, the increase in the IC50 at 32.5°C in 2780CP/Cl-16 (from 6.5 ± 0.3 to 14.3 ± 1.0 μM) or 2780CP/Cl-24 (from 6.8 ± 0.8 to 6.6 ± 0.5 μM) cell lines was substantially less (~2-fold) or minimal, respectively. Based on these IC50 values, the resistance factors of about 26- to 27-fold at the normotheric temperature were reduced to about 7.5-fold in 2780CP/Cl-16 cells and about 3.5-fold in 2780CP/Cl-24 cells at 32.5°C (Figure 6D). The data demonstrate that normalization of cisplatin-induced transcriptional function of p53 at 32.5°C was sufficient to reduce cisplatin resistance.
Figure 6.
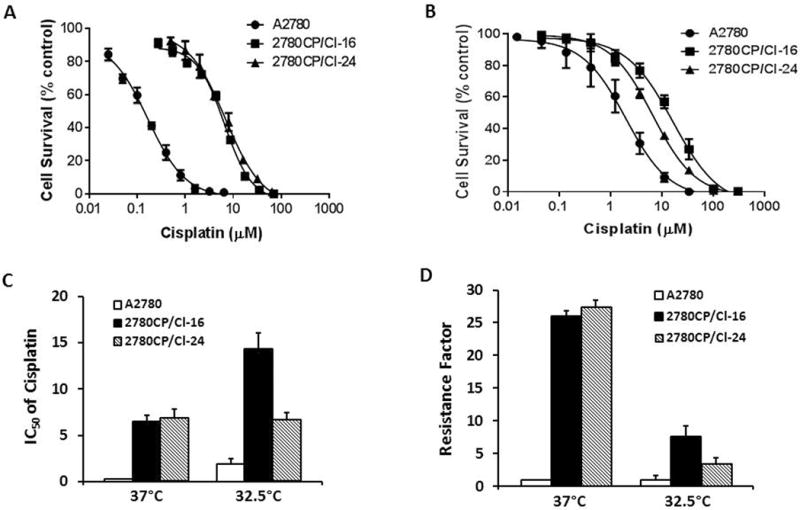
Cisplatin-resistant tumor cells demonstrate reduced resistance when grown at 32.5°C. Tumor cells grown at 37 °C (A) or 32.5 °C (B) were used to determine dose-response curves for cisplatin following growth-inhibition analysis by the MTT assay. The IC50 values (C), generated from dose-response curves in A and B, were used to determine resistance factors (D). The results are shown as mean ± SD of three independent experiments.
DISCUSSION
Cisplatin resistance may arise when the p53 tumor suppressor becomes inhibited by any one of several mechanisms, such as rapid proteosomal degradation or mutation, which prevent its function as a transcriptional activator and inducer of apoptosis.5,24 Based on the reported observation that cisplatin-resistant 2780CP and A2780/CP70 ovarian tumor cell lines harbor a heterozygous V172F mutation in p5313,14 and that the stability of p53 in A2780/CP70 cells was reduced three-fold,16 which was confirmed in 2780CP/Cl-16 cells in the present study, it was possible that either the mutation, the rapid degradation or both contributed to cisplatin resistance. In the present study, however, we have uncovered a novel mechanism of p53 inactivation for inducing cisplatin resistance whereby the heterozygous V172F missense mutation in p53 enhances MDM4 recruitment into the p53/MDM2/MDM4 complex that prevents p53-dependent transcriptional activity and apoptosis.
The V172F mutation is rare in human cancer; only 20 cases of this mutation have ever been documented, with five associated with ovarian cancer (http://p53.iarc.fr/). Therefore, its independent appearance in two disparate human tumor models of cisplatin resistance in tissues of ovarian13,14 and cervical15 origins raises the possibility that the mutation is mediated by cisplatin-induced selection pressures. Irrespective of this intriguing possibility, the mutation is of significance as it has allowed investigation of novel and potentially important mechanisms that regulate p53 levels and function in cisplatin-resistant model systems used routinely in translational and drug development research. Normally, missense mutations in the p53 gene usually hyper-stabilize the protein product, and this has led to the application of positive immunohistochemical staining of p53 as a predictive marker of mutation.24 In the present study, we also observed this phenomenon of increased stability in 2780CP/Cl-24 cells, but not in 2780CP/Cl-16 cells where the p53 was grossly unstable. The difference in p53 stability between these two tumor models from an identical source is due to p53 zygosity, with 2780CP/Cl-16 cells expressing both wild-type and V172F-mutant p53, whereas 2780CP/Cl-24 cells expressed only mutant p53 following loss of heterozygosity. Since the mutation is temperature-sensitive, we have demonstrated that the difference in p53 stability is obviated at the permissive temperature. This observation, together with other supporting data in our present investigation, strongly indicates that increased degradation of p53 in 2780CP/Cl-16 cells is due to the heteromeric conformation of the p53 complex that results in greater polyubiquitination of p53 than in 2780CP/Cl-24 or parental A2780 cells. The extent of polyubiquitination of p53 is generally ascribed to the interplay between MDM2 and MDM4 levels and, more specifically, the MDM4:MDM2 ratio.5,20 In consonance, the greater p53-bound levels of MDM2 and MDM4 in both 2780CP/Cl-16 and 2780CP/Cl-24 cells compared to parental A2780 cells resulted in MDM4:MDM2 ratios that correlated with relative p53 stability in these tumor models. Interestingly, bound levels of MDM2 and MDM4 in the three cell lines did not directly mirror total cellular levels of these p53 regulators, and this suggests that specific factors enhanced their interaction with p53. Since p53-bound MDM2 increased and MDM4 concomitantly decreased at the permissive temperature for wild-type p53 conformation, and this in parallel was associated with normalization of p53 stability in all three cell lines, it may be concluded that the V172F mutation is the major driver for greater MDM4 binding to p53 at the normothermic temperature and, thereby, MDM4 becomes the major modulator of p53 stability. The retention of relatively higher MDM2 and MDM4 levels bound to p53 in resistant cells compared to A2780 cells at the permissive temperature suggests that resistant mechanisms other than the V172F mutation may also be involved. This is consistent with residual resistance to cisplatin of about 4- to 8-fold when p53 is functioning as wild-type at 32.5°C. Part of this residual resistance (about 2-fold), however, may be attributable to reduced cisplatin uptake.13
Hypo-stability of mutant p53 as a heteromeric complex with wild-type p53 is not confined to the V172F mutation, and this is supported by observations in a mouse model engineered to express R172H-p53 gain-of-function mutant (equivalent to the “hotspot” codon 175 in human p53). Specifically, the incidence of undetectable p53 by immunohistochemistry in lymphoma arising spontaneously in mice harboring heterozygous R172H mutation was over twice that in mice expressing homozygous mutant p53R172H (40% vs, 17%).25 This indicates that hypo-stability of p53 protein in tumors harboring heterozygous p53 mutation may occur more broadly. Although murine studies suggest that the greater stability of mutant p53 is a prerequisite for the gain-of-function phenotype,25 in the present study the substantial difference in p53 stability between 2780CP/Cl-16 and 2780CP/Cl-24 tumor cells did not translate into differential sensitivity to cisplatin at normothermic temperatures, but both cell lines were equally resistant to this platinum drug. This may be consistent with the observation that hypo-stability in 2780CP/Cl-16 cells or hyper-stability in 2780CP/Cl-24 cells do not actually result in differences in basal levels of p53. Thus, the instability of p53 in 2780CP/Cl-16 is likely compensated by other mechanisms.
An important observation in our present investigation is that in contrast to A2780 cells, p53 in 2780CP/Cl-16 and 2780CP/Cl-24 cells exposed to cisplatin at normothermic temperature is unable to transactivate its target genes, p21 and MDM2. Loss of this transactivity is not due to the inability of V172F mutation to perform transactivation function, as knockdown of MDM2 or MDM4 was sufficient to induce both p53 and p21 in resistant cells. This raises an important question as regards the mechanism that impedes p53-dependent transactivation when resistant cells are exposed to cisplatin. Since MDM4 binding to p53 has been postulated to mask the transactivation domain of p53, a seminal requirement for transactivation following DNA damage is a reduction in MDM4 levels by MDM2-dependent proteosomal degradation.4,5,26 In our present study, MDM4 was significantly reduced by cisplatin at normothermic temperature in A2780 cells, but not in 2780CP/Cl-16 or 2780CP/Cl-24 cells. At the permissive temperature when p53 binding to MDM4 is reduced, however, cisplatin-induced transactivation of p21 and MDM2 was restored, and this coincided with significant reversal of cisplatin resistance seen under normothermic conditions. Thus, our data demonstrate that modulation of MDM4 interaction with mutant p53V172F regulates p53-dependent cytotoxicity of cisplatin.
In conclusion, we have demonstrated the existence of a novel mechanism of p53 regulation in cisplatin-resistant 2780CP/Cl-16 and A2780/CP70 tumor cells at the normothermic temperature. This regulation involves heterozygous p53V172F mutation, which increases binding of p53 to MDM4 that mediates MDM2-dependent proteosomal degradation of p53, but, more importantly, the increased binding induces cisplatin resistance by inhibiting p53-dependent transactivation. These and related findings from our studies at 37°C are schematically presented and summarized in Figure 7. Since tumor models derived from A2780 cells are used in drug development studies, it becomes important to determine whether novel drugs arising from such studies are achieving their desired goals of circumventing cisplatin resistance by effectively disrupting the p53/MDM2/MDM4 complex.
Figure 7.
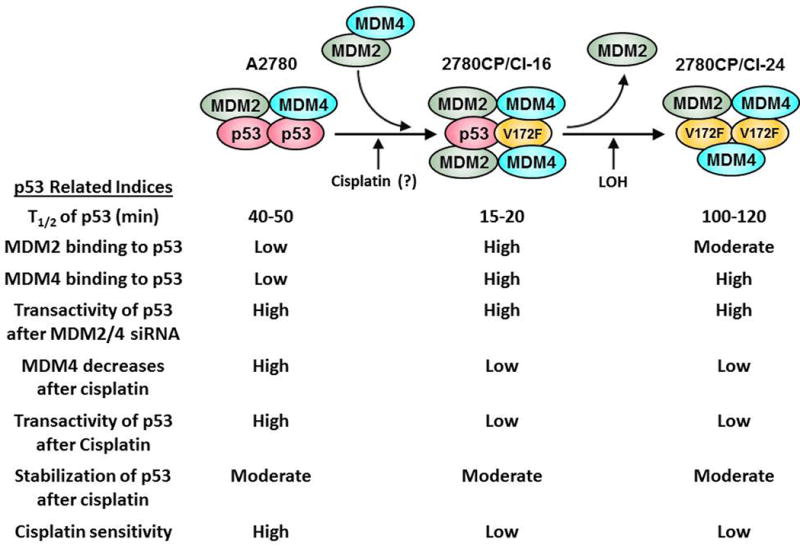
Schematic representation and summary of the effect of V172F mutation on stability, MDM2/4-binding, transactivity and cisplatin-mediated activation of p53 at the normothermic temperature in A2780 and cisplatin-resistant tumor cells. The exact stoichiometry of the p53-MDM2-MDM4 complex and the role of cisplatin in the induction of p53V172F mutation are not known. LOH, loss of heterozygosity.
Material and methods
Cell culture and reagents
A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells were maintained at either 37 or 32.5°C as monolayer cultures, as previously reported.27 The stable cisplatin-resistant clones 2780CP/Cl-16 and 2780CP/Cl-24 were isolated from 2780CP cells13 and maintained under cisplatin-free conditions. All cell lines were negative for mycoplasma and authenticated by STR DNA fingerprinting in MD Anderson Core Facilities. Protein A beads (sc-2001) and antibodies for p53 (sc-126, sc-47698, sc-6243), p21 (sc-6246), MDM2 (sc-13161), MDM4 (sc-374147) and β-actin (sc-47778) were purchased from Santa Cruz, MTT (19265) from Affymetrix, agarose (R901-01) from Invitrogen, Lipofectamine® RNAiMAX (13778150) from Life technologies, Halt™ Protease & Phosphatase inhibitor (1861280) from Thermo Scientific, QuikChange Lightning Multi Site-Directed Mutagenesis Kit (210515) from Agilent Technologies, and cisplatin (C2210000), MG132 (C2211), Ponceau S (P7170) and cycloheximide (C1988) from Sigma. Ni-NTA and His-Ubiquitin plasmid were kind gifts from Dr. Shiaw-Yih Lin (MD Anderson Cancer Center).
Western blot and immunoprecipitation
The procedures were conducted essentially as described before.27 In brief, cells were collected, pelleted by centrifugation at 1000 × g and 4°C for 1 min, and cell lysates prepared in four volumes of ice-cold lysis buffer containing 10 mM sodium β-glycerophosphate, 2 mM EDTA, 150 mM NaCl, 10 mM NaF, 50 mM Tris-HCL, 0.5% NP40 and Halt™ Protease & Phosphatase inhibitor. About 20–50 μg of lysate supernatant was used to prepare immunoblots, and the protein bands quantified by densitometry using the Image J software.
For immunoprecipitation, antibodies were added to the cell lysate and incubated at 4°C overnight, and Protein A beads were then added. Beads were centrifuged one hour later and washed three times with the immunocomplex wash buffer containing 50 mM Tris·HCl (pH 7.5), 10 mM sodium β-glycerophosphate, 10 mM NaF, 2 mM EDTA, 150 mM NaCl. SDS loading buffer (2×) was added to the beads, the mixture boiled and the supernatants subjected to immunoblot analysis, as described above.
Cell growth inhibition assay
Cells grown at 37°C or 32.5°C were trypsinized, plated into 96-well plates, and exposed to cisplatin 24 hours later. Growth inhibition was measured 5 days (37°C) or 7 days (32.5°C) later by MTT assay, as reported previously.12 The longer MTT time point at 32.5°C was necessary to compensate for the slower growth. The IC50 value was determined from dose-response data fitted to a 4-parameter sigmoidal curve using the GraphPad Prism software. Resistance factors were assessed, as reported previously.13
Ubiquitination assay
The assay was performed as described before with minor modifications.28 Briefly, cells were transfected with His-ubiquitin, and 48 h later exposed to MG132 for 1 or 6 h. Cells were then washed with ice-cold PBS, collected by scraping from dishes and pelleted by centrifugation at 1000g and 4°C for 5 min. Cells were resuspended in 1 mL Buffer A (6M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole and adjusted to pH 8.0 with NaOH) and mixed by pipetting. After sonication, resultant cell lysates were centrifuged at 14,000g and 4°C for 10 min, and the supernatants collected. Fifty μL 50% Ni-NTA agarose bead suspension was added to the supernatants and incubated for 3 h at room temperature. Beads were then pelleted by centrifugation at 14,000g for 2 min and washed twice with 1 ml buffer A, twice with buffer A/TI and once with buffer TI (25 mM Tris-HCl,20 mM imidazole and adjusted to pH 6.8 with HCl). The supernatant was aspirated immediately and 50 μL of 2× SDS sample buffer added to resuspend the beads by vortexing, centrifuged at 14,000g for 1 min and boiled for 10 min. The released proteins were then subjected to Western blot analysis, as described above.
Small interfering RNA
siRNA duplexes for MDM2 (sense, 5′-GCCUGGCUCUGΜGUGUAAUdTdT-3′; antisense, 5′-AUUACACACAGAGCCAGGCdTdT-3′), MDM4 (sense, 5′-AGATTCAGCTGGTTATTAAdTdT-3′; antisense, 5′-TTAATAACCAGCTGAATCTdTdT-3′), and control luciferase GL3 (sense, 5′-CUUACGCUGAGUACUUCGAdTdT-3′; antisense, 5′-UCGAAGUACUCAGCGUAAGdTdT-3′) were obtained from Sigma. These siRNAs have been validated previously for their specificity.7,29,30 Cells were transfected with 100 nM siRNA with RNAiMax using the manufacturer’s instruction. Cells were collected 48 hours later and processed for immunoblot analysis.
Half-life assay
Half-life of p53 was determined as described before with minor modifications.31 Essentially, cells were grown in 60-mm petri dish to 70–80% confluence, exposed to 4 μM cycloheximide (CHX), washed with ice-cold PBS and lysed with NP-40 lysis buffer, as described above. Lysate at each time point were immunoblotted for p53. The intensity of the bands was determined using the Image J software and fitted to a monoexponential decay equation with a weighting factor of 1/y using the GraphPad Prism computer program to determine the half-life.
Site-directed mutagenesis
The mutations R172V and L344P were introduced into p53 by using QuikChange Lightning Multi Site-Directed Mutagenesis Kit, following the manufacturer’s instruction. The primers for the mutagenesis are as follows:
5′-AGCACATGACGGAGTTTGTGAGGCGCTGC-3′ and 5′-GCAGCGCCTCACAAACTCCGTCATGTGCT-3′ for R172V; 5′-GATGTTCCGAGAGCCGAATGAGGCCTTGGAACTCAAGG-3′ and 5′-CCTTGAGTTCCAAGGCCTCATTCGGCTCTCGGAACATC-3′ for L344P.
Determination of p53 status
The p53 status and the temperature-sensitive nature of the p53 mutation in 2780CP/Cl-24 cells was determined exactly as previously described by us in A2780 and 2780CP/Cl-16.13
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The research support from the U.S. Public Health Service grants CA160687 to ZHS and Support Grant CA16672 to MD Anderson Cancer Center awarded by the National Cancer Institute, and in part from the Megan McBride Franz Endowed Research Fund, is gratefully acknowledged. We thank Dr. Shiaw-Yih Lin (MD Anderson Cancer Center) for the His-Ubiquitin plasmid, and also Dr. Guangan He and Michelle Martinez for helpful discussions.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest.
Author Contributions
XX, GL and ZHS designed and/or executed the experiments, and analyzed and interpreted the data. XX and ZHS wrote the initial draft of the manuscript, and GL contributed to revision for the final version.
Supplementary Information
Supplementary Information (Figures S1 and S2 and Table S1) accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Martinez-Rivera M, Siddik ZH. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem Pharmacol. 2012;83:1049–1062. doi: 10.1016/j.bcp.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toledo F, Wahl GM. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int J Biochem Cell Biol. 2007;39:1476–1482. doi: 10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry ME. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb Perspect Biol. 2010;2:a000968. doi: 10.1101/cshperspect.a000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012;586:1390–1396. doi: 10.1016/j.febslet.2012.02.049. [DOI] [PubMed] [Google Scholar]
- 5.Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012;3:228–235. doi: 10.18632/oncotarget.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, et al. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol. 2005;25:9608–9620. doi: 10.1128/MCB.25.21.9608-9620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278:45946–45953. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- 9.Kondo S, Barnett GH, Hara H, Morimura T, Takeuchi J. MDM2 protein confers the resistance of a human glioblastoma cell line to cisplatin-induced apoptosis. Oncogene. 1995;10:2001–2006. [PubMed] [Google Scholar]
- 10.Cocker HA, Hobbs SM, Tiffin N, Pritchard-Jones K, Pinkerton CR, Kelland LR. High levels of the MDM2 oncogene in paediatric rhabdomyosarcoma cell lines may confer multidrug resistance. Br J Cancer. 2001;85:1746–1752. doi: 10.1054/bjoc.2001.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam S, Lodder K, Teunisse AF, Rabelink MJ, Schutte M, Jochemsen AG. Role of Mdm4 in drug sensitivity of breast cancer cells. Oncogene. 2010;29:2415–2426. doi: 10.1038/onc.2009.522. [DOI] [PubMed] [Google Scholar]
- 12.Mujoo K, Watanabe M, Nakamura J, Khokhar AR, Siddik ZH. Status of p53 phosphorylation and function in sensitive and resistant human cancer models exposed to platinum-based DNA damaging agents. J Cancer Res Clin Oncol. 2003;129:709–718. doi: 10.1007/s00432-003-0480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siddik ZH, Mims B, Lozano G, Thai G. Independent pathways of p53 induction by cisplatin and X-rays in a cisplatin-resistant ovarian tumor cell line. Cancer Res. 1998;58:698–703. [PubMed] [Google Scholar]
- 14.Lu X, Errington J, Curtin NJ, Lunec J, Newell DR. The impact of p53 status on cellular sensitivity to antifolate drugs. Clin Cancer Res. 2001;7:2114–2123. [PubMed] [Google Scholar]
- 15.Komlodi-Pasztor E, Trostel S, Sackett D, Poruchynsky M, Fojo T. Impaired p53 binding to importin: a novel mechanism of cytoplasmic sequestration identified in oxaliplatin-resistant cells. Oncogene. 2009;28:3111–3120. doi: 10.1038/onc.2009.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yazlovitskaya EM, DeHaan RD, Persons DL. Prolonged wild-type p53 protein accumulation and cisplatin resistance. Biochem Biophys Res Commun. 2001;283:732–737. doi: 10.1006/bbrc.2001.4849. [DOI] [PubMed] [Google Scholar]
- 17.Gaglia G, Guan Y, Shah JV, Lahav G. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci U S A. 2013;110:15497–15501. doi: 10.1073/pnas.1311126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davison TS, Yin P, Nie E, Kay C, Arrowsmith CH. Characterization of the oligomerization defects of two p53 mutants found in families with Li-Fraumeni and Li-Fraumeni-like syndrome. Oncogene. 1998;17:651–656. doi: 10.1038/sj.onc.1202062. [DOI] [PubMed] [Google Scholar]
- 19.Tsvetkov P, Reuven N, Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010;17:103–108. doi: 10.1038/cdd.2009.67. [DOI] [PubMed] [Google Scholar]
- 20.Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, et al. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–19254. doi: 10.1074/jbc.C200150200. [DOI] [PubMed] [Google Scholar]
- 21.Mancini F, Gentiletti F, D’Angelo M, Giglio S, Nanni S, D’Angelo C, et al. MDM4 (MDMX) overexpression enhances stabilization of stress-induced p53 and promotes apoptosis. J Biol Chem. 2004;279:8169–8180. doi: 10.1074/jbc.M311793200. [DOI] [PubMed] [Google Scholar]
- 22.Siddik ZH. Cisplatin resistance: Molecular basis of a multifactorial impediment. In: Teicher BA, editor. Cancer Drug Resistance. Humana Press; Totowa: 2006. pp. 283–307. [Google Scholar]
- 23.Wang YV, Wade M, Wong E, Li YC, Rodewald LW, Wahl GM. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc Natl Acad Sci U S A. 2007;104:12365–12370. doi: 10.1073/pnas.0701497104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brachova P, Thiel KW, Leslie KK. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int J Mol Sci. 2013;14:19257–19275. doi: 10.3390/ijms140919257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenos K, Jochemsen AG. Functions of MDMX in the modulation of the p53-response. J Biomed Biotechnol. 2011;2011:876173. doi: 10.1155/2011/876173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuang J, He G, Huang Z, Khokhar AR, Siddik ZH. Bimodal effects of 1R,2R-diaminocyclohexane(trans-diacetato)(dichloro)platinum(IV) on cell cycle checkpoints. Clin Cancer Res. 2001;7:3629–3639. [PubMed] [Google Scholar]
- 28.Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–1138. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 30.He G, Kuang J, Khokhar AR, Siddik ZH. The impact of S- and G2-checkpoint response on the fidelity of G1-arrest by cisplatin and its comparison to a non-cross-resistant platinum(IV) analog. Gynecol Oncol. 2011;122:402–409. doi: 10.1016/j.ygyno.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He G, Kuang J, Koomen J, Kobayashi R, Khokhar AR, Siddik ZH. Recruitment of trimeric proliferating cell nuclear antigen by G1-phase cyclin-dependent kinases following DNA damage with platinum-based antitumour agents. Br J Cancer. 2013;109:2378–2388. doi: 10.1038/bjc.2013.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.