

Abstract
PEGylated liposomes have transformed chemotherapeutic use of doxorubicin by reducing its cardiotoxicity; however, it remains unclear whether liposomal doxorubicin is therapeutically superior to free doxorubicin. Here, we demonstrate a novel PEGylated liposome system, named DAFODIL (Doxorubicin And 5-Flurouracil Optimally Delivered In a Liposome) that inarguably offers superior therapeutic efficacies compared to free drug administrations. Delivery of synergistic ratios of this drug pair led to greater than 90% reduction in tumor growth of murine 4T1 mammary carcinoma in vivo. By exploiting synergistic ratios, the effect was achieved at remarkably low doses, far below the maximum tolerable drug doses. Our approach re-invents the use of liposomes for multi-drug delivery by providing a chemotherapy vehicle which can both reduce toxicity and improve therapeutic efficacy. This methodology is made feasible by the extension of the ammonium-sulfate gradient encapsulation method to nucleobase analogues, a liposomal entrapment method once conceived useful only for anthracyclines. Therefore, our strategy can be utilized to efficiently evaluate various chemotherapy combinations in an effort to translate more effective combinations into the clinic.
Keywords: Drug combinations, synergistic, chemotherapy, liposomes, nanoparticles
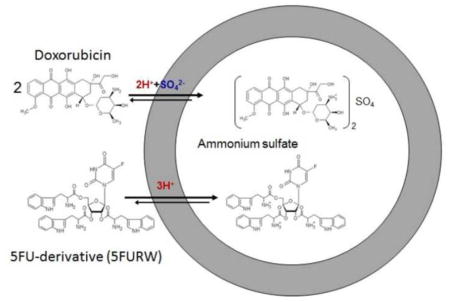
Graphical abstract
INTRODUCTION
This year marks two decades since the FDA approval of doxorubicin-entrapped liposomes (Doxil), a transformative nanoparticle formulation which has consistently reduced adverse cardiac side effects of the original free drug administration [1–3]. However, this first generation liposomal system has been confronted with challenges which ultimately question its advantages over free doxorubicin (DOX) [4–10][11–14]. Some clinical trials reported little or only comparable therapeutic efficacy [4, 5, 8–10], whereas other studies showed that liposomal DOX merely increased the tolerable dose, making it difficult to decipher whether liposomal entrapment actually enhanced antitumor efficacy [6, 7]. Attempts have been made to improve Doxil’s efficacy is tumor-targeting via conjugation of ligands with enhanced affinity towards membrane receptors over-expressed in cancer cells. Tumor-homing peptides, proteins and even small molecules have been shown to enhance cancer cell kill both in vitro and in vivo of DOX-entrapped liposomes [15–19]. Tumor-targeting has proven particularly useful against DOX-resistant cell lines exhibiting poor drug internalization of free DOX. In these drug delivery systems, however, it remains challenging to decouple the effect of enhanced drug uptake from enhanced drug potency.
Co-encapsulation of synergistic chemotherapy combinations could overcome these concerns by eliciting significant therapeutic efficacy at low doses. In this approach, the drug payload is improved by incorporating multiple drugs which can simultaneously inhibit various stages of cancer cell proliferation. Numerous in vitro studies have demonstrated that chemotherapy combinations can appreciably reduce drug doses required to inhibit significant cancer cell growth [20–22]. Here we co-encapsulate a prominent drug pair of 5-fluorouracil (5FU) and DOX in liposomes at an optimal synergistic drug ratio. 5FU and DOX have been included in nearly all combination chemotherapy regimens against gastric cancer [23–31], and has been employed in many clinical trials against breast carcinoma as well [32–34]. When included in combination chemotherapy regimens, response rates can improve from 15–38% [5, 35–37] to 40–50% against advanced gastric cancers [23–25, 28, 38] and 50–75% against advanced breast carcinoma [32–34]. However, the median survival time for patients treated with these regimens is still quite low, ranging between 7–9 months. Despite encouraging improvements in therapeutic efficacy, there still remains a clear unmet need to advance these combinations to create more successful therapeutics with more complete responses and longer survival times [24, 32].
Therapeutic potency of the combination can be improved by optimizing the ratio of drugs exposed to cancer cells, as the same drug pair can exhibit antagonistic or synergistic cancer cell kill depending on the ratio [39–41]. To our knowledge, however, there have been no reported studies that explored whether synergistic interactions exist between 5FU and DOX. Such fundamental drug interaction studies would validate therapeutic merit in combining 5FU and DOX, especially since the pair is frequently utilized in the clinic. Here we report such synergistic combination and a method of encapsulating it in a liposome to deliver it to tumors. Since liposomes and nanoparticles in general exhibit low tumor accumulation, about 1.5–2.0% initial dose per g of tumor tissue [42–44], it is essential that the combination is co-delivered in a single vehicle. The liposomal co-encapsulation of 5FU and DOX ensures that the optimally identified synergistic ratio is exposed to the tumor cells, thereby achieving the most therapeutic benefit from the combination.
MATERIALS AND METHODS
Cell culture
All cell lines were obtained by ATCC and maintained in a humidified CO2 incubator at 37°C. BT-474 human breast cancer cells were cultured in Hybri-Care medium (ATCC), 4T1 murine breast cancer cells in RPMI-1640 medium (Thermo Scientific), and bEnd.3 mouse brain endothelial cells in Dulbecco’s Modified Eagle’s medium (DMEM; ATCC). Both breast tumor cell lines investigated were DOX-sensitive. All above cells were supplemented with 10% fetal bovine serum (FBS; Thermo Scientific). 4T1 and bEnd.3 cells were supplemented with 1% penicillin/streptomycin (Thermo Scientific). MCF10A human breast epithelial cells were cultured in Mammary Epithelial Basal Medium (MEBM; Lonza) supplemented with BPE, hydrocortisone, hEGF, insulin, and 100 ng/mL cholera toxin (Sigma-Aldrich).
Chemotherapy combination studies
The Combination Index (CI) method [45], was adopted to identify ratios of 5-fluorouracil (5FU; Sigma-Aldrich) and doxorubicin (DOX; LC laboratories) which synergistically inhibit cancer cell proliferation in vitro. For cell viability studies, BT-474 cells were seeded at a density of 10,000 cells per well in 100 μL cell culture medium in a 96-well plate and grown overnight. Cell viability was assessed post drug-incubation using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Invitrogen Life Technologies) assays. Individual drug concentrations which inhibit 50% cell growth (D50) were first determined by fitting dose-effect data to the median-effect model [46]. For combinatorial studies, 5FU and DOX were simultaneously incubated with BT-474 cells at ratios of differing multiples of their D50 concentrations, and cell viability was subsequently assessed. CI values were further calculated, as previously described [45], with the designations of CI<1, CI=1, and CI> 1 respectively representing synergy, additivity, and antagonism. The same methodology was applied to assess interactions between all other chemotherapy combinations presented.
Liposome preparation
Liposomes were prepared utilizing the conventional thin film evaporation method [47]. Zwitterionic liposomes (DOX-L) were composed of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC; Avanti Polar Lipids) and cholesterol (Chol; Sigma-Aldrich) in a molar ratio of 55:45, and cationic liposomes consisted of DSPC, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP; Avanti Polar Lipids), and Chol in a ratio of 80:10:10. The lipid concentrations for zwitterionic liposomes mimic those implemented in Doxil [48], and the elevated levels of PC-lipids in cationic formulations ensures that insignificant toxicity will be induced by the presence of positively-charged lipids. PEGylated liposomes were prepared using DSPC:1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (mPEG-DSPE):DOTAP:Chol in a ratio of 75:5:10:10. Cationic liposomes designated as +DOX-L, 5FU-L, and 5FURW-L represent liposomes encapsulating DOX, 5FU or 5FURW, respectively. 5FURW refers to the ribosylated nucleoside form of 5FU (5FUR), conjugated to tryptophans (W) via esterification of 5FUR hydroxyl moieties (Fig. S3). DOX And 5FU Optimally Delivered In a Liposome, abbreviated as DAFODIL, was also made with the cationic lipid composition. Lipids were co-dissolved in chloroform and methanol in a round-bottom flask, and organic solvent was removed using a Buchi R-210 rotary evaporator at reduced pressure and 60°C (5°C above the lipid transition temperature) in order to form a thin lipid film. The film was subsequently hydrated in 125 mM ammonium sulfate at 60°C and passed 21 times through an extruder (Avestin LiposoFast Extruder) with 100 nm polycarbonate filters. A transmembrane ammonium sulfate gradient was generated by passing liposomes through a Sephadex G-25 PD-10 column (GE Healthcare Life Sciences) equilibrated with PBS [49]. Please see SI Text for optimal liposome encapsulations of each drug. To remove free drug, liposomes were finally passed through a Sephadex G-25 PD-10 column.
Liposome characterization
To measure drug encapsulation, 50 μL of drug-loaded liposomes were dissolved in 950 μL methanol through vortexing and sonication. The dissolved liposomes were centrifuged at 12000 g for 20 minutes to allow the lipids to pellet. The supernatant was collected, diluted serially, and their absorbances were measured at 480 nm, 266 nm, 264 nm, 268 nm, 268 nm, and 394 nm to quantify DOX, 5FU, 5FUR, and 5FURW concentrations, respectively. Drug encapsulation is reported as the mol% of drug with respect to lipids ± SD. Liposome sizes and ζ potentials were determined utilizing dynamic light scattering and electrophoretic light scattering, respectively, on a Malvern ZetaSizer NanoZS. Samples were diluted 100X in distilled de-ionized water immediately prior analysis, and each measurement is reported as an average of 3 independent sets of at least 13 runs each ± SD.
Cancer cell growth inhibition by liposomal drugs
In vitro anticancer efficacy of drug-loaded liposomes was determined using calcein-AM cell viability assay (Life Technologies). BT-474 cells or 4T1 cells were seeded in a 96-well cell culture plate at a density of 11,000 cells per well or 1,000 cells per well in a total volume of 100 μL media and allowed to adhere overnight. Media was then replaced with fresh media containing liposomes and incubated for 72 hours or 48 hours for BT-474 or 4T1 cells, respectively. The differences in cell seeding densities and drug incubation times between BT-474 cells and 4T1 cells is due to the much lower doubling time of 4T1 cells compared to BT-474. Parameters were optimized such that untreated cells reached 70% confluency after treatment. After incubation with liposomes, media was aspirated and replaced with 1 μM calcein-AM in PBS for 30 minutes at room temperature. Fluorescence intensity of intracellularly hydrolyzed calcein-AM was measured using excitation and emission wavelengths of 490 nm and 520 nm. Fractional cell inhibition was calculated by subtracting fluorescence of live cells in experimental wells from those of untreated cells and normalizing against untreated cells. D50 concentrations were determined by fitting experimental data to the median-effect model [46], and their error is reported as the standard error of the model fit.
DOX internalization
Confocal laser scanning microscopy was utilized to visualize DOX intracellular distribution. BT-474 cells were seeded in an 8-well chambered borosilicate coverglass at 85,000 cells/300 μL/well, and were allowed to adhere overnight. Fresh media containing DOX, DOX-L or +DOX-L at drug-equivalent concentrations of 1 μM were incubated with cells at 37°C and 5% CO2. After specified incubation times, cells were washed three times with warmed PBS, followed by nuclear staining with 25 μg/mL Hoechst for 30 minutes at 37°C and 5% CO2. Excessive Hoechst was removed by washing with PBS and cells were then cultured in culture medium. Live cells were immediately imaged with an Olympus Fluoview 1000 spectral confocal microscope. Hoechst and DOX were excited by 405 nm 50 mW diode and 488 nm 10 mW Argon gas lasers, respectively. Optical filters of 425–475 nm and 574–674 nm emission wavelengths were used to view fluorescence from Hoechst and DOX, respectively. Z-stacks of 10 μm were captured and averaged. Fluorescence intensity is reported as the raw integrated density of DOX fluorescence divided by number of cells, as determined by Hoechst labeling.
Synthesis of 5FURW
A 5FU prodrug was synthesized to modify the native drug’s chemical properties and facilitate incorporation in liposomes bearing a transmembrane ammonium sulfate gradient. See SI Text for complete synthetic steps.
In vivo tumor growth inhibition
All experiments were performed according to approved protocols by the Institutional Animal Care and Use Committee of the University of California, Santa Barbara. To assess tumor growth inhibition in vivo, 1 × 105 4T1 cells in 100 μL saline were subcutaneously injected into the mammary fat pad of female BALB/c mice six to eight weeks in age (Charles River Laboratories). Post-inoculation, mice were randomized into experimental and control groups, and were monitored daily for tumor growth and body weight changes. Mice were injected intravenously with either DAFODIL or free 5FURW combined with free DOX, at drug-equivalent doses of 3 mg/kg DOX and 0.62 mg/kg 5FURW diluted in sterile saline. For the small tumor model, treatments began on day 3 post tumor inoculation when volumes were approximately 10 mm3 (estimated based on extrapolation of a typical growth curve) and were repeated every other day for a total of 4 injections. For the large tumor model, treatments began when volumes were 300 mm3, on day 15 post inoculation. Tumor volumes were calculated as V = 1/2 (l) x (w)2, where l and w correspond to the longest and shortest tumor diameters, respectively, as measured by a digital caliper. Body weights were also measured daily to assess overall health.
In vivo biodistribution
Biodistribution studies were conducted by embedding the nonexchangeable and nonmetabolizable lipid marker [3H]-cholesterylhexadecyl ether (CHDE; Perkin Elmer, Waltham, MA) [50] in the liposome membrane. CHDE was dissolved at a ratio of 0.5 μCi/μmol phospholipid prior to thin film formation, and liposome fabrication proceeded as described above. [3H]-liposomes at drug equivalent doses of 6 mg/kg DOX + 1.2 mg/kg 5FURW were injected intravenously to BALB/c mice bearing approximately 100 mm3 4T1 tumors and circulated for 6 hours. Solvable (Perkin Elmer, Waltham, MA), 5 mL, was added to harvested organs, and incubated overnight at 60°C, followed by 10 mL Ultima Gold (Perkin-Elmer). Radioactive content was measured in a Packard TriCarb 2100TR scintillation counter. Results were reported as organ disintegrations per minute (DPM) relative to initial dose DPM, normalized to organ weight.
Statistical analyses
Statistical significance was assessed via the two-tailed, unpaired Student’s t-test in Microsoft Excel. One star indicates significance with p < 0.05, and two starts indicate p < 0.01.
RESULTS
Synergistic inhibitory effect on tumor cell proliferation by optimal ratio of 5FU:DOX
BT-474 human breast cancer cells were exposed to 5FU+DOX at various ratios and evaluated for synergistic growth inhibition utilizing the Combination Index (CI) method [46]. At a constant 5FU concentration, the ratio which yielded the lowest CI, and hence greatest synergy, was R=819:1 ± 64 5FU:DOX (Fig 1a). R<819 caused additivity (CI=1) and R>819 induced antagonistic effects (CI>1). Moreover, the pair consistently outperformed individual drugs when combined at R=819 (Figs. S1a–b). On the contrary, 5FU+DOX at an antagonistic ratio R=6551 ± 170 consistently led to underperformance compared to 5FU alone. 5FU+DOX led to elevated reactive oxygen species (ROS) generation, a nontraditional mechanism by which DOX induces cancer cell death, and was a probable contributor to the synergistic activity (Fig. S2).
Fig. 1. Dependence of synergy between DOX and 5FU on molar ratio R and verification that synergistic combination exhibits increased cancer specificity.

(a) Combination indices calculated for various ratios of 5FU+DOX exposed to BT-474 human breast cancer cells. For all ratios, 5FU concentration was kept constant at 487 μM. Data expressed as mean ± SD (n≥8). (b) Ratio of D50 for BT-474 cells relative to MCF 10A breast epithelial cells for 5FU (black bars) and DOX (white bars) treated as single drugs or in combinations of R=819, R=6551. D50s were determined for experimental fits of cell inhibition data to the ME model, and error is propagated from the standard error of the model fit.
The synergistic combination (R=819) also enhanced drug specificity towards BT474 cells over non-cancerous MCF 10A epithelial cells (Fig. 1b). The ratio of D50 between BT474 and MCF 10A, an indicator of cancer specificity, was significantly greater than 1 for free 5FU and DOX, indicating lack of cancer specificity. This ratio was reduced by 36-fold and 5-fold for 5FU and DOX, respectively when 5FU+DOX was administered at the synergistic ratio, and reached a value less than 1, indicating higher cancer specificity of the combination. Additional cell inhibition studies verified that 5FU+DOX at both R=819 and R=6551 is less toxic than single drug treatments in endothelial and epithelial cell lines bEnd.3 and MCF 10A, respectively (Fig. S1c–f).
Incorporation of DOX in liposomes
DOX was initially encapsulated in zwitterionic liposomes (DOX-L) using an ammonium sulfate gradient. DOX activity was significantly reduced in zwitterionic liposomes (Fig. 2a), with 100-fold greater D50 concentration relative to free DOX. In order to recover drug activity, a small fraction of cationic lipids (DOTAP) was incorporated (properties listed in Table 1). Cationic lipids can combat poor release from liposomes after ammonium sulfate gradient-mediated loading [51–53]. Moreover, cationic liposomes preferentially accumulate in tumor endothelium compared to neutral and negatively-charged vesicles [54–56], and can provide tumor-homing complementary to the enhanced permeation and retention (EPR) effect. Drug activity was significantly enhanced in cationic liposomes (+DOX-L)(Fig. 2a).
Fig. 2.

Dependence of DOX efficacy on its encapsulation in neutral (DOX-L) and cationic (+DOX-L) liposomes. (a) In vitro cell growth inhibition of BT-474 cells exposed to free DOX (red circles), DOX in neutral liposomes (blue triangles), and DOX in cationic liposomes (green squares) for 72 hrs. Data expressed as mean ± SD (N≥6). Dashed lines represent dose-effect curves fit to the ME model, with D50 concentrations of DOX, DOX-L and +DOX-L corresponding to 0.3 μM, 33.5 μM, and 6.7 μM, respectively. BT-474 cells imaged via confocal microscopy after 24 hr incubation with (b) no drug, or 1 μM drug-equivalent concentration of DOX in (c) free solution (d) neutral liposomes (e) cationic liposomes. (f) DOX fluorescence (red) intensity is reported as mean ± SD (N=3). Representative images are shown as an average of 10 μm z-stacks. Scale bar=20 μm. *, P < 0.05; **, P < 0.01 performed by two-tailed Student’s t-test.
Table 1.
Physical and chemical properties of single drug-loaded liposomes.
| Liposome Formulation | Liposome Composition | Size (nm) | Zeta Potential (mV) | Drug Incorporation (mol%) |
|---|---|---|---|---|
| DOX-L | 55:45 DSPC:Chol | 154.5 ± 5.0 | −9.55 ± 3.56 | 1.08 ± 0.16 |
| +DOX-L | 80:10:10 DSPC:DOTAP:Chol | 155.6 ± 5.3 | 39.93 ± 4.81 | 0.96 ± 0.13 |
| 5FU-L | 80:10:10 DSPC:DOTAP:Chol | 173.5 ± 43 | 41.7 ± 9.8 | 0.7 ± 0.2 |
| 5FURW-L | 80:10:10 DSPC:DOTAP:Chol | 163.8 ± 17.2 | 32.2 ± 6.5 | 26.6 ± 2.4 |
Data expressed as mean ± SD (N≥3)
The D50 concentration was found to be 5-fold lower than DOX-L. This was likely due to enhanced DOX internalization in +DOX-L, which was 12-fold greater than that of DOX-L (Fig. 2b–f). Both DOX-L and +DOX-L were less active than free DOX (D50 = 0.3 μM), which is expected since liposomes must first overcome the added barrier of active cell internalization before DOX can reach its intracellular target. All liposome formulations thereafter included 10% cationic lipids in order to facilitate drug release and to better preserve anticancer activity.
Enhanced incorporation of 5FU in liposomes by tryptophan esterification
The transmembrane ammonium sulfate gradient, which was specifically developed for incorporating amphipathic anthracyclines, presents difficulty for the entrapment of drugs from different classes. Specifically, 5FU could only be incorporated at 0.7 mol% relative to lipids using the ammonium sulfate gradient (Table 1), congruent with previous studies [57]. To incorporate 5FU and DOX in the same liposome, an analogue of 5FU was developed by improving its compatibility with the transmembrane ammonium sulfate gradient. The analogue, designated as 5FURW, consists of the ribosylated nucleoside form of 5FU (5FUR), conjugated to tryptophans (W) via esterification of 5FUR hydroxyl moieties (Fig. S3).
Contrary to unmodified 5FU, 5FURW exhibits extensive aromaticity and basicity: functionalities responsible for high liposomal DOX encapsulations. DOX’s free amine governs its ability to form a salt in the presence of ammonium sulfate, and Π electron stacking of planar aromatic rings drives intra-liposomal DOX oligomerization [58]. These functionalities, carried by tryptophan, were integrated in 5FURW to improve encapsulation, and were also designed to be cleavable in order to preserve 5FU+DOX synergy. 5FURW can be hydrolyzed to 5FUR, which is further metabolized by uridine phosphorylase to 5FU [59–61]. Furthermore, 5FURW itself exhibits the same anticancer activity as 5FUR (Fig. S4). Thus, 5FUR is projected to behave similarly to 5FU, including its synergistic interaction with DOX. 5FURW yields a 38-fold enhancement in encapsulation to 26.6 mol% (Table 1), a number similar to DOX encapsulation (~14–24%) [49].
Synergistic activity of 5FURW and DOX
Since 5FURW hydrolyzes directly to its precursor, 5FUR, synergistic interactions were investigated between both 5FURW+DOX and 5FUR+DOX. High synergy was observed when high doses of 5FURW were combined with low doses of DOX (R≥75), and slight synergy occurred at the opposite regime, when low doses of 5FURW were combined with high doses of DOX (R=0.1) (Fig. 3a). 5FUR and DOX exhibited synergy at extreme ratios (R≤1 and R=600), as well. These synergistic regimes are similar to the original synergistic interactions observed with unmodified 5FU and DOX, which occurred at R≤0. 5 and R=819. While the exact synergistic ratios are slightly different for each 5FU analogue, the regimes of synergy are similar, which attests to the potent interactions between this particular combination. It was also verified that the cleavable moiety, tryptophan, was non-toxic for 5FURW doses utilized in these studies (Fig. S5); therefore, synergy is attributed to 5FUR+DOX interactions.
Fig. 3.

Verification of synergy between DOX and 5FU derivatives in the solution (a) and liposomal (b) form. (a) CIs for 5FUR+DOX (hatched bars) or 5FURW+DOX (black bars). BT-474 cells were exposed to various ratios of each combination for 72 hours. Drug concentrations (μM) for 5FUR and DOX, respectively, corresponding to each ratio were: 0.1 (0.06, 0.60), 0.5 (0.15, 0.3), 1.0 (0.3, 0.3), 5.0 (1.5, 0.3), 19 (2.8,0.15), 75 (11.25, 0.15), 150 (22.5, 0.15), 300 (22.5, 0.075), 600 (45, 0.075). Drug concentrations (μM) for 5FURW and DOX, respectively, corresponding to each ratio were: 0.1 (0.06, 0.60), 0.5 (0.15, 0.30), 1 (0.30, 0.30), 5 (1.50, 0.30), 19 (5.60, 0.30), 75 (22.50, 0.30), 150 (45.00, 0.30), 300 (45.00, 0.15), 600 (90.0, 0.15). (b) CI calculated for various ratios of 5FURW-L and +DOX-L exposed to BT-474 cells for 72 hours. Drug concentrations (μM) for 5FURW and DOX, respectively, corresponding to each ratio were: 0.1 (0.30, 2.40), 0.5 (0.60, 1.20), 1 (1.20, 1.20), 5 (22.50, 4.70), 19 (45.00, 2.40), 75 (180.00, 2.40), 150 (180.00, 1.20), 300 (180.00, 0.60), 600 (360.00, 0.60). Data shown as average ± SD (N≥6).
Single-drug loaded liposomes were next tested for synergy preservation after encapsulation. Contrary to their free drug counterparts, single drug-loaded liposomes synergistically inhibited cancer cell growth only at one regime, R≤1. Although free drugs were synergistic for R>75, liposome-encapsulated forms were highly antagonistic (CI ≫ 1), with CIs two orders of magnitude greater than those of liposomal synergistic ratios. While a surprising finding, it is not unusual since liposomal encapsulation can impact drug internalization and intracellular concentrations, which further alter drug-drug interactions. Since 5FU+DOX consistently elicited synergistic cancer cell kill at R≤1, regardless of delivery method, this regime was chosen for their dual encapsulation in liposomes.
Co-incorporation of 5FURW and DOX in liposomes
Two ratios of 5FURW+DOX, one synergistic and one antagonistic, were incorporated in liposomes, and a third formulation of PEGylated synergistic liposomes was synthesized for in vivo studies (physical and chemical properties listed in Table 2). Co-loaded liposomes were first assessed in vitro against 4T1 cells, the same cell line ultimately used for in vivo studies. The 4T1 model was chosen to challenge formulations against aggressive tumors in immunocompetent mice. Synergistic 5FURW+DOX liposomes (R=0.18) exhibit superior cell kill compared to either 5FURW- or DOX-loaded liposomes (Fig. 4a–b) (CI=0.31 ± 0.24). Antagonistic combinations of the same drugs (R=12.2) exhibit expected behavior (CI=1.92 ± 1.21).
Table 2.
Physical and chemical properties of liposomes containing both 5FURW and DOX.
| Liposome Formulation | R (5FURW:DOX) | DOX Incorporation (mol%) | 5FURW Incorporation (mol%) | Size (nm) | Zeta Potential (mV) |
|---|---|---|---|---|---|
| ant DAFODIL | 12.2 | 0.45 ± 0.02 | 5.47 ± 0.94 | 156.9 ± 5.7 | 36.2 ± 0.5 |
| -PEG DAFODIL | 0.18 | 7.75 ± 0.09 | 1.41 ± 0.47 | 149.8 ± 15.1 | 35.7 ± 4.3 |
| DAFODIL | 0.15 | 14.82 ± 0.69 | 2.17 ± 0.23 | 168.8 ± 18.7 | −23.0 ± 3.0 |
Lipid composition was 80:10:10 DSPC:DOTAP:Chol, except for DAFODIL, which consisted of 75:5:10:10 DSPC:mPEG-DSPE:DOTAP:Chol. Drug loadings and DLS measurements are reported as mean ± SD (N≥3).
Fig. 4.

Verification of efficacy of 5FURW- and DOX- co-encapsulated liposomes against 4T1 cells. (a) Comparison of 5FURW-liposomes (black circles) to DAFODIL with antagonistic drug ratio (R=12.2, red circles), DAFODIL without PEG (R=0.18, blue squares), and DAFODIL (R=0.15, green diamonds). Significance is provided between 5FURW-L and –PEG DAFODIL at 2.8 μM 5FURW. (b) The same co-loaded liposomes in (a) are compared to DOX-liposomes (black circles), antagonistic DAFODIL with R=12.2 (red circles), DAFODIL without PEG (blue squares) and DAFODIL (green diamonds). Dashed lines represent fits to the ME model. Average calculated CI for the cell inhibition data of antagonistic DAFODIL (R=12.2) and DAFODIL without PEG are 1.92 ± 1.21 and 0.31 ± 0.24, respectively. Data reported as average ± SD (N≥6). (c) Drug release of 5FURW (purple squares) and DOX (black circles) from DAFODIL in PBS at pH 5.5 (open marks) or pH 7.4 (closed marks). Data shown as average ± SD (N=3). Lines represent exponential fits to release profiles (t1/2 = 14.1, 27.7, 26.9, and 35.3 for 5FURW pH 5.5, 5FURW pH 7.4, DOX pH 5.5, and DOX pH 7.4 respectively).
PEGylation of cationic liposomes is necessary for prolonged systemic circulation in order to prevent opsonization [62, 63]. A small fraction (5mol%) PEG2000-DSPE was incorporated in the lipid bilayer, and resulted in a slightly larger size of 168. 8 nm compared to non-PEGylated version (149.8 nm), as well as a change in ζ potential. Upon PEGylation, the cationic lipids became shielded, as was evident in the ζ potential of −23.0 mV. Drug encapsulations and ratios were only slightly altered. The inclusion of PEG allowed twice as much DOX retention compared to non-shielded liposomes, and may be a result of the added barrier of long, hydrophilic polymeric chains which must be overcome for drug leakage. Therefore, R for encapsulated combination shifted from 0.18 to 0.15, in favor of greater drug synergy. The anticancer activity of PEGylated synergistic liposomal formulation surpassed that of non-PEGylated version (Fig. 4a–b), and was therefore labeled as DAFODIL (DOX And 5FU Optimally Delivered In Liposomes). 5FURW is released slightly faster than DOX, and acidic conditions accelerated drug release (Fig. 4c). Therefore, the effective free drug R exposed to cancer cells is slightly higher than the R encapsulated in liposomes, but is evidently still potent at inhibiting cell growth. Delayed DOX release relative to 5FURW may account for the abolishment of high R synergy in liposomes, particularly if a threshold DOX concentration is necessary for synergistic interactions.
Inhibitory effect of 5FURW+DOX liposomes on tumor growth in vivo
Potent multi-drug loaded liposomes were challenged against a highly metastatic and aggressive 4T1 mouse breast cancer model in vivo. This model was also chosen for its robust tumor formation in immuno-competent BALB/c mice, which allows for a more accurate depiction of nanoparticle clearance and efficacy compared to models in immuno-incompetent mice. PEGylated liposomes (DAFODIL) were administered intravenously to mice bearing 4T1 murine breast carcinoma, at drug-equivalent doses of 3 mg/kg DOX + 0.62 mg/kg 5FURW for a total of four i.v. injections. Tumors of both small and large sizes were investigated, where injections started when tumor volumes were ~10 mm3 (see methods) or ~300 mm3 respectively. Assuming similar tumor weight to body mass, a 10 mm3 murine tumor corresponds to a human tumor diameter of 2 cm, whereas the large model corresponds to a 6.3 cm tumor diameter. Clinical tumor diameters of 1 cm encompass 58% of breast cancer cases, whereas tumor diameters > 5 cm are present in ~11% of patients and corresponds to poorer prognosis [64]. Significant tumor reduction was achieved by DAFODIL (Fig 5a). By day 20, when survival of untreated mice dropped to 50%, DAFODIL elicited 91% (77 ± 11 vs. 904 ± 98 mm3) tumor growth inhibition, whereas free 5FURW+DOX at the same doses were only capable of inhibiting only 39% tumor growth (547 ± 49 vs. 904 ± 98 mm3). Moreover, all tumors treated with free 5FURW+DOX eventually grew to the same sizes as control mice, and hence were only able to extend average survival of untreated mice by 4 days (25 vs. 21days). By contrast, DAFODIL treatments extended median survival rates (Fig. 5b) by at least 44 days. Free drug combinations as well as liposomal combination were well tolerated as judged by the body weight (Fig. 5c).
Fig. 5.

In vivo efficacy of DAFODIL in 4T1 tumor bearing mice. (a) 4T1 tumor volumes of untreated BALB/c mice (black circles), mice treated with DAFODIL (green squares), or free 5FURW and DOX in saline (red triangles) at drug-equivalent doses of 0.61 mg/kg 5FURW and 3 mg/kg DOX (R=0.15). Data shown until the first DAFODIL-treated mouse is sacrificed. Significance is provided for the last day when untreated mice survival > 50%. (b) Survival rates for all experimental groups. (c) Corresponding body weight changes of tumor-bearing mice are provided for all groups. Data shown as mean ± SEM (Initial N=8, and varies according to survival).
Contrary to free drug-treated mice, most DAFODIL-treated mice reached a peak tumor volume between 50–80 mm3 on Day 25, followed by tumor regression and finally, complete disappearance in 5 out of 8 mice. Only 3 of 8 tumors from this group eventually grew. No detectable tumors in 5 of 8 mice were observed for the remainder of the study. Tumors in 3 DAFODIL-treated mice that showed detectable growth grew significantly slower than those in untreated mice. Whereas all untreated mice reached 1000 mm3 tumor volumes by Day 26, the first DAFODIL-treated mouse to reach 1000 mm3 tumor volume occurred on Day 44. The average survival was significantly extended by at least 40 days (>160 % over untreated mice) when treated with the liposomal drug combination (Fig. 5b). To the best of our knowledge, this is the first time that 4T1 tumor growth was inhibited by >90% at a cumulative DOX dose <15 mg/kg, either used as a single agent or combined with another chemotherapy drug [44, 65–72]. Biodistribution studies were conducted to see if antitumor efficacy was dictated by high uptake in tumors (Fig. S9b). Liposomes exhibited biodistributions and clearance (Fig. S9a) typical of PEGylated liposomes [43, 44, 73], with greatest accumulation occurring in the liver and spleen, and 2.3% initial dose per g (ID/g) present in tumors. Therefore, the enhanced antitumor efficacy is likely attributed to the potent synergistic interactions between 5FURW and DOX and not enhanced tumor accumulation.
Liposomal treatment of large (300 mm3) tumors also exhibited significant antitumor effects. By Day 11, 48% tumor reduction (530 vs. 1018 mm3) was achieved with DAFODIL treatments of the same dose as the small tumor study (Fig. S10a, b). The median survival was extended by 190% (19 vs. 10 days, Fig. S10c) and demonstrated the potential of synergistic liposomes for treating aggressive and late stage cancers.
DISCUSSION
This study shows that low dose chemotherapy, if co-delivered in synergistic ratios, is in fact capable of completely regressing small tumors. Chemotherapy efficacy is hindered by poor specificity, so nanoparticle delivery is imperative in order to alleviate distribution to healthy organs. Nanoparticles promote accumulation in tumors via the EPR effect. Although liposomes have been applied for chemotherapy delivery since 1995, overall response rates have not significantly improved compared to free drug administration [5, 74]. The new generation liposomes reported here not only significantly improve therapeutic efficacy of free chemotherapy administrations, but also maintain the paramount safety benefit of liposomal formulations. 5FU+DOX combination in particular has been utilized in numerous clinical chemotherapy combinations, but this is the first investigation, to our knowledge, which aimed at identifying and delivering synergistic ratios of the pair to tumors. This widely-used pair has been repeatedly reported to improve partial response rates, however complete response rates have been rare and median survival times are still typically less than a year. Utilizing synergy, we demonstrated the immense therapeutic potential of 5FU+DOX at low doses. Commonly regarded as inactive, low dose chemotherapy elicited remarkable tumor disappearance due to co-delivery of optimal ratios of 5FU and DOX in liposomes.
Drug ratios have long been determined to dictate combination chemotherapy potency in vitro, but FDA-approved combinations have yet to incorporate this important factor. Our results confirm enhanced cytotoxic activity at synergistic ratios. Studies with healthy breast epithelial cell line MCF 10A also introduced another advantage of ratiometric delivery; selective cancer cell toxicity. At synergistic R, much less total drug dose is required to kill cancer cells compared to MCF 10A. The reverse was also true; at antagonistic R or even free drug incubations, less drug is required to inhibit healthy MCF 10A cell growth compared to malignant cells. Thus, the specificity of chemotherapy treatment is improved simply by combining the two drugs in a proper, optimized ratio. The same trends were observed for a control endothelial cell line, bEnd.3 (Fig. S1c–d).
The mechanism of synergy between 5FU and DOX may be multi-faceted. As seen in Fig. S2, the presence of 5FU amplifies DOX-generated ROS in cancer cells, and likely contributes to enhanced cell death. However, concerted cell cycle phase arrests may be another plausible reason for 5FU+DOX anticancer synergy. 5FU is a cell cycle-specific drug which causes accumulation of cells in the G1/S phases [75–78], whereas the cell cycle-specificity of DOX is controversial. One report by Ling and co-workers demonstrated that DOX-induced cytotoxicity can be significantly enhanced when cells are previously synchronized in the S phase [79]. By first imposing a thymidine block on cancer cells, the researchers were able to accumulate cells in the S phase and subsequently reduce DOX D50 concentrations 3.3-fold relative to concentrations needed for asynchronized cells. This enhanced DOX potency also correlated with significant accumulation of cancer cells in the G2/M phases. In this report, it is likely that 5FU, a thymidine analogue, synchronizes cancer cells in the G1/S phases and further allows DOX to arrest cells in the G2/M phases. Consequently, the combination elicits significantly greater cancer cell death when combined rather than when used alone. However, it is clear that this effect does not always occur, particularly when 5FU and DOX are introduced at R=6551. It is likely that at very large R, not enough DOX is present relative to 5FU in order to promote further progression of cancer cells to the G2/M phases, and their synergistic cell cycle effects may not be realized.
Selectivity of synergistic combination towards cancer cells over non-malignant cells may be attributed to inherently different cytotoxicity mechanisms the drugs elicit depending on the cell type. For instance, susceptibility for cell death after 5FU treatment greatly depends on the activity of various enzymes responsible for RNA and DNA incorporation, such as uridine phosphorylase and thymidylate synthase. The activities of these enzymes have been shown to differ between various cells and seemed to correlate with drug sensitivity [80]. Altered levels of these enzymes can influence the ability of 5FU to synchronize cells in the G1/S phases, and is one plausible reason for reduced combination activity in bEnd.3 and MCF 10A cells. Moreover, DOX has been previously reported to trigger apoptosis in endothelial cells and cancer cells via different pathways. DOX-mediated generation of ROS has been shown to play a pivotal role in endothelial cell apoptosis, but not in tumor cell apoptosis [81]. Hence, DOX activity in endothelial cells may not promote the progression of S-synchronized cells to the G2/M phases in the same way it can in cancer cells. Such differences in drug sensitivity may prevent cell cycle effects between 5FU and DOX that are seen in cancer cells, and provide a probable cause for reduction in combination activity. Results from these studies suggest that the enhanced cell inhibition of 5FU+DOX at R=819 is specific for cancer cells. In fact, the combination may not be any more toxic than either 5FU or DOX when administered as a single agent because the combination, at best, only inhibits as much cell growth as DOX alone. This is a remarkable finding, since amplified adverse side effects due to co-localization of multiple drugs in essential organs is a major challenge with combination chemotherapies. Our results suggest that this challenge can potentially be overcome by optimizing drug pair and ratio, so that synergistic cell kill occurs in malignant cells, but not in healthy cells. Further in vivo or ex vivo studies, wherein 5FU+DOX are exposed to tissues in their natural environments, would need to be executed to assess this hypothesis.
To incorporate a synergistic ratio of 5FU+DOX in a single, effective carrier, an optimal liposome formulation and 5FU entrapment method was developed. A small fraction of cationic lipids (10 mol%) enhanced DOX delivery and activity relative to completely zwitterionic liposomes. This is likely due to enhanced ionic interactions between liposome and cell membranes. It is in fact thought that the positively-charged liposomes are first associated with cell membranes via ionic interactions, and then are capable of fusing with cells by flip-flopping with anionic phospholipids [51, 82]. This fusion allows liposome drug payloads to escape and release into the cytoplasm or even nucleus, where chemotherapy drugs can then attack their targets. The small fraction of cationic lipids in the liposome was non-toxic at relevant concentrations for drug delivery (Fig. S6).
The novel prodrug reported here, 5FURW, allowed high incorporation in liposomes, to the same extent as DOX (~26 mol% relative to lipids). To the best of our knowledge, this is the first time 5FU was successfully incorporated in liposomes bearing a pH gradient. Regardless of initial drug loading, time of drug loading, and temperature, unmodified 5FU was demonstrated incapable of measurable integration in liposomes with an acidic core [57]. In fact, it was previously reported that the exact opposite approach, utilizing basic media (~pH 8.6), is required for liposomal encapsulation of 5FU [83]. A possible reason for this is that 5FU lacks a charge, and can therefore easily pass through the lipophilic bilayer to escape the liposome. 5FU has only been reported to exist in anionic forms, which occur at neutral pH [84], and hence not in liposomes bearing an acidic pH gradient. Thus, for the purpose of simultaneously co-delivering 5FU+DOX, it was imperative that we develop a new method. Free amine and aromatic modifications to 5FUR helped overcome poor encapsulation issues, and were also designed to be cleavable in order to restore the active drug which is synergistic with DOX. While this approach was demonstrated only for 5FU, it can be theoretically applied to the entire class of nucleobase analogue chemotherapies, such as cytarabine, gemcitabine, and decitabine, all of which contain pendant hydroxyl groups for tryptophan conjugation. Tryptophan modification has implications not only for delivering agents that previously could not be liposomally-entrapped, but also for combination co-delivery of the many drugs that can be compatible with this encapsulation method.
Similar to co-administered single-drug loaded liposomes, 5FURW and DOX co-encapsulated in the same liposome at R<1 inhibited greater cancer cell growth than either single-drug loaded liposome (Fig. 4a–b). This was evident for both non-PEGylated and PEGylated liposomes. However, the true merit of PEGylated 5FURW+DOX co-encapsulated liposomes (DAFODIL) was captured in vivo when challenged against a 4T1 murine mammary carcinoma tumor. Non-PEGylated liposomes were unable to prolong survival rates (Fig. S7), while DAFODIL was able to completely reverse tumor growth and cause tumor disappearance in 62.5% of treated mice. This is not surprising, as PEGylation prevents opsonization and prolongs systemic circulation. What is surprising, however, is the low doses that were sufficient to achieve immense antitumor effects. Mice were treated with 4 total injections of 3 mg/kg DOX and 0.62 mg/kg 5FURW, for a total DOX and 5FURW dose of 12 mg/kg and 2.5 mg/kg, respectively. To the best of our knowledge, never before has cumulative DOX doses <15 mg/kg been able to inhibit aggressive 4T1 tumor growth by >90%, regardless of its use as a single agent or in combination with another chemotherapy drug [44, 65–72]. Doxil alone is required at doses of 20 mg/kg to provide significant 4T1 tumor reduction, as reported by previous studies [44]. This is also the first instance of 5FU-liposomes which were both well-tolerated and effective at inhibiting tumor growth, let alone at remarkably low doses [55, 85]. Moreover, tumor disappearance was maintained for the remainder of the study, and resulted in median survival rates greater than 65 days post-treatment. DAFODIL was also effective in treating large tumors at the same low-dose and dosing frequency, resulting in 48% reduction in tumor growth 12 days post-treatment. Through these studies, it was evident that nanoparticle co-delivery is crucial to manifest the potency of 5FU+DOX in vivo, as free drug equivalents were only able to prolong median survival rates from 21 to 25 days, and were largely ineffective. This is likely due to uncoordinated drug pharmacokinetics and fast plasma clearance of the small molecule drugs when injected intravenously as free solutions. Clinical studies have shown that 5FU and DOX exhibit elimination half-lives of 8–22 minutes [86] and 4–5 minutes [87], respectively. These rapid clearance rates demonstrate a clear need for nanoparticle delivery to ensure concurrent delivery to tumors.
Collectively, studies investigated here demonstrate the vast, untapped therapeutic potential harbored by low dose chemotherapy. Although liposome-entrapment can alleviate chemotherapy side effects, it has not necessarily improved patient survival or response rates. By identifying and co-encapsulating synergistic chemotherapy combinations in liposomes, it was possible to deliver effective therapies that were also safe. Ratiometric drug delivery has been largely explored in vitro, yet clinical translation of synergistic combinations is still rare. Furthermore, encapsulation of multiple drugs in liposomes, apart from a few exceptions [39, 40], achieving therapeutic doses of both drugs has been a challenging problem. Novel methods for nucleobase entrapment described here provide a means to manifest 5FU+DOX synergy in vivo. More notably, findings here suggest a transformative methodology for evaluating chemotherapy clinically. Instead of dose pushing, chemotherapy can be engineered to maximize tumor growth inhibition and minimize off-target toxicity through the use of synergistic combinations. High encapsulation methods and tumor-homing can further achieve these effects with low cumulative doses which were previously regarded as therapeutically inactive.
Chemotherapy is conventionally administered at the maximum dose possible, risking serious adverse effects in exchange for hope of maximum tumor reduction. Despite dose pushing and numerous efforts to improve chemotherapy, tumor eradications are rare. Drug combinations are widely proposed to improve therapeutic efficacies, encompassing 25% of all oncology clinical trials since 2008 [88]. Until now, however, little emphasis has been devoted towards fundamentally understanding and optimizing combination chemotherapy efficacy. Our results demonstrate that complete tumor disappearance can be achieved, contrary to convention, at remarkably low chemotherapy doses if the combination is administered at an optimal synergistic ratio. This study provides a basis for transforming combination treatments and capturing their full therapeutic potential by delivering the correct drug ratio to tumors.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the use of the Biological Nanostructures Laboratory within the California NanoSystems Institute, supported by the University of California, Santa Barbara and the University of California, Office of the President, the Materials Research Laboratory (MRL) Shared Experimental Facilities, supported by the MRSEC Program of the NSF under Award No. DMR 1121053; a member of the NSF-funded Materials Research Facilities Network, as well as the NRI-MCDB Microscopy Facility, funded by NIH Grant No. 1 S10 OD010610-01A1.D.R.V. was supported by NSF Graduate Research Fellowship under Grant No. DGE-1144085. Authors acknowledge partial support from UCSB Duncan and Suzanne Mellichamp cluster and Department of Defense CDMRP Breast Cancer Research Program (W81XWH-11-1-0110).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lyass O, et al. Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer. 2000;89(5):1037–47. doi: 10.1002/1097-0142(20000901)89:5<1037::aid-cncr13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 2.Safra T, et al. Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Annals of Oncology. 2000;11(8):1029–1033. doi: 10.1023/a:1008365716693. [DOI] [PubMed] [Google Scholar]
- 3.Muggia FM, et al. Phase II study of liposomal doxorubicin in refractory ovarian cancer: antitumor activity and toxicity modification by liposomal encapsulation. Journal of Clinical Oncology. 1997;15(3):987–993. doi: 10.1200/JCO.1997.15.3.987. [DOI] [PubMed] [Google Scholar]
- 4.Judson I, et al. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL®/CAELYX®) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: a study by the EORTC Soft Tissue and Bone Sarcoma Group. European Journal of Cancer. 2001;37(7):870–877. doi: 10.1016/s0959-8049(01)00050-8. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien MER, Wigler N, Inbar M. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYXTM/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Annals of Oncology. 2004;15(3):440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 6.Mayer LD, et al. Influence of vesicle size, lipid composition, and drug-to-lipid ratio on the biological activity of liposomal doxorubicin in mice. Cancer research. 1989;49:5922–5930. [PubMed] [Google Scholar]
- 7.Cabanes A, Tzemach D, Goren D. Comparative study of the antitumor activity of free doxorubicin and polyethylene glycol-coated liposomal doxorubicin in a mouse lymphoma model. Clinical Cancer Research. 1998;4:499–505. [PubMed] [Google Scholar]
- 8.Ellerhorst J, et al. Phase II trial of doxil for patients with metastatic melanoma refractory to frontline therapy. Oncology reports. 1999;6(5):1097–1106. doi: 10.3892/or.6.5.1097. [DOI] [PubMed] [Google Scholar]
- 9.Halford S, et al. A phase II study evaluating the tolerability and efficacy of CAELYX (liposomal doxorubicin, Doxil) in the treatment of unresectable pancreatic carcinoma. Annals of Oncology. 2001;12(10):1399–1402. doi: 10.1023/a:1012522120294. [DOI] [PubMed] [Google Scholar]
- 10.Muggia FM, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. Journal of clinical oncology. 2002;20(9):2360–2364. doi: 10.1200/JCO.2002.08.171. [DOI] [PubMed] [Google Scholar]
- 11.Barenholz Y. Doxil®--the first FDA-approved nano-drug: lessons learned. Journal of Controlled Release. 2012;160(2):117–34. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 12.Barenholz Y. Liposome application: problems and prospects. Current opinion in colloid & interface science. 2001;6(1):66–77. [Google Scholar]
- 13.Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Progress in lipid research. 2003;42(6):463–478. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 14.Andresen TL, Jensen SS, Jørgensen K. Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Progress in lipid research. 2005;44(1):68–97. doi: 10.1016/j.plipres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell MJ, et al. E-selectin liposomal and nanotube-targeted delivery of doxorubicin to circulating tumor cells. Journal of controlled release. 2012;160(3):609–617. doi: 10.1016/j.jconrel.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang FY, et al. Focused ultrasound and interleukin-4 receptor-targeted liposomal doxorubicin for enhanced targeted drug delivery and antitumor effect in glioblastoma multiforme. Journal of controlled release. 2012;160(3):652–658. doi: 10.1016/j.jconrel.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 17.Schiffelers RM, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. Journal of Controlled Release. 2003;91(1):115–122. doi: 10.1016/s0168-3659(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 18.Gabizon A, et al. In Vivo Fate of Folate-Targeted Polyethylene-Glycol Liposomes in Tumor-Bearing Mice. 2003;9:6551–6559. [PubMed] [Google Scholar]
- 19.Gupta B, V, Torchilin P. Monoclonal antibody 2C5-modified doxorubicin-loaded liposomes with significantly enhanced therapeutic activity against intracranial human brain U-87 MG tumor xenografts in nude mice. Cancer immunology, immunotherapy. 2007;56(8):1215–23. doi: 10.1007/s00262-006-0273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Budman DR, Calabro A, Kreis W. In vitro evaluation of synergism or antagonism with combinations of new cytotoxic agents. Anti-cancer drugs. 1998;9(8):697–697. doi: 10.1097/00001813-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Leu KM, et al. Laboratory and clinical evidence of synergistic cytotoxicity of sequential treatment with gemcitabine followed by docetaxel in the treatment of sarcoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22(9):1706–12. doi: 10.1200/JCO.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 22.Soma CE, et al. Reversion of multidrug resistance by co-encapsulation of doxorubicin and cyclosporin A in polyalkylcyanoacrylate nanoparticles. Biomaterials. 2000;21(1):1–7. doi: 10.1016/s0142-9612(99)00125-8. [DOI] [PubMed] [Google Scholar]
- 23.Wils JA, Klein HO. Methotrexate and fluorouracil combined with doxorubicin--a step ahead in the treatment of advanced gastric cancer: a trial of the European Organization for Research. Journal of Clinical …. 1991;9(5):827–831. doi: 10.1200/JCO.1991.9.5.827. [DOI] [PubMed] [Google Scholar]
- 24.Murad aM, et al. Modified therapy with 5-fluorouracil, doxorubicin, and methotrexate in advanced gastric cancer. Cancer. 1993;72(1):37–41. doi: 10.1002/1097-0142(19930701)72:1<37::aid-cncr2820720109>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 25.Macdonald JS. 5-Fluorouracil, doxorubicin, and mitomycin (FAM) combination chemotherapy for advanced gastric cancer. Annals of internal medicine. 1980;93:533–536. doi: 10.7326/0003-4819-93-4-533. [DOI] [PubMed] [Google Scholar]
- 26.Levi JA, et al. Analysis of a prospectively randomized comparison of doxorubicin versus 5-fluorouracil, doxorubicin, and BCNU in advanced gastric cancer: implications for future studies. Journal of Clinical Oncology. 1986;4(9):1348–1355. doi: 10.1200/JCO.1986.4.9.1348. [DOI] [PubMed] [Google Scholar]
- 27.Cazap E, et al. Phase II trials of 5-FU, doxorubicin, and cisplatin in advanced, measurable adenocarcinoma of the lung and stomach. Cancer treatment reports. 1986;70(6):781–783. [PubMed] [Google Scholar]
- 28.Klein HO, Buyse M, Wils JA. Prospective randomized trial using 5-fluorouracil, adriamycin and methotrexate (FAMTX) versus FAM for treatment of advanced gastric cancer. Oncology Research and Treatment. 1992;15:364–367. [Google Scholar]
- 29.Vanhoefer U, Rougier P. Final results of a randomized phase III trial of sequential high-dose methotrexate, fluorouracil, and doxorubicin versus etoposide, leucovorin, and fluorouracil versus. Journal of Clinical Oncology. 2000;18(14):2648–2657. doi: 10.1200/JCO.2000.18.14.2648. [DOI] [PubMed] [Google Scholar]
- 30.Webb A, Cunningham D. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. Journal of Clinical Oncology. 1997;15(1):261–267. doi: 10.1200/JCO.1997.15.1.261. [DOI] [PubMed] [Google Scholar]
- 31.Cullinan SA, et al. A comparison of three chemotherapeutic regimens in the treatment of advanced pancreatic and gastric carcinoma: fluorouracil vs fluorouracil and doxorubicin vs fluorouracil, doxorubicin, and mitomycin. Jama. 1985;253(14):2061–2067. [PubMed] [Google Scholar]
- 32.Hortobagyi GN, Bodey GP. Evaluation of high-dose versus standard FAC chemotherapy for advanced breast cancer in protected environment units: a prospective randomized study. Journal of Clinical …. 1987;5(3):354–364. doi: 10.1200/JCO.1987.5.3.354. [DOI] [PubMed] [Google Scholar]
- 33.Buzzoni R, et al. Adjuvant chemotherapy with doxorubicin plus cyclophosphamide, methotrexate, and fluorouracil in the treatment of resectable breast cancer with more than three positive axillary nodes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1991;9(12):2134–40. doi: 10.1200/JCO.1991.9.12.2134. [DOI] [PubMed] [Google Scholar]
- 34.Gabra H, Cameron DA, Lee LE. Weekly doxorubicin and continuous infusional 5-fluorouracil for advanced breast cancer. British journal of cancer. 1996;74:2008–2012. doi: 10.1038/bjc.1996.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krakoff IH. Chemotherapy of gastrointestinal cancer. Cancer. 1972;30(6):1600–1603. doi: 10.1002/1097-0142(197212)30:6<1600::aid-cncr2820300628>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 36.Hansen R, et al. Continuous 5-fluorouracil infusion in refractory carcinoma of the breast. Breast Cancer Research and Treatment. 1987;10(2):145–149. doi: 10.1007/BF01810577. [DOI] [PubMed] [Google Scholar]
- 37.Gordon AN, et al. Recurrent Epithelial Ovarian Carcinoma: A Randomized Phase III Study of Pegylated Liposomal Doxorubicin Versus Topotecan. Journal of Clinical Oncology. 2001;19(14):3312–3322. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- 38.Cascinu S, et al. Pegylated liposomal doxorubicin, 5-fluorouracil and cisplatin versus mitomycin-C, 5-fluorouracil and cisplatin for advanced gastric cancer: a randomized phase II trial. Cancer Chemotherapy and Pharmacology. 2010;68(1):37–43. doi: 10.1007/s00280-010-1424-8. [DOI] [PubMed] [Google Scholar]
- 39.Mayer LD, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Molecular Cancer Therapeutics. 2006;5(7):1854–63. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]
- 40.Tardi P, et al. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leukemia research. 2009;33(1):129–39. doi: 10.1016/j.leukres.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 41.Camacho KM, et al. Synergistic Antitumor Activity of Camptothecin-Doxorubicin Combinations and their Conjugates with Hyaluronic Acid. Journal of Controlled Release. 2015;210:198–207. doi: 10.1016/j.jconrel.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gabizon A, et al. Effect of liposome composition and other factors on the targeting of liposomes to experimental tumors: biodistribution and imaging studies. Cancer research. 1990;50(19):6371–6378. [PubMed] [Google Scholar]
- 43.Chang YJ, et al. Biodistribution, pharmacokinetics and microSPECT/CT imaging of 188Re-bMEDA-liposome in a C26 murine colon carcinoma solid tumor animal model. Anticancer research. 2007;27(4B):2217–2225. [PubMed] [Google Scholar]
- 44.Liu CM, et al. Comparison of the therapeutic efficacy of 188Rhenium-liposomes and liposomal doxorubicin in a 4T1 murine orthotopic breast cancer model. Oncology reports. 2012;27(3):678–684. doi: 10.3892/or.2011.1557. [DOI] [PubMed] [Google Scholar]
- 45.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58(3):621–621. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 46.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 47.Szoka F, Jr, Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes) Annual review of biophysics and bioengineering. 1980;9(1):467–508. doi: 10.1146/annurev.bb.09.060180.002343. [DOI] [PubMed] [Google Scholar]
- 48.Tardi PG, Boman NL, Cullis PR. Liposomal doxorubicin. Journal of drug targeting. 1996;4(3):129–40. doi: 10.3109/10611869609015970. [DOI] [PubMed] [Google Scholar]
- 49.Haran G, et al. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochimica et biophysica acta. 1993;1151(2):201–15. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 50.Pool G, et al. Use of radiolabeled hexadecyl cholesteryl ether as a liposome marker. Lipids. 1982;17(6):448–452. doi: 10.1007/BF02535225. [DOI] [PubMed] [Google Scholar]
- 51.Zelphati O, Szoka FC. Mechanism of oligonucleotide release from cationic liposomes. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(21):11493–8. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett CF, et al. Cationic lipids enhance cellular uptake and activity of phosphorothioate antisense oligonucleotides. Molecular Pharmacology. 1992;41(6):1023–1033. [PubMed] [Google Scholar]
- 53.Friend DS, Papahadjopoulos D, Debs RJ. Endocytosis and intracellular processing accompanying transfection mediated by cationic liposomes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1996;1278(1):41–50. doi: 10.1016/0005-2736(95)00219-7. [DOI] [PubMed] [Google Scholar]
- 54.Thurston G, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. The Journal of clinical investigation. 1998;101(7):1401–13. doi: 10.1172/JCI965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalra AV, Campbell RB. Development of 5-FU and doxorubicin-loaded cationic liposomes against human pancreatic cancer: Implications for tumor vascular targeting. Pharmaceutical research. 2006;23(12):2809–17. doi: 10.1007/s11095-006-9113-3. [DOI] [PubMed] [Google Scholar]
- 56.Campbell RB, Fukumura D, Brown EB. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Research. 2002;62:6831–6836. [PubMed] [Google Scholar]
- 57.Costa CAMD, Moraes AM. Encapsulation of 5-fluorouracil in liposomes for topical administration. Acta Sci Technol. 2003;(1):53–61. [Google Scholar]
- 58.Barenholz Y, et al. Stability of liposomal doxorubicin formulations: problems and prospects. Medicinal research reviews. 1993;13(4):449–491. doi: 10.1002/med.2610130404. [DOI] [PubMed] [Google Scholar]
- 59.Longley DB, Johnston PG. 5-Fluorouracil: Molecular Mechanisms of Cell Death. Human Press Inc; Totawa, NJ: 2007. pp. 263–278. [Google Scholar]
- 60.Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nature reviews Cancer. 2003;3(5):330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 61.Ishitsuka H, et al. Role of uridine phosphorylase for antitumor activity of 5′-deoxy-5-fluorouridine. Gann= Gan. 1980;71(1):112–123. [PubMed] [Google Scholar]
- 62.Levchenko TS, Rammohan R. Liposome clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. International journal of …. 2002;240:95–102. doi: 10.1016/s0378-5173(02)00129-1. [DOI] [PubMed] [Google Scholar]
- 63.Klibanov AL, Maruyama K. Activity of amphipathic poly (ethylene glycol) 5000 to prolong the circulation time of liposomes depends on the liposome size and is unfavorable for immunoliposome. Biochimica et Biophysica Acta (BBA) 1991;1062:142–148. doi: 10.1016/0005-2736(91)90385-l. [DOI] [PubMed] [Google Scholar]
- 64.Carter CL, Allen C, Henson DE. Relation of tumor size, lymph node status, and survival in 24,740 breast cancer cases. Cancer. 1989;63(1):181–187. doi: 10.1002/1097-0142(19890101)63:1<181::aid-cncr2820630129>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 65.Sun Y, et al. Bioreducible PAA-g-PEG graft micelles with high doxorubicin loading for targeted antitumor effect against mouse breast carcinoma. Biomaterials. 2013;34(28):6818–28. doi: 10.1016/j.biomaterials.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 66.Charrois GJR, Allen TM. Multiple injections of pegylated liposomal doxorubicin: pharmacokinetics and therapeutic activity. Journal of Pharmacology and Experimental Therapeutics. 2003;306(3):1058–1067. doi: 10.1124/jpet.103.053413. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, et al. Codelivery of Doxorubicin and Paclitaxel by Crosslinked Multilamellar Liposome Enables Synergistic Antitumor Activity. Molecular pharmaceutics. 2014;11:1651–1661. doi: 10.1021/mp5000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Du G, et al. Quercetin greatly improved therapeutic index of doxorubicin against 4T1 breast cancer by its opposing effects on HIF-1α in tumor and normal cells. Cancer chemotherapy and pharmacology. 2010;65(2):277–287. doi: 10.1007/s00280-009-1032-7. [DOI] [PubMed] [Google Scholar]
- 69.Bandyopadhyay A, et al. Doxorubicin in combination with a small TGFbeta inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLOS ONE. 2010;5(4):e10365–e10365. doi: 10.1371/journal.pone.0010365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang H, et al. Dexamethasone as a chemosensitizer for breast cancer chemotherapy: potentiation of the antitumor activity of adriamycin, modulation of cytokine expression, and pharmacokinetics. International journal of oncology. 2007;30(4):947–953. [PubMed] [Google Scholar]
- 71.Wang J, et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33(28):6877–6888. doi: 10.1016/j.biomaterials.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 72.Mastria EM, et al. Doxorubicin-conjugated polypeptide nanoparticles inhibit metastasis in two murine models of carcinoma. Journal of Controlled Release. 2015;208:52–58. doi: 10.1016/j.jconrel.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrington K, et al. Biodistribution and pharmacokinetics of111In-DTPA-labelled pegylated liposomes in a human tumour xenograft model: implications for novel targeting strategies. British journal of cancer. 2000;83(2):232. doi: 10.1054/bjoc.1999.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jehn C, et al. First safety and response results of a randomized phase III study with liposomal platin in the treatment of advanced squamous cell carcinoma of the head and neck (SCCHN) Anticancer research. 2008;28(6B):3961–3964. [PubMed] [Google Scholar]
- 75.Lewin F, et al. Effect of 5-fluorouracil on the cell growth and cell cycle kinetics of a mouse ascites tumor growing in vivo. Acta Oncologica. 1987;26(2):125–131. doi: 10.3109/02841868709091751. [DOI] [PubMed] [Google Scholar]
- 76.Mirjolet JF, et al. G(1)/S but not G(0)/G(1)cell fraction is related to 5-fluorouracil cytotoxicity. Cytometry. 2002;48(1):6–13. doi: 10.1002/cyto.10087. [DOI] [PubMed] [Google Scholar]
- 77.Li MH, et al. Effect of 5-fluorouracil on G1 phase cell cycle regulation in oral cancer cell lines. Oral oncology. 2004;40(1):63–70. doi: 10.1016/s1368-8375(03)00136-2. [DOI] [PubMed] [Google Scholar]
- 78.Yoshiba S, et al. Hypoxia induces resistance to 5-fluorouracil in oral cancer cells via G 1 phase cell cycle arrest. Oral oncology. 2009;45(2):109–115. doi: 10.1016/j.oraloncology.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 79.Ling YH, et al. Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Molecular pharmacology. 1996 [PubMed] [Google Scholar]
- 80.Peters GJ, et al. Sensitivity of human, murine, and rat cells to 5-fluorouracil and 5′-deoxy-5-fluorouridine in relation to drug-metabolizing enzymes. Cancer research. 1986;46(1):20–28. [PubMed] [Google Scholar]
- 81.Wang S, et al. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. The Journal of biological chemistry. 2004;279(24):25535–43. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]
- 82.Wrobel I, Collins D. Fusion of cationic liposomes with mammalian cells occurs after endocytosis. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1995;1235(2):296–304. doi: 10.1016/0005-2736(95)80017-a. [DOI] [PubMed] [Google Scholar]
- 83.Kaiser N, et al. 5-Fluorouracil in vesicular phospholipid gels for anticancer treatment: entrapment and release properties. International Journal of Pharmaceutics. 2003;256(1–2):123–131. doi: 10.1016/s0378-5173(03)00069-3. [DOI] [PubMed] [Google Scholar]
- 84.Markova N, Enchev V, Ivanova G. Tautomeric equilibria of 5-fluorouracil anionic species in water. The journal of physical chemistry A. 2010;114(50):13154–62. doi: 10.1021/jp1063879. [DOI] [PubMed] [Google Scholar]
- 85.Thomas AM, et al. Development of a liposomal nanoparticle formulation of 5-fluorouracil for parenteral administration: Formulation design, pharmacokinetics and efficacy. Journal of Controlled Release. 2011;150(2):212–9. doi: 10.1016/j.jconrel.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 86.Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clinical pharmacokinetics. 1989 doi: 10.2165/00003088-198916040-00002. [DOI] [PubMed] [Google Scholar]
- 87.Gabizon A, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Research. 1994;54:987–992. [PubMed] [Google Scholar]
- 88.WUM, et al. Biocomputing 2015. WORLD SCIENTIFIC; 2014. CHARACTERISTICS OF DRUG COMBINATION THERAPY IN ONCOLOGY BY ANALYZING CLINICAL TRIAL DATA ON CLINICALTRIALS.GOV; pp. 68–79. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.