

Abstract
Steady state alveolar macrophages (AM) are long-lived lung-resident macrophages with sentinel function. Evidence suggests that AM precursors originate during embryogenesis and populate lungs without replenishment by circulating leukocytes. However, their presence and persistence are unclear following human lung transplantation (LTx). Our goal was to examine donor AM longevity and evaluate whether AM of recipient origin seeds the transplanted lungs. Origin of AM was accessed using donor-recipient HLA mismatches. We demonstrate that 94–100% of AM present in bronchoalveolar lavage (BAL) were donor derived and importantly AM of recipient origin were not detected. Further, analysis of BAL cells up to 3.5 yrs post-LTx revealed that majority of AM (>87%) was donor derived. Elicitation of de novo donor specific antibody (DSA) is a major post-LTx complication and a risk factor for development of chronic rejection. The donor AM responded to anti-HLA framework Ab with secretion of inflammatory cytokines. Further, in an experimental murine model, we demonstrate that adoptive transfer of allogeneic AM stimulated humoral and cellular immune responses to alloantigen and lung-associated self-antigens and led to bronchiolar obstruction. Therefore, donor derived AM play an essential role in the DSA induced inflammatory cascade leading to obliterative airway disease of the transplanted lungs.
Introduction
Tissue resident macrophages (TRM) represent distinct cell populations in terms of phenotype and function (1, 2). Despite heterogeneity, TRM are adapted to their tissue of residence and participate in various tissue specific immunologic and physiologic functions (3). The requirement at lung microenvironment is unique, as during gas exchange the respiratory surface to be protected from potential airborne infectious and polluting agents. Alveolar macrophages (AM) and interstitial macrophages (IM) are two recognized lung TRM that respectively constitute 80% and 20% of lung resident macrophage pool. Further, vast majority of AM occur in the lumen of alveoli and bronchioles, and comprise up to 95% of the leukocytes present in mouse bronchoalveolar lavage (BAL) under steady state (4). Mature mouse lungs contain up to 3×106 AM while that of humans contain up to 6×109 AM (5). AM are professional phagocytes and have been implicated in homeostatic maintenance of the respiratory surface, and airway immunity and inflammation (1). Furthermore, AM are stationary cells and are found attached to the alveolar epithelium during steady state as well as inflammatory challenges (6). A selective loss of AM results in lung failure, fatal hypoxia, severe morbidity and mortality due to viral infections (7). Functional impairment of AM has been linked to pulmonary alveolar proteinosis (8). While role of AM in gaseous exchange and immune surveillance at respiratory interface has been recognized, cellular origin, persistence and/or turnover rates of AM particularly following lung transplantation (LTx) remains undefined. AM were earlier considered as a component of the lung mononuclear phagocytic system with circulating monocytes as central precursor for AM differentiation and replenishment (9, 10). A number of recent studies have examined the progenitors and phenotypes of mature TRM (reviewed in (3, 11, 12)). Opposed to the earlier held view, TRM including AM (and Kupffer cells, microglia, Langerhans cells, cardiac macrophages and osteoclasts) have prenatal origins from yolk-sac erythro-myeloid progenitors (EMP) (4, 13–17). These studies have demonstrated a negligible contribution by bone marrow (BM) derived monocytes in AM recruitment and sustenance. Further, development and differentiation of AM from EMP requires both cell-intrinsic and cell-extrinsic signals and is granulocyte macrophage colony-stimulating factor (GM-CSF) and peroxisome proliferator-activated receptor gamma dependent (2, 4, 15). AM are long-lived TRM and locally maintained independent of replenishment by BM cells (4, 18). In this background, we evaluated AM persistence following human LTx. Though, LTx remains as the viable treatment option for various end-stage lung diseases, >50% of lung allografts undergo chronic rejection in the first five years post-transplantation, clinically diagnosed as bronchiolitis obliterans syndrome (BOS). Development of immune responses to mismatched donor HLA and immune responses directed to lung-associated self-antigens are significant risk factors for development of BOS (19–22). Studies have demonstrated that depletion of donor specific antibody (DSA) as well as Abs to lung-associated self-antigens lowers the hazard ratio for the development of BOS (23–26). In view of poor long-term success following human LTx, we postulated that post-LTx persistence of donor AM can influence immunologic responses directed to donor antigens.
Materials and Methods
Collection, analysis and culture of human AM
Human LTx recipients (LTxR) that underwent bilateral LTx at Barnes Jewish Hospital/Washington University School of Medicine were enrolled in the study. The LTxR gave informed consent and Washington University Institutional Review Board approved the studies. Recipient and donor HLA phenotypes and DSA status were obtained from histocompatibility laboratories. Cells were isolated by centrifugation of BAL fluid at 500xg for 10min at 4°C and AM were identified by flow cytometry. For AM isolation, stained live cells were sorted by a BD FACS Aria II cell sorter and cultured in RPMI 1640 supplemented with 10% FBS and 20% L-929 culture supernatant.
Functional assessment of human AM
Isolated AM were stimulated either with toll-like receptor (TLR) ligands (Human TLR 1-9 agonists, InvivoGen) or 2 phorbol 12-myristate 13-acetate (PMA) and ionomycin or mAb to framework-HLA (W6/32) or isotype control (C.1.18.4). Culture supernatants were analyzed by a 25-Plex Human Cytokine/Chemokine luminex Panel (ThermoFisher Scientific). AM endocytosis was accessed by incubation with pH sensitive pHrodoRed dextran beads and analyzed by EVOS FL Cell Imaging System (ThermoFisher Scientific).
Mice, AM reconstitution and histopathological assessment
Wild-type C57BL/6 (H-2Kb) and Balb/c (H-2Kd) mice (6–8 wks, male) were purchased from Charles River Laboratories. C57BL/6 mice that express H-2Kd transgene on macrophages (huCD68-Kd Tg C57BL/6) were described earlier (27). All animals were maintained at Washington University School of Medicine according to institutional guidelines and approved protocols. Mice were anesthetized with Ketamine/Xylazine, lungs perfused with 1×10 mL phosphate-buffered saline (PBS) via right ventricle and BAL collected via a 22G catheter installed into trachea followed by 5×1 mL washes with ice-chilled PBS supplemented with 0.5mM ethylenediamine tetraacetic acid. BAL cells were pelleted by centrifugation at 250xg for 10 min at 4°C. For reconstitution studies, AM (CD11c+ and Siglec-F+ BAL cells) were flow sorted and administered intrabronchially. Since our average yield was 6×105 AM (10 wk, male), we transferred 4×105 AM/mouse at a harvestable ratio of 60:40. Lung tissue was fixed with 10% neutral buffered formalin, paraffin embedded and sectioned for pathologic evaluations by H&E and Trichrome staining.
Detection of donor specific immune responses
Elicitation of H-2Kd specific Abs in BAL and serum following AM reconstitution were evaluated by staining mouse T cells (CD4+ T cell isolation kit, Stemcell Technologies) by BD FACSCanto II cell analyzer. Enumeration of H-2Kd and H-2Kb reactive T cells was conducted by stimulating splenocytes from recipient mice at 16 wk post AM-transfer with irradiated (30Gy) T cells from Balb/c and C57BL/6 respectively in ELISPOT assays (BD Biosciences).
Detection of lung-associated self-antigen specific immune responses
Detection of mouse de novo Abs to lung-associated Ags (Kα1Tubulin (Kα1T) and Collagen V (Col V)) was conducted by an ELISA assay detailed in our previous publication (28). Briefly, wells were coated with recombinant Col V or Kα1T proteins (100ng/well) and quantitation of the specific Abs in serum was done by probing with HRP conjugated goat anti-mouse Ig (Jackson Immunoresearch Laboratories). Analysis of donor specific HLA Abs, and Col V or Kα1T auto-Abs in LTx patient sera were earlier described (29). Enumeration of lung infiltrating Kα1T and Col V specific Th1 and Th17 cells was conducted by quantifying cytokine (IFN-γ and IL-17A) secreting T cells in ELISPOT assays following our optimized protocol (28).
Results
Donor and recipient HLA mismatch is associated with de novo DSA
We enrolled15 human LTxR that received first-time bilateral lung transplant where the HLA mismatches between the lung donor and LTxR encompassed at least one of the HLA-A2 or HLA-A3 allele. In this cohort, donor and recipient mismatched at >1 HLA-A locus that was correlated with development de novo DSA (Table 1). All of the subjects underwent an Ab directed immunosuppressive regimen following which DSA was resolved in 40% (6/15) of LTxR. Additionally, Kα1T and Col V specific Ab were detected at varying levels. Clinical history included at least one episode of acute rejection and 12 out of 15 (~70%) LTxR developed BOS.
Table 1.
Summary of HLA-A mismatches between lung allograft donors and recipients, and clinical features following human lung transplantation.
| I.D. | Diseasea | LTxb | HLA-A allele: Donor | HLA-A allele:Recipient | DSA-Ac | Auto Abd | BOSe | ARf | ||
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | CF | 1 | A2 | A33 | A3 | A68 | + | +++ | 2 | 2 |
| P2 | IPF | 1 | A3 | A33 | A2 | A24 | + | ++ | 3 | 2 |
| P3 | IPF & RA | 1 | A3 | A3 | A2 | A24 | +/− | + | 0-p | 1 |
| P4 | COPD | 1 | A2 | A26 | A3 | A11 | +/− | +/− | Neg | 2 |
| P5 | A1AD | 1 | A2 | A2 | A25 | A32 | + | ++ | 1 | 1 |
| P6 | IPF | 1 | A2 | A3 | A24 | A25 | +/− | ++ | 1 | 2 |
| P7 | IPF | 1 | A2 | A30 | A66 | A69 | +/− | + | Neg | 1 |
| P8 | COPD | 1 | A3 | A68 | A43 | A68 | +/− | ++ | Neg | 2 |
| P9 | CF | 1 | A2 | A29 | A11 | A30 | + | +/− | 0-p | 1 |
| P10 | CTD | 1 | A2 | A34 | A69 | A80 | + | +++ | 2 | 2 |
| P11 | CF | 1 | A11 | A33 | A3 | A24 | + | ++ | Neg | 2 |
| P12 | COPD | 1 | A11 | A33 | A2 | A3 | + | ++ | 0 | 1 |
| P13 | IPF | 1 | A30 | A32 | A3 | A68 | + | +++ | 1 | 3 |
| P14 | CF | 1 | A24 | A66 | A2 | A31 | +/− | + | 0-p | 2 |
| P15 | PAH | 1 | A69 | A80 | A3 | A26 | + | +/− | Neg | 2 |
CF, cystic fibrosis; IPF, idiopathic pulmonary fibrosis; RA, rheumatoid arthritis; COPD, chronic obstructive pulmonary disease; A1AD, alpha 1 anti-trypsin deficiency; CTD, connective tissue disease; PAH, pulmonary arterial hypertension
1, primary transplantation (bilateral); 2, re-transplantation (bilateral)
DSA determination performed by LABScreen Single Antigen assay (One Lambda Inc): +, persistent/ recurrent DSA; +/−, resolved DSA; −, DSA negative
Ab to Collagen V or Kα1Tubulin measured by ELISA: +++, strong positive; ++, moderate positive; +, weak positive; −, negative
Bronchiolitis Obliterans Syndrome (BOS) grade at clinical diagnosis; Neg, negative for BOS
Grade of acute rejection (AR)
Donor derived AM persist following human LTx
Until recently, lack of a definitive identification and characterization of AM was the major impediment to fully understand role of AM in fibrosis, immunity and inflammation of the lungs. Studies by Misharin et al. (30), and Yu et al. (31) validated by Bharat et al. (32) have utilized a set of unified surface markers to define murine and human AM respectively. Based on Bharat et al. (32), we identified human AM as larger and granular BAL cells with high auto fluorescence and being CD45+, HLA-DR+, CD15−, CD11b+, CD206+, CD169+ and CD163+ (Figure 1). This analysis eliminated lymphocytes, monocytes, neutrophils and other granulocytes that occur in clinical BAL samples. Furthermore, it allowed us to distinguish between AM from donor or recipient origin by differential HLA staining between LTxR and graft donor. Relative composition of the AM in BAL cells is presented (Table 2). Based on the above morphometric and phenotypic characteristics, AM constituted 6.87% of the total BAL cells isolated from human LTxR (Table 2) with 97% purity (Figure 1). In the LTxR cohort, donor and recipient HLA mismatches were either HLA-A2 or HLA-A3 (Table 1). We analyzed BAL samples encompassing three possible donor→recipient combinations i.e. A2/A3→A3/A2 (P1–P4), A2/A3→X (P5–P10) and X→A2/A3 (P11–P15) where X being a non-HLA-A2 or non-HLA-A3 allele). The analyzed BAL samples ranged between 302 to 1265 days post-LTx. Vast majority (94–100%) of identifiable AM in BAL cells from A2/A3→A3/A2 group were positive for the donor-HLA (Figure 2). Significantly, no AM carrying unique recipient HLA-A were detected. Further, in A2/A3→X group, 87–97% of the total AM population was positive for donor HLA. Likewise, we did not detect any AM in X→A2/A3 group that expressed recipient HLA. Since our probes encompassed markers for both donor and recipient HLA in A2/A3→A3/A2, it was feasible to assess donor vs recipient contributions into the AM pool. Significantly, majority of AM in A2/A3→A3/A2 group were donor derived with no detectable contribution from the recipient. The two other groups (A2/A3→X and X→A2/A3) allowed us to estimate, respectively, the persistence of donor AM and their replacement by cells of recipient origin. Since MHC class I is ubiquitously expressed on nucleated cells and clone BB7.2 (anti-HLA-A2) and clone GAP.A3 (anti-HLA-A3) have no known cross-reactivity, we chose to focus on these two HLA-A locus mismatches. Collectively, in our experimental settings, a significant proportion (>87%) of detectable AM were donor derived indicating a long-term persistence of AM without being replaced by recipient’s cells up to 3.5 years post-LTx (Figure 2).
Figure 1. Detection of AM in BAL fluid from human LTxR.

Strategy to identify AM by cell surface staining in a flow cytometry analysis. Freshly collected BAL cells were incubated with human BD Fc Block (BD Biosciences) and human TruStain FcX (Biolegend) to inhibit Fc specific binding followed by staining with following Abs (obtained from Biolegend and eBioscience): CD45-Alexa Fluor 488 (HI30), CD11b-PE/Cy7 (M1/70), HLA DR-APC/Cy7 (L243), CD169-APC (7-239), CD15-Brilliant Violet 510 (W6D3), CD163-Brilliant Violet 421 (GHI/61) and CD206-PE (15-2). Stained cells were fixed with neutral buffered paraformaldehyde (2%) before being analyzed by BD FACSCanto II cell analyzer (BD BioSciences) and flow data were analyzed by FlowJo 10.1 (Tree Star). Small and less-granular cells and cell clumps were excluded from analysis. Large and cells with high-granularity were selected and further enriched for high autofluorescence based of FSC and SSC criteria. The sequential flow chart of analysis is depicted. AM were defined as autofluorescent BAL cells being positive for CD45, CD11b, HLA-DR, CD169, CD206 and CD163. Frequency of cells in each gate or quadrant is indicated.
Table 2.
Frequency of cells observed in clinical BAL samples from human LTx recipients.
| Cell type | Frequency (% total ± SD) |
|---|---|
| Bronchoalveolar lavage cells | 100 ± 0 |
| Large and granular BAL cells | 25.86 ± 8.51 |
| Autofluorescent cells | 23.28 ± 7.43 |
| Leukocytes | 20.49 ± 4.31 |
| MHC class II high myelocytes (HLA-DRhigh and CD11b+) | 15.21 ± 2.94 |
| Mannose receptor (CD169)+ and sialoadhesin (CD206)+ cells | 9.34 ± 1.56 |
| Scavenger receptor (CD163)+ cells | 7.22 ± 0.45 |
| Alveolar macrophages | 6.87 ± 0.34 |
Figure 2. Persistence of donor derived AM following human LTx.

Assessment of donor and recipient contribution to the post-LTx AM pool was analyzed based on donor-recipient HLA-A mismatches. AM were identified as described in Figure 1 and were further analyzed by staining with anti-HLA-A2 (BB7.2) and anti-HLA-A3 (GAP.A3) to distinguish their cellular origin. Tracking of AM in eight representative BAL specimens is presented. HLA-A mismatches (donor→recipient), patient I.D. (Table 1) and time of post-LTx BAL collection are indicated for the respective specimens. Frequency of cells in each quadrant is indicated.
The persistent donor derived AM respond to TLR and anti-MHC stimuli
We next determined post-LTx AM responses to various inflammatory stimuli (TLR ligands) and donor specific HLA Abs. We used highly purified (>99%) AM isolated via flow sorting to determine their responsiveness to a panel of human TLR agonists, mitogen (PMA+Ionomycin) and Abs (anti-HLA and isotype control). Cytokine production was expressed as fold increase in stimulated cells over that of unstimulated cells. Overall, AM responded to stimuli, albeit, differentially in range and magnitude (Figure 3). The stimuli mimicked infections by virus, gram positive and negative bacteria, and induced status of an intracellular infection. An up-regulation in inflammatory cytokines including IL-1β, IL-6, IL-12 and TNFα were prominent. Additionally, chemokines including macrophage inflammatory proteins (MIP-1α, MIP-1β), RANTES, IP-10 and MIG involved in chemotaxis of T cell and other inflammatory cells were also elicited. These cytokine/chemokine signatures are part of a broader immune response by cells including but not limited to macrophages and indicate a preservation of AM responsiveness to various infectious and inflammatory stimuli during post-LTx period. Furthermore, TLR stimuli induced IL-8 (a macrophage produced chemokine important in neutrophil recruitment and degranulation) up to 9.3 fold (Table S1). By contrast, as expected, the cytokine signature lacked any T cell specific cytokines (IL-2 and IFNγ) indicating an absence of contaminating T cells in the AM purification protocol. The most striking finding was the AM responsiveness to anti-HLA. W6/32 induced a unique signature with stimulation of IL-1β (19.47 fold), IL-6 (4.86 fold), IL-12 (8.48 fold), RANTES (4.31 fold), MIP-1α (3 fold), MIP-1β (3.3 fold), MCP-1 (3 fold), TNFα (2.75 fold) and MIG (1.88 fold) over that of isotype control (Figure 3A). Importantly, W6/32 also induced IL-8 >3 fold compared to isotype treatment (Table S1). Taken together, our data demonstrate that donor AM purified from the BAL following LTx are not only able to respond to infections but also react to MHC ligation with Abs resulting in secretion of inflammatory cytokines.
Figure 3. Preservation of AM functionality in donor derived post-LTx AM.

(A) Elicitation of inflammatory cytokines and chemokines was evaluated in response to a panel of TLR agonists: TLR1/2- Pam3CysSerLys4 (Pam3CSK4); TLR2- heat-killed Listeria monocytogenes (HKLM); TLR3- Poly (I:C) high molecular weight 1.8–8 kb(HMW), Poly (I:C) low molecular weight 0.2–1 kb(LMW); TLR4-Lipopolysaccharide from Escherichia coli K12 (LPS); TLR5- Salmonella typhimurium Flagellin (ST-FLA); TLR6/2- N-terminal part of the 44-kDa lipoprotein LP44 of Mycoplasma salivarium, Pam2CGDPKHPKSF (FSL-1); TLR7- R837 (Imiquimod); TLR8-ssRNA40 and TLR9-ODN2006 type B. PMA and Inonomycin were used at 5 ng/mL and 500 ng/mL respectively and HLA specific mAb (W6/32) or isotype control (C.1.18.4) were used at 100 μg/mL to study effect of MHC I crosslinking. Flow sorted AM were seeded in a 12-well plate at 5×105 cells/well and release of cytokines after 24 hr stimulation at 37°C was measured in culture supernatant by a 25-plex luminex panel. The mean fluorescence intensity (MFI) of an individual analyte is expressed as fold increase in stimulated cells compared to that in un-stimulated cells. Data on 12 selected analytes from a representative AM culture are presented and computed values (pg/mL) for all (25) analytes are given in Table S1. (B) Particle endocytosis by AM was accessed by incubation with pH sensitive pHrodoRed dextran (10,000 MW) beads (50 μg/mL) for 20 min at 37°C. Cells were stained with NucBlue live ready probe to visualize nucleus and analyzed by EVOS FL Cell Imaging System. A representative merged image from bright field, and red and blue fluorescence is given. Scale bar is 200 μm.
We then assessed AM endocytosis by pH sensitive dextran beads. The beads following internalization into acidic endosomal and lysosomal compartments fluoresce that allowed their detection. Following a 20-min pulsing, >95% of AM (Figure 3B) demonstrated bead uptake indicating preservation of particle endocytosis in post-LTx AM.
Adoptive transfer of AM with mismatched MHC class I induces alloimmunity in murine model
The murine AM in BAL cells were identified by being CD45+, CD11c+ and Siglec-F+ (Figure 4A). We utilized a novel huCD68-Kd Tg C57BL/6 mouse, which expresses a full length H-2Kd transgene in C57BL/6 macrophages (27) to evaluate AM reconstitution. Nearly all (~98%) of AM from huCD68-Kd Tg expressed both H-2Kd and H-2Kb (Figure 4A). This unique expression of surface H-2Kd transgene in huCD68-Kd Tg AM enabled us to examine effect of AM restricted donor specific MHC mismatches in elicitation of donor specific alloimmunity. We evaluated whether transfer of huCD68-Kd Tg (Tg) AM into C57BL/6 (B6) recipient will induce H-2Kd specific immune responses. In parallel, we also carried out allogeneic transfer of B6 AM into Tg recipients and syngeneic transfer of Tg AM into Tg recipients, and transfer of B6 AM into B6 recipients. Development of H-2Kd (alloantigen) reactive lung-resident T cells (Figure 4B) and alloAbs in BAL fluid and serum (Figure 4C) were analyzed. Adoptive transfer of Tg into B6 stimulated alloantigen specific T cell responses and elicited detectable H-2Kd Abs both in BAL fluid as well as in serum. Since the donor AM expressed H-2Kd as a transgene, it is being recognized as a foreign antigen. It is important to note that both the AM donors (huCD68-Kd Tg) and recipients (C57BL/6) had identical MHC class II (I-Ab). Therefore the donor AM was capable of priming I-Ab restricted H-2Kd reactive CD4 T cells. Further, the engrafted AM expressed equivalent levels of surface MHC II present on native AM indicating an optimal antigen presentation ability (Figure 4D). Taken together, demonstration of H-2Kd specific Ab and T cell responses suggest that mismatched MHC I antigen on AM is sufficient to elicit donor specific immune responses.
Figure 4. Induction of donor specific immunity following allogeneic AM reconstitution.

(A) Gating strategy and validation of H-2Kd transgene expression on huCD68-Kd Tg C57BL/6 AM (27). The mouse BAL cells were Fc blocked (clones 2.4G2 and 93) to saturate CD16/CD32 mediated binding and were characterized by staining with CD45-PE/Cy7 (I3/2.3), CD11c-Alexa Fluor 488 (N418), Siglec-F- Brilliant violet 421 (E50-2440) and the H-2Kd transgene expression was compared by staining with H-2Kb-APC (AF6-88.5), H-2Kd-PE (SF1-1.1). Mouse AM were defined as large granular BAL cells being CD45+, CD11c+ and Siglec-F+. (B) Stimulation of allogeneic T cell response following intrabronchial AM transfer. Development of H-2Kd and H-2Kb reactive T cells was enumerated by stimulating splenocytes (1×106) from indicated recipient mice at 16 wk post AM-transfer with irradiated (30Gy) T cells (1×105) from Balb/c or C57BL/6 in a IL-2 ELISPOT assay (BD Biosciences). Five mice were used per experimental group, data represent mean±SEM and multiple t-test was applied to evaluate statistical significance (p value indicated). (C) Induction of H-2Kd specific Abs following AM transfer was evaluated by staining CD4+ T cells from Balb/c mice with cell free BAL fluid (neat) or serum (1:50) at 16 wk post-AM transfer or naïve mice (from panel B) followed by staining with goat anti-mouse IgG APC. Five-six mice were used in each experiment. (D) AM reconstitution at 16 weeks post-intrabronchial transfer of huCD68-Kd Tg AM in to C57BL/6 recipients. Native AM in recipient mice were H-2Kb+ and H-2Kd− while donor AM were double positive for H-2Kb and H-2Kd. Expression of MHC class II on donor and recipient AM was evaluated by staining with I-A/I-E-Brilliant Violet 421 (M5/114.15.2) and presented as overlaid image.
Allogeneic AM transfer induces small airway obstruction and fibrosis
We have earlier developed a murine obliterative airway disease model where administrations of exogenous MHC Ab into native lungs induce bronchiolitis, a clinical correlate of human BOS (33, 34). In the present study of AM transfer model, we evaluated pathologic effect of de novo DSA on small airway obstruction and fibrosis. Increased peribronchiolar leukocyte clusters, and greater epithelial hypertrophy and hyperplasia were evident in Tg→B6 compared to B6→Tg group (Figure 5A). Additionally, higher depositions of collagenous extracellular matrix proteins and higher occurrence of small airway obstruction in Tg→B6 mice indicated a respiratory impairment. Histopathology assessment for the AM transfer groups is presented in Figure 5B and allogeneic AM transfer (Tg→B6) that stimulated DSA resulted in a significant loss of bronchiolar architecture with concomitant increase in small airway inflammation and obstruction.
Figure 5.

Histopathologic assessment of lungs following adoptive AM transfer. (A) Paraffin embedded tissue sections were analyzed for leukocytic infiltrations (H&E) and fibrosis (Masson’s Trichrome). (B) The slides were scored according to (34) and average score from five fields is presented. Three mice per group were analyzed. Multiple t-test was applied and statistical significance was determined using Holm-Sidak method (p value indicated). Scale bars are 40 μm.
Allogeneic AM transfer induces immune responses directed to lung-associated self-antigens
In the view of Tg→B6 eliciting de novo DSA (Figure 4) and bronchiolar obstruction (Figure 5), we examined stimulation of lung-associated self-antigen (Kα1T and Col V) specific autoimmunity following AM transfer. Mean serum titers for anti-Kα1T and anti-Col V in Tg→B6 AM transfer mice were 105 μg/mL and 183 μg/mL respectively and were significantly higher than that in other transfer groups measured at <40 μg/mL (Figure 6A). Furthermore, Tg→B6 stimulated significantly greater Kα1T and Col V reactive IL-17 and IFNγ secreting T cells (Figure 6B). The mean Kα1T specific IL-17 and IFNγ secreting T cells were 75 and 55 (spots/106 cells) respectively while those of Col V reactive T cells were 97 and 84 (spots/106 cells) respectively. The self-antigen reactive T cells in other AM transfer groups remained <20 spots/106 cells. Taken together, stimulation of T cells and development of Abs directed to lung-associated antigens indicate that MHC I mismatched AM were sufficient to induce humoral and cellular immune responses to lung-associated self-antigens.
Figure 6.
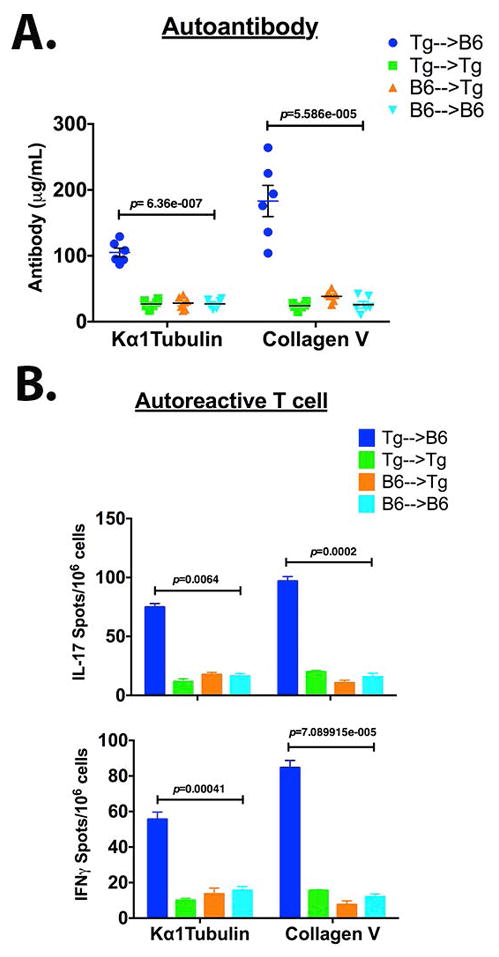
Allogeneic AM reconstitution induces lung-associated antigen specific autoimmunity. (A) Elicitation of serum Ab to Kα1T and Col V at 16 wk post AM transfer was measured by ELISA (33). (B) Accumulation of Kα1T and Col V specific T cells was evaluated by IL-17A and IFNγ ELISPOT assays (BD Biosciences). Single cell suspension was prepared from lungs by digestion with Liberase TL (300μg/mL, Roche Life Science) and DNaseI (5U/mL, Sigma-Aldrich) at 37°C for 25 min. Lung leukocytes were isolated from the single cell suspension by Ficoll-Paque (Sigma-Aldrich) gradient centrifugation. One million lung leukocytes were added per well and were supplemented with 5×104 C57BL/6 splenocytes (irradiated at 30 Gy) as antigen presenting cells. The cells were stimulated with Col V or Kα1T protein at 1μg/mL and cytokine producing spots were enumerated. Individual serum titer from five mice is plotted (in A) and stimulation of T cells (in B) is expressed as mean±SEM (n=5). Multiple t-tests were applied and statistical significance was determined using Holm-Sidak method (p value indicated).
Discussion
Despite improvements in surgical techniques and post-operative care, incidence of BOS continues to be the primary obstacle in long-term success of LTx (35). Development of de novo DSA to mismatched donor HLA and Ab to lung-associated self-antigens are considered major risk factors for BOS development (20, 36, 37). Role of AM in DSA induced lung inflammation and BOS is largely unknown. This is partly due to lack of a uniform experimental approach and definitive AM characterization. AM are terminally differentiated TRM and express distinctive surface markers compared to other tissue resident and circulatory macrophages (3). The recently described flow cytometric analyses (30–32) allow unambiguous identification of human and mouse AM and enable them to be distinguished from “passenger leukocytes”. It is generally believed that recipient stem cells seed the transplanted organ, and endothelial cells and macrophages repopulate the graft with potential to minimize host immune detection (39–46). However, the dichotomy of immunologic complications and poor prognosis associated with LTx intrigued us to investigate post-LTx persistence of donor AM and its contribution to chronic rejection. AM are the major lung TRM and constitute professional phagocytes at blood-air interface (47). We, for the first time, demonstrate that donor AM persist following LTx for a long period (up to 3.5 yr) and respond to HLA specific Ab resulting in secretion of pro-inflammatory cytokines. This finding has two-fold implications. First, long-term survival of donor AM (bearing donor-HLA) in transplanted lungs may serve as a repository of mismatched HLA antigens. Second, our finding demonstrating pro-inflammatory responsiveness of AM to MHC ligation indicate a putative role for AM in mediating DSA induced pathogenesis. Our findings of a long-term sustenance of AM following human LTx and our inability to detect recipient derived cellular chimerism in AM compartment is in contrast to earlier published studies (39, 40, 44, 48). The disagreement is due to differences in experimental methods and target cells studied. For example, report by Kjellström et al. (40) used DNA microsatellite described a rapid turnover of donor CD14+ BAL cells. By lineage analysis, human AM lack CD14 expression while monocytes are CD14+ (31, 49, 50). The study by Judson et al. (39) analyzed pleural cell chimerism primarily in unilateral LTx patients. Cellular composition in pleural lavage fluid includes lymphoid and myeloid cells (51) but lacks AM. Nakata et al. (44) and Kennedy et al. (48) while studying reconstitution and proliferation of BAL cells following BM transplantation (BMT) reported appearance of BM derived AM in BAL around 60 days post-BMT. These authors based their conclusions on lymphocyte-depleted monocytes and macrophages but not on purified AM following irradiation and BMT. Therefore, due to lack of a consistent analysis on AM, it is difficult to interpret these results in recipient AM reconstitution. Our approach to identify AM is based on surface markers that enabled us to distinguish the AM from IM, monocytes, neutrophils, lymphocytes and other granulocytes (30–32). In addition, using HLA allele we are able to identify AM as either of donor or recipient origin. Until recently, AM have been consistently misidentified and misrepresented with regards to their phenotype and lineage specificity. Recent studies have concluded that yolk-sac derived fetal monocyte but not circulating hematopoietic stem cell (HSC) derived monocyte is precursor for AM development (4, 15). Genetic reconstitution and parabiosis experiments have also confirmed that circulating monocytes minimally contribute to the homeostatic AM pool (4, 15, 16, 52). Further, constitutive and inducible fate mapping of embryonic cells indicated that yolk-sac EMPs accumulated in fetal liver give rise to AM while HSC driven hematopoiesis minimally replaced the AM compartment (13). Moreover, transplantation of fetal liver cells following AM depletion by clodronate liposomes or lethal irradiation resulted in an efficient reconstitution of AM cells (53). IM, on the other hand, are blood monocyte derived and demonstrate a high turnover rate (54). The impact of an ischemia reperfusion injury (IRI) associated with LTx on AM is currently unknown. In a rat LTx model, viable AM were isolated from BAL cells following transplantation suggesting a minimal effect of IRI, if any, on AM survival (55). Though we did not specifically test the influence of IRI on AM in this study, our results suggest that human AM endured IRI stress and persisted up to 3.5 years post-LTx. While in experimental models a long-term replacement of steady state AM by HSC cells was negligible, a slow (~1yr) and partial (~40–48%) turnover of AM has been reported in BMT following myeloablative irradiation (18, 48), infection with influenza virus (56), high dose endotoxin challenge (57) and due to old age (13) that depleted the homeostatic AM pool. It remains unclear though the phenotypic and transcriptional differences between yolk-sac derived native AM and neo-differentiated AM from BM hematopoiesis. Our cohort of LTxR received no irradiation treatment and remained free from a documented viral infection during the follow-up period. So existence of a negative pressure on donor AM survival was unlikely. Further, donor AM responded to TLR agonists and MHC ligation. Responsiveness to TLR stimuli indicates AM’s functional preservation as “first line of innate defense” at respiratory interface (50). Further, we demonstrate inflammatory responses by donor AM following MHC I ligation, which is in agreement with the reported dominant role for donor endothelial cells in anti-HLA induced inflammation (58). In addition, we conclusively demonstrated that the donor AM in an ex vivo assay stimulated inflammatory cytokines in response to anti-HLA. Extracorporeal removal of DSA has been shown to reduce inflammatory cytokines and increase anti-inflammatory cytokines (23). This has a significant clinical implication as AM may contribute to DSA induced pathogenesis in fibrotic and inflammatory lung diseases.
We investigated whether AM reconstitution is viable with adoptive transfer of allogeneic AM (with a single MHC I mismatch). Intrabronchial administration of naïve Tg AM resulted in AM persistence with a stable reconstitution rate (Figure S1). The donor AM were localized to alveoli and detectable even after four months post-transfer suggesting a viable engraftment. Since the Tg AM carried surface H-2Kd, an alloantigen, we evaluated whether Tg→B6 led to alloimmunity. We evaluated elicitation of local as well as systemic alloAb. Following AM transfer, Tg→B6 stimulated Ab and T cell responses to alloantigen (Figure 4B–C) and lung-associated self-antigens (Figure 6A–B) concomitant with bronchiolar obstruction and fibrosis (Figure 5A–B). AM serve as antigen presenting cells in natural infection (59) and vaccination (60, 61). Earlier studies reported persistence of donor AM and induction of alloAb (in BAL and serum) and T cells following allogeneic AM transfers (62, 63). The genetic mismatches between donor and recipient in those studies were major encompassing both MHC (I and II) and non-MHC loci. By contrast, AM transfer experiments performed in our study included one defined allelic mismatch that helped us to determine immunogenicity of MHC I mismatched AM.
A limitation to our study is narrow probe range that permitted analysis of human BAL cells only when the organ donor and recipient’s genetic mismatches included at least HLA-A2 or HLA-A3. This excluded a number of potential BAL samples resulting in a small sample size. Nevertheless, our finding of donor derived AM in engrafted lungs is the first demonstration of a long-term persistence of native AM following human LTx. In this study we did not evaluate causality and/or consequence of donor AM in the development of BOS. It is possible that additional cellular and molecular processes participate in the development of DSA and bronchiolar obstruction leading to BOS. However, ability of AM to induce DSA and be stimulated by MHC Ab demonstrate an important role for AM in the DSA induced immunopathogenesis leading to chronic lung allograft rejection.
Supplementary Material
(A) A representative H&E stained tissue section of lungs from huCD68-Kd Tg AM transfer into C57BL/6 (Figure 4B). AM in alveolar space are marked with arrows. (B) Engraftment of donor AM following huCD68-Kd Tg AM transfer into C57BL/6 recipients. Frozen tissue sections (10 μm) from four-month reconstitution study (Figure 4B) were stained with CD11c-Alexa Fluor 594, H-2Kb- Alexa Fluor 488, H-2Kd- Alexa Fluor 647 and photomicrographs were captured by Zeiss Observer.Z1 microscope with Axiovision 4.8.2. Engrafted AM are marked with arrows and native recipient AM are marked with arrowheads. Scale bars are 100 μm. (C) Maintenance of AM reconstitutive chimerism following a single intrabronchial transfer of 4×105 naïve huCD68-Kd Tg AM into C57BL/6 lungs (at an initial recipient:donor reconstitution rate at 60:40) was evaluated. Native AM in recipient mice and the transplanted AM were distinguished by staining with anti-H-2Kb (AF6-88.5) and anti-H-2Kd (SF1-1.1).
Acknowledgments
This work was supported by grants from NIH (R01HL056643 and R01HL092514) and BJC foundation to TM, and a grant from NIH (R01AI102891) to MT. FZ was supported by State Scholarship Fund from China Scholarship Council (201406260136).
Abbreviations
- Ab
antibody
- AM
alveolar macrophages
- BAL
bronchoalveolar lavage
- BM
bone marrow
- BMT
bone marrow transplant
- BOS
bronchiolitis obliterans syndrome
- Col V
Collagen V
- DSA
donor specific antibody
- EMP
erythro-myeloid progenitors
- GM-CSF
granulocyte macrophage colony-stimulating factor
- HSC
hematopoietic stem cell
- IRI
ischemia reperfusion injury
- IM
interstitial macrophages
- Kα1T
K alpha 1 Tubulin
- LTx
lung transplantation
- LTxR
lung transplant recipient
- PBS
phosphate-buffered saline
- PMA
2 phorbol 12-myristate 13-acetate
- Tg
transgenic
- TLR
toll-like receptor
- TRM
tissue resident macrophages
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–95. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol. 2015;16(1):36–44. doi: 10.1038/ni.3052. [DOI] [PubMed] [Google Scholar]
- 4.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. The Journal of experimental medicine. 2013;210(10):1977–92. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone KC, Mercer RR, Gehr P, Stockstill B, Crapo JD. Allometric relationships of cell numbers and size in the mammalian lung. Am J Respir Cell Mol Biol. 1992;6(2):235–43. doi: 10.1165/ajrcmb/6.2.235. [DOI] [PubMed] [Google Scholar]
- 6.Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, Bhattacharya J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. 2014;506(7489):503–6. doi: 10.1038/nature12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 2014;10(4):e1004053. doi: 10.1371/journal.ppat.1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Happle C, Lachmann N, Skuljec J, Wetzke M, Ackermann M, Brennig S, Mucci A, Jirmo AC, Groos S, Mirenska A, et al. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci Transl Med. 2014;6(250):250ra113. doi: 10.1126/scitranslmed.3009750. [DOI] [PubMed] [Google Scholar]
- 9.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 10.van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. The Journal of experimental medicine. 1968;128(3):415–35. doi: 10.1084/jem.128.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon SB, Read RC. Macrophage defences against respiratory tract infections. Br Med Bull. 2002;61:45–61. doi: 10.1093/bmb/61.1.45. [DOI] [PubMed] [Google Scholar]
- 12.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science (New York, NY) 2013;342(6161):1242974. doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- 13.Perdiguero GE, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–78. doi: 10.1016/j.immuni.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 2014;15(11):1026–37. doi: 10.1038/ni.3005. [DOI] [PubMed] [Google Scholar]
- 16.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (New York, NY) 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 17.Yamane T, Kunisada T, Yamazaki H, Era T, Nakano T, Hayashi SI. Development of osteoclasts from embryonic stem cells through a pathway that is c-fms but not c-kit dependent. Blood. 1997;90(9):3516–23. [PubMed] [Google Scholar]
- 18.Murphy J, Summer R, Wilson AA, Kotton DN, Fine A. The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38(4):380–5. doi: 10.1165/rcmb.2007-0224RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bharat A, Saini D, Steward N, Hachem R, Trulock EP, Patterson GA, Meyers BF, Mohanakumar T. Antibodies to self-antigens predispose to primary lung allograft dysfunction and chronic rejection. Ann Thorac Surg. 2010;90(4):1094–101. doi: 10.1016/j.athoracsur.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Girnita AL, Duquesnoy R, Yousem SA, Iacono AT, Corcoran TE, Buzoianu M, Johnson B, Spichty KJ, Dauber JH, Burckart G, et al. HLA-specific antibodies are risk factors for lymphocytic bronchiolitis and chronic lung allograft dysfunction. Am J Transplant. 2005;5(1):131–8. doi: 10.1111/j.1600-6143.2004.00650.x. [DOI] [PubMed] [Google Scholar]
- 21.Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, Steward N, Aloush A, Hachem R, Trulock E, et al. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2011;30(6):624–31. doi: 10.1016/j.healun.2011.01.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder LD, Wang Z, Chen DF, Reinsmoen NL, Finlen-Copeland CA, Davis WA, Zaas DW, Palmer SM. Implications for human leukocyte antigen antibodies after lung transplantation: a 10-year experience in 441 patients. Chest. 2013;144(1):226–33. doi: 10.1378/chest.12-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baskaran G, Tiriveedhi V, Ramachandran S, Aloush A, Grossman B, Hachem R, Mohanakumar T. Efficacy of extracorporeal photopheresis in clearance of antibodies to donor-specific and lung-specific antigens in lung transplant recipients. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2014;33(9):950–6. doi: 10.1016/j.healun.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hachem RR, Yusen RD, Meyers BF, Aloush AA, Mohanakumar T, Patterson GA, Trulock EP. Anti-human leukocyte antigen antibodies and preemptive antibody-directed therapy after lung transplantation. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2010;29(9):973–80. doi: 10.1016/j.healun.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ius F, Sommer W, Tudorache I, Kuhn C, Avsar M, Siemeni T, Salman J, Hallensleben M, Kieneke D, Greer M, et al. Preemptive treatment with therapeutic plasma exchange and rituximab for early donor-specific antibodies after lung transplantation. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015;34(1):50–8. doi: 10.1016/j.healun.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 26.Tinckam KJ, Keshavjee S, Chaparro C, Barth D, Azad S, Binnie M, Chow CW, de Perrot M, Pierre AF, Waddell TK, et al. Survival in sensitized lung transplant recipients with perioperative desensitization. Am J Transplant. 2015;15(2):417–26. doi: 10.1111/ajt.13076. [DOI] [PubMed] [Google Scholar]
- 27.Huang J, Li X, Kohno K, Hatano M, Tokuhisa T, Murray PJ, Brocker T, Tsuji M. Generation of tissue-specific H-2Kd transgenic mice for the study of K(d)-restricted malaria epitope-specific CD8+ T-cell responses in vivo. J Immunol Methods. 2013;387(1–2):254–61. doi: 10.1016/j.jim.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Fukami NRS, Saini D, Walter M, Chapman W, Patterson GA, Mohanakumar T. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182(1):309–18. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hachem RR, Tiriveedhi V, Patterson GA, Aloush A, Trulock EP, Mohanakumar T. Antibodies to K-alpha 1 tubulin and collagen V are associated with chronic rejection after lung transplantation. Am J Transplant. 2012;12(8):2164–71. doi: 10.1111/j.1600-6143.2012.04079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49(4):503–10. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu YA, Hotten DF, Malakhau Y, Volker E, Ghio AJ, Noble PW, Kraft M, Hollingsworth JW, Gunn MD, Tighe RM. Flow Cytometric Analysis of Myeloid Cells in Human Blood, Bronchoalveolar Lavage, and Lung Tissues. Am J Respir Cell Mol Biol. 2015 doi: 10.1165/rcmb.2015-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bharat A, Bhorade SM, Morales-Nebreda L, Mc Quattie-Pimentel AC, Soberanes S, Ridge K, DeCamp MM, Mestan KK, Perlman H, Budinger GR, et al. Flow Cytometry Reveals Similarities Between Lung Macrophages in Humans and Mice. Am J Respir Cell Mol Biol. 2015 doi: 10.1165/rcmb.2015-0147LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukami N, Ramachandran S, Saini D, Walter M, Chapman W, Patterson GA, Mohanakumar T. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. Journal of immunology (Baltimore, Md : 1950) 2009;182(1):309–18. doi: 10.4049/jimmunol.182.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takenaka M, Tiriveedhi V, Subramanian V, Hoshinaga K, Patterson AG, Mohanakumar T. Antibodies to MHC class II molecules induce autoimmunity: critical role for macrophages in the immunopathogenesis of obliterative airway disease. PLoS One. 2012;7(8):e42370. doi: 10.1371/journal.pone.0042370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotloff RM, Thabut G. Lung transplantation. Am J Respir Crit Care Med. 2011;184(2):159–71. doi: 10.1164/rccm.201101-0134CI. [DOI] [PubMed] [Google Scholar]
- 36.Jaramillo A, Smith MA, Phelan D, Sundaresan S, Trulock EP, Lynch JP, Cooper JD, Patterson GA, Mohanakumar T. Development of ELISA-detected anti-HLA antibodies precedes the development of bronchiolitis obliterans syndrome and correlates with progressive decline in pulmonary function after lung transplantation. Transplantation. 1999;67(8):1155–61. doi: 10.1097/00007890-199904270-00012. [DOI] [PubMed] [Google Scholar]
- 37.Palmer SM, Davis RD, Hadjiliadis D, Hertz MI, Howell DN, Ward FE, Savik K, Reinsmoen NL. Development of an antibody specific to major histocompatibility antigens detectable by flow cytometry after lung transplant is associated with bronchiolitis obliterans syndrome. Transplantation. 2002;74(6):799–804. doi: 10.1097/00007890-200209270-00011. [DOI] [PubMed] [Google Scholar]
- 38.Hayes D, Jr, Black SM, Tobias JD, Kopp BT, Kirkby SE, Mansour HM, Whitson BA. Influence of human leukocyte antigen mismatching on bronchiolitis obliterans syndrome in lung transplantation. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015 doi: 10.1016/j.healun.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 39.Judson MA, Sahn SA, Hahn AB. Origin of pleural cells after lung transplantation: from donor or recipient? Chest. 1997;112(2):426–9. doi: 10.1378/chest.112.2.426. [DOI] [PubMed] [Google Scholar]
- 40.Kjellstrom C, Ichimura K, Chen XJ, Riise GC, Collins VP. The origin of alveolar macrophages in the transplanted lung: a longitudinal microsatellite-based study of donor and recipient DNA. Transplantation. 2000;69(9):1984–6. doi: 10.1097/00007890-200005150-00046. [DOI] [PubMed] [Google Scholar]
- 41.Kleeberger W, Rothamel T, Glockner S, Flemming P, Lehmann U, Kreipe H. High frequency of epithelial chimerism in liver transplants demonstrated by microdissection and STR-analysis. Hepatology (Baltimore, Md) 2002;35(1):110–6. doi: 10.1053/jhep.2002.30275. [DOI] [PubMed] [Google Scholar]
- 42.Kleeberger W, Versmold A, Rothamel T, Glockner S, Bredt M, Haverich A, Lehmann U, Kreipe H. Increased chimerism of bronchial and alveolar epithelium in human lung allografts undergoing chronic injury. The American journal of pathology. 2003;162(5):1487–94. doi: 10.1016/S0002-9440(10)64281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagaaij EL, Cramer-Knijnenburg GF, van Kemenade FJ, van Es LA, Bruijn JA, van Krieken JH. Endothelial cell chimerism after renal transplantation and vascular rejection. Lancet (London, England) 2001;357(9249):33–7. doi: 10.1016/S0140-6736(00)03569-8. [DOI] [PubMed] [Google Scholar]
- 44.Nakata K, Gotoh H, Watanabe J, Uetake T, Komuro I, Yuasa K, Watanabe S, Ieki R, Sakamaki H, Akiyama H, et al. Augmented proliferation of human alveolar macrophages after allogeneic bone marrow transplantation. Blood. 1999;93(2):667–73. [PubMed] [Google Scholar]
- 45.Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart. The New England journal of medicine. 2002;346(1):5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- 46.Rothmeier C, Roux E, Spiliopoulos A, Gerbase M, Nicod LP. Early chimerism of macrophages and lymphocytes in lung transplant recipients is predictive of graft tolerance. Transplantation. 2001;71(9):1329–33. doi: 10.1097/00007890-200105150-00026. [DOI] [PubMed] [Google Scholar]
- 47.MacLean JA, Xia W, Pinto CE, Zhao L, Liu HW, Kradin RL. Sequestration of inhaled particulate antigens by lung phagocytes. A mechanism for the effective inhibition of pulmonary cell-mediated immunity. The American journal of pathology. 1996;148(2):657–66. [PMC free article] [PubMed] [Google Scholar]
- 48.Kennedy DW, Abkowitz JL. Kinetics of central nervous system microglial and macrophage engraftment: analysis using a transgenic bone marrow transplantation model. Blood. 1997;90(3):986–93. [PubMed] [Google Scholar]
- 49.Strauss-Ayali D, Conrad SM, Mosser DM. Monocyte subpopulations and their differentiation patterns during infection. J Leukoc Biol. 2007;82(2):244–52. doi: 10.1189/jlb.0307191. [DOI] [PubMed] [Google Scholar]
- 50.Tomlinson GS, Booth H, Petit SJ, Potton E, Towers GJ, Miller RF, Chain BM, Noursadeghi M. Adherent human alveolar macrophages exhibit a transient pro-inflammatory profile that confounds responses to innate immune stimulation. PLoS One. 2012;7(6):e40348. doi: 10.1371/journal.pone.0040348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noppen M. Normal volume and cellular contents of pleural fluid. Paediatric respiratory reviews. 2004;5(Suppl A):S201–3. doi: 10.1016/s1526-0542(04)90038-3. [DOI] [PubMed] [Google Scholar]
- 52.Hoeffel G, Ginhoux F. Ontogeny of Tissue-Resident Macrophages. Front Immunol. 2015;6(486) doi: 10.3389/fimmu.2015.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Everhart MB, Han W, Parman KS, Polosukhin VV, Zeng H, Sadikot RT, Li B, Yull FE, Christman JW, Blackwell TS. Intratracheal administration of liposomal clodronate accelerates alveolar macrophage reconstitution following fetal liver transplantation. J Leukoc Biol. 2005;77(2):173–80. doi: 10.1189/jlb.1203647. [DOI] [PubMed] [Google Scholar]
- 54.Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, Kuroda MJ. In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. Journal of immunology (Baltimore, Md : 1950) 2014;192(6):2821–9. doi: 10.4049/jimmunol.1302269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakamura T, Abu-Dahab R, Menger MD, Schafer U, Vollmar B, Wada H, Lehr CM, Schafers HJ. Depletion of alveolar macrophages by clodronate-liposomes aggravates ischemia-reperfusion injury of the lung. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2005;24(1):38–45. doi: 10.1016/j.healun.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 56.Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. Journal of immunology (Baltimore, Md : 1950) 2013;191(3):1250–9. doi: 10.4049/jimmunol.1300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, Seeger W, Welte T, Lohmeyer J. Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol. 2006;35(2):227–35. doi: 10.1165/rcmb.2005-0241OC. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Rozengurt E, Reed EF. HLA class I molecules partner with integrin beta4 to stimulate endothelial cell proliferation and migration. Sci Signal. 2010;3(149):ra85. doi: 10.1126/scisignal.2001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hartwig SM, Holman KM, Varga SM. Depletion of alveolar macrophages ameliorates virus-induced disease following a pulmonary coronavirus infection. PLoS One. 2014;9(3):e90720. doi: 10.1371/journal.pone.0090720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benoit A, Huang Y, Proctor J, Rowden G, Anderson R. Effects of alveolar macrophage depletion on liposomal vaccine protection against respiratory syncytial virus (RSV) Clinical and experimental immunology. 2006;145(1):147–54. doi: 10.1111/j.1365-2249.2006.03114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macdonald DC, Singh H, Whelan MA, Escors D, Arce F, Bottoms SE, Barclay WS, Maini M, Collins MK, Rosenberg WM. Harnessing alveolar macrophages for sustained mucosal T-cell recall confers long-term protection to mice against lethal influenza challenge without clinical disease. Mucosal Immunol. 2014;7(1):89–100. doi: 10.1038/mi.2013.27. [DOI] [PubMed] [Google Scholar]
- 62.Heidler KM, Baker K, Woods K, Schnizlein-Bick C, Cummings OW, Sidner R, Foresman B, Wilkes DS. Instillation of allogeneic lung antigen-presenting cells deficient in expression of major histocompatibility complex class I or II antigens have differential effects on local cellular and humoral immunity and on pathology in recipient murine lungs. Am J Respir Cell Mol Biol. 2000;23(4):499–505. doi: 10.1165/ajrcmb.23.4.4172. [DOI] [PubMed] [Google Scholar]
- 63.Wilkes DS, Heidler KM, Bowen LK, Quinlan WM, Doyle NA, Cummings OW, Doerschuk CM. Allogeneic bronchoalveolar lavage cells induce the histology of acute lung allograft rejection, and deposition of IgG2a in recipient murine lungs. Journal of immunology (Baltimore, Md : 1950) 1995;155(5):2775–83. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) A representative H&E stained tissue section of lungs from huCD68-Kd Tg AM transfer into C57BL/6 (Figure 4B). AM in alveolar space are marked with arrows. (B) Engraftment of donor AM following huCD68-Kd Tg AM transfer into C57BL/6 recipients. Frozen tissue sections (10 μm) from four-month reconstitution study (Figure 4B) were stained with CD11c-Alexa Fluor 594, H-2Kb- Alexa Fluor 488, H-2Kd- Alexa Fluor 647 and photomicrographs were captured by Zeiss Observer.Z1 microscope with Axiovision 4.8.2. Engrafted AM are marked with arrows and native recipient AM are marked with arrowheads. Scale bars are 100 μm. (C) Maintenance of AM reconstitutive chimerism following a single intrabronchial transfer of 4×105 naïve huCD68-Kd Tg AM into C57BL/6 lungs (at an initial recipient:donor reconstitution rate at 60:40) was evaluated. Native AM in recipient mice and the transplanted AM were distinguished by staining with anti-H-2Kb (AF6-88.5) and anti-H-2Kd (SF1-1.1).