

Abstract
We developed a microarray-based system for screening small molecules in mammalian cells. This system is compatible with image-based screens and requires fewer than 100 cells per compound. Each compound is impregnated in a 200-μm-diameter disc composed of biodegradable poly-(d),(l)-lactide/glycolide copolymer. Cells are seeded on top of these discs, and compounds slowly diffuse out, affecting proximal cells. In contrast with microtiter-based screening, this system does not involve the use of wells or walls between each compound-treated group of cells. We demonstrate detection of the effects of a single compound in a large microarray, that diverse compounds can be released in this format, and that extended release over several days is feasible. We performed a small synthetic lethal screen and identified a compound (macbecin II) that has reduced activity in cells with RNA interference-mediated decrease in the expression of tuberous sclerosis 2. Thus, we have developed a microarray-based screening system for testing the effects of small molecules on mammalian cells by using an imaging-based readout. This method will be useful to those performing small-molecule screens to discover new chemical tools and potential therapeutic agents.
Microarrays are widely used tools in the postgenomic era: They can be used to generate a vast amount of data in a short period with highly parallel experimental methods and analyses (1-4). Early microarrays consisted of regularly repeating arrays of DNA (5, 6) and were used to measure changes in gene expression by converting cellular mRNA to a fluorescently labeled nucleic acid and hybridizing it to a DNA microarray (2). In addition, microarrays with features containing expression plasmids or small interfering RNAs (siRNAs) can be used to transfect mammalian cells to study the consequences of perturbing, in parallel, the expression of a large number of genes (7-9).
More recently, microarrays have been extended to proteins and small molecules (4, 10-20). Protein microarrays have been created for measuring protein activities, interactions between proteins, and ligand binding to proteins (21-24); small molecule microarrays have been used for detecting the interactions between proteins and many small molecules (10, 25-29). These previously reported small-molecule microarrays have been used exclusively to detect in vitro interactions; it has not been possible to test the effects of small molecules on cells in a microarray format.
We sought to develop a microarray format for performing high-throughput screens of small molecules in mammalian cells. We envisioned that such a technology would be valuable for chemical genetic screens and for drug discovery efforts. Chemical genetics involves testing thousands of small molecules for their effects on a cellular or organismal phenotype; subsequently, the proteins targeted by these phenotype-modifying compounds are identified. This process can reveal proteins that regulate biological processes of interest (30-35).
Here, we describe the creation of microarrays consisting of small molecules impregnated in a biodegradable polymer and printed on a standard microscope slide. A monolayer of cells is grown over the array, allowing each compound to affect proximal cells. To localize compounds on a slide surface, we used a biodegradable poly-(d),(l)-lactide/glycolide copolymer (PLGA) after assessing more than two dozen different polymer materials. It was not clear, a priori, that any polymer would be useful in a microarray format, which requires that the polymer matrix exhibit a sufficiently large barrier to diffusion such that compounds are slowly released from a flat disc with a volume on the order of 1 nl. The nontoxic and slow-release properties of PLGA have been documented for a variety of purposes (36), ranging from the delivery of insulin-like growth factor for the treatment of osteoporosis (37) to that of paclitaxel for the treatment of tumors (38). PLGA and other poly(lactic acid) polymers enable the controlled release of proteins and small molecules through a combination of drug diffusion and polymer erosion in the context of large macroscopic amounts of polymer (36, 39).
By using PLGA as a scaffold, we developed a small-molecule cell microarray format that is compatible with existing chemical libraries, in which compounds are typically stored as frozen solutions in dimethyl sulfoxide. We have performed proof-of-principle experiments to evaluate this screening format.
Materials and Methods
Reagent Preparation. In a 2-ml microfuge tube, 100 mg of PLGA (lactide/glycolide ratio, 50:50; inherent viscosity, 0.5-0.68; catalog no. 48300-025, Polysciences) was added to 1 ml of methyl salicylate (catalog no. M-2047, Sigma). The tube was vortex mixed (minivortex at setting 10; Sigma) for 30 min, aliquoted into 1-ml tubes, and stored at -20°C. All compounds were from Sigma, Calbiochem, and the National Cancer Institute and were dissolved in microfuge tubes at a concentration of 80 mM in DMSO (catalog no. D-5879, Sigma). Compounds were further diluted in DMSO and stored at -20°C. To prepare fibronectin (catalog no. 354008, BD Biosciences), 1 ml of sterile H2O was added to a vial containing 1 mg of fibronectin, and the solution was incubated at room temperature (RT) for 45 min, aliquoted, and stored at -20°C.
Microarray Printing. A microarraying robot (Pixsys 5500, Genomic Solutions, Ann Arbor, MI) with SMP4 and SMP10 pins (Arrayit SMP4 or SMP10, Telechem, Sunnyvale, CA) was used to deposit reagent on Ni-chelated glass slides (catalog no. MNC00010, Xenopore, Hawthorne, NJ). Ni-chelated slides were used because the surface allowed for the printing of small, well-formed polymer spots and mammalian cell attachment. Before printing, 20 μl of printing solution was loaded into a round-bottom, polypropylene 384-well plate and centrifuged with a Beckman GS-6R centrifuge with a GH3.8A horizontal rotor at 1,000 rpm for 30 sec. All printing was performed at 23°C with 55% humidity. The small-molecule microarray spots were built by layering different solutions in the same position. Polymer (100 mg·ml-1 PLGA in methyl salicylate) was printed with an SMP4 pin by using a pins-down slide time of 50 msec. After a 10-min drying time, an SMP10 pin was used to deposit one to five cycles of compound/DMSO (20 nl total) by using a pins-down slide time of 1,000 msec. Each cycle was separated by at least 8 min of drying time to allow for DMSO evaporation. Finally, spots were topped by using an SMP4 pin to deposit methyl salicylate with a pins-down slide time of 300 msec. Once printed, arrays were desiccated for 30 min at RT and stored at 4°C or used immediately. Spot sizes were ≈200 μm. The spot-to-center distances featured in each figure is as follows: Fig. 1, 500 μm; Fig. 2, >3 mm; Fig. 3, 4 mm; and Fig. 4, 1.12 mm for the initial screen and 4.5 mm for the retest.
Fig. 1.

Robot-generated arrays of biodegradable polymer. (a) An array of 441 spots was printed on a slide. The center spot was loaded with 50 pmol of PAO, and all other spots were loaded with DMSO. In the photographs, nuclei are blue (Hoechst stain) and filamentous actin is green (phalloidin-FITC stain). (b) Pictures of an array of spots loaded with podophyllotoxin (Left) and PAO (Right) showing a dose-response effect on A549 cells. (c) Quantitation of cell density from the images in b for each concentration of drug at different distances from the spot (the units of the vertical axis are cells per unit of area for all graphs in this figure). (d) Replicates of the arrays used in c assayed with HeLa cells. (e) Replicate arrays of b that were stored at 4°C for 48 h and then assayed by using A549 cells.
Fig. 2.

Extended release of rapamycin. (a) These arrays are stained with Hoechst 33342 and a cy3-anti-pS6 antibody [a downstream effector of mTOR (cells affected by rapamycin show a lack of S6 phosphorylation)]. For each spot, the largest radius of effect was determined by measuring pS6 per unit of area. Regions with average pS6 levels below a threshold were considered affected. Each line in the graph represents the concentration of rapamycin in relation to the radius of effect for each storage condition. (b) The quantitation of pS6 intensity and the number of cells per unit of area at 50-μm intervals from the center of a spot loaded with 7.5 μM rapamycin. The level of pS6 increases with increasing distance from the center of the spot, suggesting that a concentration gradient has been established. Consistent with the known effects of rapamycin, the spots have a mild antiproliferative effect on cells in a dose-dependent manner.
Fig. 3.

Evaluation of 12 compounds in the microarray format. (a) The graph compares a calcein assay performed in microtiter plates with a cell-density assay performed on a microarray by using verrucarin A [502.5 atomic mass units (amu)] in A549 cells. (b) Each graph represents the effect of a different compound known to be active against A549 cells assayed in the microarray format (n = 3). Each graph shows five different concentrations of compound and a “no compound” control (far right). (c) Images of cells affected by different compounds. Nuclei are blue (Hoechst stain), and filamentous actin is green (phalloidin-FITC stain). We presume that the distance over which compounds act is determined by potency; intrinsic activity; partitioning of each compound between the slide surface, cell surfaces, and the medium; and the rate of diffusion of each compound out of the polymer, through the medium, and along the slide surface.
Fig. 4.

Synthetic lethal screen of 70 compounds. We used siRNAs in either A549 or HeLa cells to knock down one of seven target genes and then seeded each set of knocked-down cells on identical microarrays loaded with 70 different compounds. (a) To confirm that each siRNA was effective, we performed Western blots on small aliquots of cells that were simultaneously seeded on the microarrays. The right lane of the blots was loaded with lysate from cells transfected with siRNA for a target gene, and the left lane was loaded with lysate from cells treated with the same reagents without the siRNA. All gels were loaded with samples normalized for total protein. (b)We found four compound-siRNA pairs in which there was a significant difference in activity in the microarray format. Student's t tests were performed on cell density measurements taken at each distance from the center of the spots, and significant changes in cell density are highlighted in yellow. (c) We confirmed the macbecin II/TSC2 synthetic lethal effect by using a calcein acetoxymethyl ester viability assay in microtiter plates.
Small-Molecule Microarray Assay. If previously printed and stored, slides containing arrays were desiccated at RT for 30 min then placed in a Petri dish at RT. One milliliter of fibronectin (diluted 1:30 in DMEM from frozen aliquots) was deposited on the array-containingslides. After 1 h of incubation at RT, slides were rinsed in sterile H2O and placed in a 100 × 100 × 10-mm, square tissue-culture dish. Actively growing cells in 25 ml of culture medium (A549 cells, DMEM with 10% FBS/50 units·ml-1 penicillin/50 μg·ml-1 streptomycin; HeLa cells, DMEM with 10% inactivated FCS/50 units·ml-1 penicillin/50 μg·ml-1 streptomycin) were seeded on arrays and incubated at 37°C in 5% CO2. When performing longer assays, fewer cells were seeded to achieve similar final cell densities. For 14-h incubation, we seeded eight million cells; for 38-h incubations, we seeded four million cells; and for 62-h incubations, we seeded two million cells. After growth for the specified amount of time, slides were fixed by using 3.7% paraformaldehyde and 4% sucrose in PBS for 20 min at RT.
siRNA Transfection. We seeded two million cells in 5 ml of medium in a 10-cm, round tissue-culture dish for 14 h in culture medium. We rinsed the cells twice with 5 ml of Optimem (Invitrogen). After the final aspiration, we added 4 ml of Optimem to each dish and set the dish in an incubator during the preparation of the siRNA transfection reagents. The cells were transfected with siRNA [BCL2, M-003307-00-05; BRCA1, P-002111-01-05; EGFR, M-003114-00-05; MDM2, M-003279-00-05; p53, M-003557-00-05; PTEN, M-003023-00-05; tuberous sclerosis complex gene 2 (TSC2), M-003029-00-05; Dharmacon] by using the Effectene transfection kit (catalog no. 301427, Qiagen, Valencia, CA). We mixed 300 μl of EC buffer with 25 μl of a 20 μM siRNA solution. After the addition of 16 μl of enhancer, the solution was vortex mixed on high for 1 sec. After a 5 min of incubation at RT, 40 μl of Effectene reagent was added. The solution was vortexed for 10 sec on high and incubated at RT for 10 min. Optimem (700 μl) was added, mixed, and placed (total preparation, ≈1 ml) into a 10-cm dish of cells. Cells were incubated at 37°C in 5% CO2 for 24 h and removed with trypsin/EDTA for use on microarrays.
Synthetic Lethal Screen. Arrays were seeded with four million cells (previously transfected with siRNA reagents as described above) in 25 ml of cell culture media and incubated for 38 h. At the time of microarray seeding, in a parallel experiment, one million cells of the same type in 4 ml of medium were seeded in 6-cm, round tissue-culture dishes and incubated for the same 38 h. After incubation, cells on arrays were fixed and stained; cells in 6-cm dishes were prepared for Western blot analysis of protein levels. The complete experiment required the analysis of 78 slides. By using parameters defined below in the image analysis section, it took ≈7 min to acquire the images for each slide (9 h total). We measured cell density at six different distances from the center of the 528 different spots on each slide. The analysis of one slide of images took ≈40 min (52 h total) by using a standard Pentium 4, 2-Ghz processor with 2 gigabytes of random access memory.
Western Blots. After 38 h, cells in 6-cm dishes were lysed and Western blots performed (40). Equal protein was loaded in each lane, normalized by using a BSA standard. Primary mouse monoclonal antibodies were used at the following concentrations: anti-p53, 1:500 (Ab-6, Oncogene Science); anti-PTEN, 1:1,000 (MS-1250-P0, Lab Vision/Neomarkers, Fremont, CA); anti-BCL2, 1:500 (sc-7382, Santa Cruz Biotechnology); anti-MDM2, 1:1,000 [sc-965 (SMP14), Santa Cruz Biotechnology]. Primary rabbit polyclonal antibodies were used at the following concentrations: anti-BRCA1, 1:100 (sc-642, Santa Cruz Biotechnology); anti-TSC2, 1:500 (Santa Cruz Biotechnology); anti-EGFR, 1:1,000 (sc-03, Santa Cruz Biotechnology). Secondary antibodies were horseradish peroxidase-labeled anti-mouse or anti-rabbit antibodies produced in donkeys (Jackson ImmunoResearch) and were used at a 1:5,000 dilution.
Immunofluorescence. All cells on arrays in Figs. 1, 3, and 4 were stained with phalloidin-FITC (F432, Molecular Probes) and Hoechst 33342 (H3570, Molecular Probes). After fixation, cells were permeabilized with 0.2% Triton X-100 for 20 min and placed in a humidity chamber with cell side up. Ten units of phalloidin in 1 ml of PBS was deposited on each slide surface and incubated at RT for 40 min. Cells were dipped in PBS, placed in a humidity chamber and incubated with 1 ml of Hoechst 33342 (1:10,000 dilution in PBS) for 30 min. For Fig. 2, cells were fixed as above, permeabilized with 0.2% Triton X-100 for 40 min and probed with primary and secondary antibodies. The primary rabbit polyclonal antibody used was anti-pS6 (ser240/4, Cell Signaling); 500 μl of antibody per slide was used at a dilution of 1:500. The secondary antibody used was anti-rabbit cy3 (The Jackson Laboratory) at a dilution of 1:1,000. After completion of secondary antibody probe, slides were stained with Hoechst 33342.
Image Capture and Analysis. We captured images of all slides by using both light and fluorescence microscopy with a Zeiss Axiovert 200 at ×50 magnification and custom software designed on the ks400 software platform. Unless otherwise mentioned, we measured the number of Hoechst-stained nuclei divided by area in a region of interest (ROI). We used a finite set of circular regions centered on a spot with radii increasing in increments of 150 μm to define ROIs. All ROIs were labeled for the radius of their outer boundary, and all ROIs, except the first, were shaped like a ring. The smallest ROI measured for each spot was a circle centered on the spot with a radius of 150 μm. The next largest ROI (radius of 300 μm) was shaped like a ring and was calculated by subtracting a circle with a radius of 150 μm from a circle with a radius of 300 μm. We divided the number of nuclei counted in each ROI by the area (μm2) of each ROI to get the number of cells per area (CPA). For each slide, we averaged all of the spots loaded with DMSO (negative controls) to get a normal CPA for each ROI. We divided all measurements of CPA taken from each ROI of an experimental spot with the average normal CPA measurements to get a percentage of CPA. All data analysis was performed on values of percent CPA. We used a two-sample, heteroskedastic t test to compare percent CPA values generated by compounds affecting cells transfected with siRNA versus without siRNA. The values of CPA reported in Figs. 1 and 3 are the average of the percent CPA for all replicates and the error bars are the standard deviation. In Fig. 4, error bars are standard error of the mean (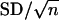 ). Fig. 2 was analyzed by using the same ROI format, but instead of measuring the number of nuclei, we measured the total cy3-pS6 signal for each ROI and divided that by the area of each ROI. This calculation gave the average signal per unit area (SPA). These values were normalized to spots with no activity and a percentage of SPA was calculated. A value of 100% equals pS6 in a normal cell population and 0% equals no detectable pS6. We set 80% as a threshold such that an ROI with a value below 80% was considered affected. For each concentration of rapamycin, we reported the outer radius of the largest ROI that had a percent SPA (an average value of three replicates) below our threshold. All percent SPA of smaller ROIs of an affected spot were below the threshold but not reported.
). Fig. 2 was analyzed by using the same ROI format, but instead of measuring the number of nuclei, we measured the total cy3-pS6 signal for each ROI and divided that by the area of each ROI. This calculation gave the average signal per unit area (SPA). These values were normalized to spots with no activity and a percentage of SPA was calculated. A value of 100% equals pS6 in a normal cell population and 0% equals no detectable pS6. We set 80% as a threshold such that an ROI with a value below 80% was considered affected. For each concentration of rapamycin, we reported the outer radius of the largest ROI that had a percent SPA (an average value of three replicates) below our threshold. All percent SPA of smaller ROIs of an affected spot were below the threshold but not reported.
Microtiter Plate Assay. Assays were performed according to the methods described in ref. 41. Four or eight thousand A549 cells were seeded per well for 14 h before compound was added. After the addition of compound to each well, the cells were incubated for 62, 38, or 14 h, respectively, and then assayed by using a calcein acetoxymethyl ester viability assay (Molecular Probes).
Results
We evaluated numerous polymer matrices, solvents, and arraying conditions to create a small-molecule-compatible, cell-microarray screening system. In the optimal system, we deposited picomoles of each compound on a solid surface encased in a biodegradable polymer, providing sustained release. We grew cells on the surface of the microarray and imaged the resulting adherent cells to identify cellular responses specific to the compound present in each spot.
We discovered that PLGA, a biodegradable polymer, is capable of releasing small molecules in a controlled fashion in a microarray format. This polymer dissolves in numerous liquid solvents at RT to form a solution with a viscosity and volatility that is compatible with printing by using a miniaturized arraying pin. We deposited at each position on a glass microscope slide ≈1 nl of PLGA solution that dried to form an ≈200-μm diameter spot. These polymer spots typically formed doughnut-like shapes when they dried, with a thicker edge and a thinner center. Polymer spots were printed in the desired array configuration and, once dried, loaded with ≈20 nl of a compound of interest dissolved in DMSO. To load a feature, we deposited a compound solution on top of each polymer spot by using an arraying pin. Subsequently, we spotted ≈1 nl of the solvent methyl salicylate onto each spot, allowing for the mixing of the compound into the polymeric matrix. This method allows us (i) to print an array of control polymer spots on a slide lacking test compounds (i.e., treated only with PLGA, DMSO, and methyl salicylate), (ii) to print a library of compounds previously dissolved in DMSO, and (iii) to arrange the test compounds and spots in any desired configuration.
As an initial test of the utility of this system, we printed an array of 441 spots and loaded all spots except the center spot with DMSO. The center spot was loaded instead with phenylarsine oxide (PAO), a cytotoxic compound that inhibits protein-tyrosine phosphatases and bis-thiol-containing proteins and is cytotoxic to A549 human lung carcinoma cells (41-45). Subsequently, we seeded A549 cells onto this microarray and incubated the cells on the array for 38 h. The cells grew to confluence throughout the array except around the area containing the PAO-loaded polymer. Longer or shorter incubation times did not result in a significantly larger or smaller ring of effect (data not shown). These results suggest that the PAO was released from the polymer spot and affected the cells growing within several hundred micrometers of this spot (Fig. 1).
As compounds were released from the polymer matrix, they diffused into surrounding culture medium; therefore, we expected to see a decrease in compound concentration and the corresponding cellular effect with an increase in distance from the spot. We measured cell density (i.e., CPA) at different distances from each spot center and found that all spots loaded with DMSO had distance-independent densities similar to normal cell densities, ≈0.7 cells per unit of area (Fig. 5, which is published as supporting information on the PNAS web site). The area occupied by the spot loaded with PAO had the lowest cellular density, ≈0.04 cells per unit of area. We observed that PAO has an IC50 of 2 μM in typical 384-well-plate-based experiments (data not shown). When testing in the microarray format, we found that spotting ≈200 pmol of PAO in a DMSO/PLGA solution gave a half-maximal effect on the spot. Thus, we estimate that these conditions produce a localized concentration of ≈2 μM PAO around such a spot. In addition, cellular densities increased with increasing distance from the spot center (Fig. 1). This finding suggests that a concentration gradient of PAO was established around the spot and the cells were affected in a dosage-dependent fashion at increasing distances from the PAO-loaded spot.
Next, we tested the effect of loading different amounts of a compound onto a small-molecule cell microarray. We created a polymer microarray containing a dilution series of either podophyllotoxin (PTX) or PAO and cultured A549 cells on it (Fig. 1). Similar to previous experiments, we found that spots loaded with PTX or PAO affected cells in a dose-dependent manner. In every case, the cell density was lowest on the spot with one of these cytotoxic compounds and increased with increasing distance. Spots loaded with a larger amount of compound tended to have a more profound and widespread effect. For example, spots loaded with ≈400 pmol of PAO affected a larger area and resulted in lower cell densities at similar distances when compared with spots loaded with 200 pmol of PAO (Figs. 1 b and c). Both PAO and PTX are cytotoxic but have different potencies, maximal activities, and mechanisms of action. PTX is more potent (i.e., active at lower concentrations) but has a weaker effect on cell density and cell viability than PAO. Of the spots loaded with PAO, only spots loaded with 400 or 200 pmol exhibited a visible effect on cell density. Moreover, the effect of PAO on these cells was local and striking. In contrast, spots loaded with all amounts of PTX reduced cell density. Although not as striking as the PAO-loaded spots, at similar concentrations, the PTX-loaded spots affected the cell density of larger surface areas. The results suggest that compound-specific effects occur in a dosage-dependant fashion on the microarrays.
By using plate-based methods, compounds show different potencies in different cell types (41). We tested the small-molecule cell microarray method on multiple cell types by comparing the effects of PTX and PAO in HeLa and A549 cells. Both compounds were more potent in HeLa cells compared with A549 cells, demonstrating that this microarray format is not limited to the use of A549 cells and that the activity in the small-molecule microarray system can depend on cell type (Fig. 1). In addition to A549 and HeLa cells, we have successfully tested the microarrays by using five other cell lines: BJ, BJELR, MEF, 293T, and DU145 (data not shown).
To determine the stability of a printed, PLGA-impregnated, small-molecule microarray, we took replicate slides containing microarrays loaded with a dilution-series of PTX and PAO and stored them at 4°C for 48 h. After storage, the slides were assayed as before by measuring the density of A549 cells throughout the slide (Fig. 1e) and compared with the results obtained on a freshly printed slide. The potencies of PTX and PAO appeared similar in the stored array and the freshly printed array, demonstrating that the arrays can be stored for at least 48 h with little reduction in effectiveness.
We tested arrays loaded with PAO and PTX assayed for time periods upwards of 10 days to evaluate compound diffusion. As expected, these longer preincubation times produce similar or slightly reduced phenotypes and ring sizes when compared with 72-h preincubation times (data not shown). This result indicates that the compound is being depleted from the spots creating a stable concentration gradient that will not affect an increasing area over time.
We sought to determine whether we could detect cellular phenotypes other than death in the microarray format. Toward this end, we tested the effects of the natural product rapamycin on the phosphorylation status of S6 in A549 cells. Rapamycin inhibits the mammalian target of rapamycin (mTOR) pathway, causing the dephosphorylation of its downstream effector, the ribosomal S6 protein. To evaluate the release profile of rapamycin from PLGA spots over extended periods of time, we performed the following experiment. We seeded A549 cells on four identical arrays loaded with rapamycin and incubated the arrays at 37°C with 5% CO2 for 24, 48, 72, and 168 h. After this initial “presoak” period, we stripped the cells from the arrays, reseeded them with fresh A549 cells, and quantitated the pS6 levels 48 h after the second seeding. The purpose of the presoaking period was to enable the normal processes of biodegradation and bioerosion of the PGLA polymer that occur in the presence of cells. If most of the rapamycin was released from the PLGA spots as an initial bolus, most presoaked arrays should have a dramatic reduction in pS6 when compared with a nonpresoaked array. If the rapamycin was released slowly, presoaking an array should reduce the amount of pS6 proportional to length of presoaking time. We classified affected areas as having <80% of the mean pS6 signal when compared with the mean pS6 of normal A549 cells. By using this threshold, we calculated the size of the ring of affected area for each spot on the array (Fig. 2a).
For each array, we found that the size of the area of affected cells increased with increasing amounts of deposited rapamycin. In general, all rapamycin-containing spots were still active after presoaking; they have smaller radii of effect with lower rapamycin concentrations, suggesting that, as the time of contact with cells increases, the rate of release might decrease. One possible explanation is that the release of compound from the polymer obeys first-order kinetics; i.e., the release of compound is proportional to the amount of compound present the polymer matrix.
To control for possible degradation of compounds or polymer unrelated to soaking with cells, we incubated an array at 4°C for 168 h. This array was somewhat less effective than the freshly printed array but more effective than the array that had been incubated with cells for the same 168-h time period.
The spots loaded with the smallest amount of rapamycin were still active under all presoaking conditions, demonstrating that a minimum level of release sufficient to affect pS6 levels is maintained. These results indicate that PLGA provides sustained release of compounds and demonstrates the usefulness of the method in assays up to at least 72 h. (Fig. 6, which is published as supporting information on the PNAS web site).
The biodegradable polymers used for clinical drug delivery are optimized around a single compound and rate of release. We required a polymer that allowed for the release of many different compounds, independent of their physical properties. To test the breadth of utility of PLGA, we assayed a set of 12 compounds known to be lethal to A549 cells and ranging in molecular mass from 167 to 1,255 atomic mass units (41). A dilution series of each compound was deposited in the microarray format, and cell density was measured. These 12 compounds had some discernable effect on the density of A549 cells, demonstrating that compounds with a range of physical properties can be used in this system (Fig. 3). An attempt was made to assess whether the potencies of these compounds correlated with their physical properties. Although each compound had a specific inhibitory effect on A549 cells, the compounds' potencies did not correlate with any molecular descriptors we tested (Fig. 3).
We performed a small-scale screen to test the ease of by using small-molecule cell microarrays in a screening process. Toward this end, we performed a synthetic lethal screen of known compounds against a set of cancer-related target genes. Our goal was to find compounds that are not generally lethal to cells but display increased or decreased activity in the presence or absence of specific cancer-related genes. Such compounds may be candidate therapeutic agents, or they may be useful probes of the functions of the tested cancer-related genes (46-49).
For this synthetic lethal screen, we used two cell types (A549 and HeLa), each separately transfected with one of seven different siRNAs that targeted for destruction mRNAs encoding p53, PTEN, MDM2, EGFR, TSC2, BCL2, and BRCA1. Thus, we had 16 cell conditions under which to test each compound (no siRNA plus seven siRNAs in two cell types). We assayed a set of 70 known biologically active compounds that we selected from Sigma, Calbiochem, and the National Cancer Institute. Each compound was tested in triplicate at three concentrations in each cell condition for its effect on cellular density. This small pilot experiment involved the collection of ≈50,000 data points.
Knockdown of mRNAs by using siRNAs is transient and can be of variable effectiveness; thus, it is important to measure directly the change in the concentration of the target protein in siRNA-mediated knockdown experiments. When we seeded each cell sample on the microarrays, we also grew up a small amount of the cell sample in parallel for Western blot analysis of target protein level (Fig. 4).
Compounds that caused a significant change (P < 0.05) in cell density under a specific set of conditions were considered putative hits. The putative hits were retested with more replicates under the same conditions to confirm their effects. We discovered that the effectiveness of three of the 70 compounds (cisplatin, macbecin II, and nocodazole) was altered in some of the siRNA-transfected cells (Fig. 4). In HeLa cells with reduced levels of p53, macbecin II, and cisplatin had altered effects. In HeLa cells with reduced levels of TSC2 or PTEN, macbecin II and nocodazole, respectively, had reduced effects. Thus, of 980 compound-siRNA cell-type combinations tested, we found four in which knockdown of a specific mRNA altered the sensitivity of tumor cells to a specific compound in the microarray format. We retested these potential synthetic lethal effects in a conventional calcein acetoxymethyl ester viability assay in 384-well plate format and found that one was confirmed: Reduction of TSC2 caused a detectable level of resistance to macbecin II (Fig. 4c).
Discussion
Macbecin II is a DNA antimetabolite that induces breaks in double-stranded DNA, possibly by means of p53-mediated apoptosis (50). Reduction in TSC2 has been shown to activate mTOR-mediated cellular hyperplasia and hypertrophy (51). Our results show that cells with reduced levels of TSC2 are less susceptible to macbecin II. In some systems, loss of mTOR activity is associated with p53-dependent apoptosis (52). Our results support this finding by suggesting that enhancement of cellular growth through loss of TSC2 may help overcome the apoptotic effects of DNA damage. Furthermore, TSC2 knockdown affected the activity of macbecin II and not cisplatin, further implicating distinct p53-related cellular responses.
Although the small-molecule cell microarray method shows promise, there are technical limitations that require attention. Currently, printing densities are limited to prevent one spot from affecting cells on another nearby spot. We have experimented with decreasing the effect of compound crosscontamination by using simple methods, such as overhead stirring (data not shown). In addition, it would be advantageous to have a polymer that could be induced to release compounds at a specified time, which would add a new level of elegance, whereby one could stabilize cellular states, such cell cycle progression, before release of the compounds.
The microarray format uses a small amount of each compound in each test and requires fewer cells than plate-based experiments. Small-molecule cell microarrays may be advantageous when testing a rare and precious natural product. The method we have developed for making these microarrays is compatible with most large chemical libraries, which are typically dissolved in DMSO.
In summary, we have reported the construction of a cell microarray that enables the screening of small-molecule libraries in mammalian cells. These well-less arrays are compatible with current cell-based assays and high-resolution imaging techniques. This microarray is a high-throughput, high-content method of screening small molecules that uses small amounts of compound, few cells, imaging-based readouts, and the potential to screen many compounds with minimal automation. This method will be useful to those performing small-molecule screens to discover new chemical tools and potential therapeutic agents.
Supplementary Material
Acknowledgments
We thank the Ira W. DeCamp Foundation and the Edith C. Blum Foundation for financial support. This research was supported in part by a Career Award at the Scientific Interface from the Burroughs Wellcome Fund (to B.R.S.).
Author contributions: D.M.S. and B.R.S. designed research; S.N.B., D.M.S., and B.R.S. performed research; S.N.B., D.M.S., and B.R.S. analyzed data; and S.N.B., D.M.S., and B.R.S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PLGA, poly-(d),(l)-lactide/glycolide copolymer; PAO, phenylarsine oxide; PTX, podophyllotoxin; siRNA, small interfering RNA; ROI, region of interest; CPA, cells per area; RT, room temperature; TSC2, tuberous sclerosis complex gene 2; mTOR, mammalian target of rapamycin.
References
- 1.Root, D. E., Kelley, B. P. & Stockwell, B. R. (2002) Curr. Opin. Drug Discovery Dev. 5, 355-360. [PMC free article] [PubMed] [Google Scholar]
- 2.Brown, P. O. & Botstein, D. (1999) Nat. Genet. 21, 33-37. [DOI] [PubMed] [Google Scholar]
- 3.Marton, M. J., DeRisi, J. L., Bennett, H. A., Iyer, V. R., Meyer, M. R., Roberts, C. J., Stoughton, R., Burchard, J., Slade, D., Dai, H., et al. (1998) Nat. Med. 4, 1293-1301. [DOI] [PubMed] [Google Scholar]
- 4.Mills, J. C., Roth, K. A., Cagan, R. L. & Gordon, J. I. (2001) Nat. Cell Biol. 3, E175-E178. [DOI] [PubMed] [Google Scholar]
- 5.Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. (1995) Science 270, 467-470. [DOI] [PubMed] [Google Scholar]
- 6.Schena, M., Shalon, D., Heller, R., Chai, A., Brown, P. O. & Davis, R. W. (1996) Proc. Natl. Acad. Sci. USA 93, 10614-10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziauddin, J. & Sabatini, D. M. (2001) Nature 411, 107-110. [DOI] [PubMed] [Google Scholar]
- 8.Wu, R. Z., Bailey, S. N. & Sabatini, D. M. (2002) Trends Cell Biol. 12, 485-488. [DOI] [PubMed] [Google Scholar]
- 9.Bailey, S. N., Wu, R. Z. & Sabatini, D. M. (2002) Drug Discovery Today 7, S113-S118. [DOI] [PubMed] [Google Scholar]
- 10.Kuruvilla, F. G., Shamji, A. F., Sternson, S. M., Hergenrother, P. J. & Schreiber, S. L. (2002) Nature 416, 653-657. [DOI] [PubMed] [Google Scholar]
- 11.MacBeath, G. & Schreiber, S. L. (2000) Science 289, 1760-1763. [DOI] [PubMed] [Google Scholar]
- 12.Haab, B. B. (2001) Curr. Opin. Drug Discovery Dev. 4, 116-123. [PubMed] [Google Scholar]
- 13.Stoll, D., Templin, M. F., Schrenk, M., Traub, P. C., Vohringer, C. F. & Joos, T. O. (2002) Front. Biosci. 7, C13-C32. [DOI] [PubMed] [Google Scholar]
- 14.Templin, M. F., Stoll, D., Schrenk, M., Traub, P. C., Vohringer, C. F. & Joos, T. O. (2002) Trends Biotechnol. 20, 160-166. [DOI] [PubMed] [Google Scholar]
- 15.Wilson, D. S. & Nock, S. (2003) Angew. Chem. Int. Ed. 42, 494-500. [DOI] [PubMed] [Google Scholar]
- 16.Glokler, J. & Angenendt, P. (2003) J. Chromatogr. 797, 229-240. [DOI] [PubMed] [Google Scholar]
- 17.Xu, Q. & Lam, K. S. (2003) J. Biomed. Biotechnol. 2003, 257-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falsey, J. R., Renil, M., Park, S., Li, S. & Lam, K. S. (2001) Bioconjugate Chem. 12, 346-353. [DOI] [PubMed] [Google Scholar]
- 19.MacBeath, G. (2001) Nat. Biotechnol. 19, 828-829. [DOI] [PubMed] [Google Scholar]
- 20.MacBeath, G. (2002) Nat. Genet. 32, Suppl., 526-532. [DOI] [PubMed] [Google Scholar]
- 21.Huang, R. P. (2001) J. Immunol. Methods 255, 1-13. [DOI] [PubMed] [Google Scholar]
- 22.Morozov, V. N., Gavryushkin, A. V. & Deev, A. A. (2002) J. Biochem. Biophys. Methods 51, 57-67. [DOI] [PubMed] [Google Scholar]
- 23.Espejo, A., Cote, J., Bednarek, A., Richard, S. & Bedford, M. T. (2002) Biochem. J. 367, 697-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, G. Y., Uttamchandani, M., Zhu, Q., Wang, G. & Yao, S. Q. (2003) ChemBioChem 4, 336-339. [DOI] [PubMed] [Google Scholar]
- 25.Zhu, Q., Uttamchandani, M., Li, D., Lesaicherre, M. L. & Yao, S. Q. (2003) Org. Lett. 5, 1257-1260. [DOI] [PubMed] [Google Scholar]
- 26.Lin, B., Qiu, J., Gerstenmeier, J., Li, P., Pien, H., Pepper, J. & Cunningham, B. (2002) Biosens. Bioelectron. 17, 827-834. [DOI] [PubMed] [Google Scholar]
- 27.Winssinger, N., Ficarro, S., Schultz, P. G. & Harris, J. L. (2002) Proc. Natl. Acad. Sci. USA 99, 11139-11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam, K. S. & Renil, M. (2002) Curr. Opin. Chem. Biol. 6, 353-358. [DOI] [PubMed] [Google Scholar]
- 29.Bedalov, A., Gatbonton, T., Irvine, W. P., Gottschling, D. E. & Simon, J. A. (2001) Proc. Natl. Acad. Sci. USA 98, 15113-15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stockwell, B. R. (2000) Nat. Rev. Genet. 1, 116-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stockwell, B. R. (2000) Trends Biotechnol. 18, 449-455. [DOI] [PubMed] [Google Scholar]
- 32.Stockwell, B. R. (2002) Neuron 36, 559-562. [DOI] [PubMed] [Google Scholar]
- 33.Schreiber, S. L. (1998) Bioorg. Med. Chem. 6, 1127-1152. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber, S. L. (2000) Science 287, 1964-1969. [DOI] [PubMed] [Google Scholar]
- 35.Schreiber, S. L. (2003) Chem. Eng. News 81, 51-61. [Google Scholar]
- 36.Langer, R. (1998) Nature 392, 5-10. [PubMed] [Google Scholar]
- 37.Cai, L., Okumu, F. W., Cleland, J. L., Beresini, M., Hogue, D., Lin, Z. & Filvaroff, E. H. (2002) Osteoarthritis Cartilage 10, 692-706. [DOI] [PubMed] [Google Scholar]
- 38.Kim, S. C., Kim, D. W., Shim, Y. H., Bang, J. S., Oh, H. S., Wan Kim, S. & Seo, M. H. (2001) J. Controlled Release 72, 191-202. [DOI] [PubMed] [Google Scholar]
- 39.Ulbrich, K., Pechar, M., Strohalm, J., Subr, V. & Rihova, B. (1997) Ann. N.Y. Acad. Sci. 831, 47-56. [DOI] [PubMed] [Google Scholar]
- 40.Kim, D. H., Sarbassov dos, D., Ali, S. M., Latek, R. R., Guntur, K. V., Erdjument-Bromage, H., Tempst, P. & Sabatini, D. M. (2003) Mol. Cell 11, 895-904. [DOI] [PubMed] [Google Scholar]
- 41.Root, D. E., Flaherty, S. P., Kelley, B. P. & Stockwell, B. R. (2003) Chem. Biol. 10, 881-892. [DOI] [PubMed] [Google Scholar]
- 42.Estrov, Z., Manna, S. K., Harris, D., Van, Q., Estey, E. H., Kantarjian, H. M., Talpaz, M. & Aggarwal, B. B. (1999) Blood 94, 2844-2853. [PubMed] [Google Scholar]
- 43.Gerhard, R., John, H., Aktories, K. & Just, I. (2003) Mol. Pharmacol. 63, 1349-1355. [DOI] [PubMed] [Google Scholar]
- 44.Bogumil, R. & Ullrich, V. (2002) Methods Enzymol. 348, 271-280. [DOI] [PubMed] [Google Scholar]
- 45.Bogumil, R., Namgaladze, D., Schaarschmidt, D., Schmachtel, T., Hellstern, S., Mutzel, R. & Ullrich, V. (2000) Eur. J. Biochem. 267, 1407-1415. [DOI] [PubMed] [Google Scholar]
- 46.Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. (2003) Cancer Cell 3, 285-296. [DOI] [PubMed] [Google Scholar]
- 47.Stockwell, B. R., Haggarty, S. J. & Schreiber, S. L. (1999) Chem. Biol. 6, 71-83. [DOI] [PubMed] [Google Scholar]
- 48.Bunz, F., Hwang, P. M., Torrance, C., Waldman, T., Zhang, Y., Dillehay, L., Williams, J., Lengauer, C., Kinzler, K. W. & Vogelstein, B. (1999) J. Clin. Invest. 104, 263-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torrance, C. J., Agrawal, V., Vogelstein, B. & Kinzler, K. W. (2001) Nat. Biotechnol. 19, 940-945. [DOI] [PubMed] [Google Scholar]
- 50.Nelson, W. G. & Kastan, M. B. (1994) Mol. Cell. Biol. 14, 1815-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inoki, K., Zhu, T. & Guan, K. L. (2003) Cell 115, 577-590. [DOI] [PubMed] [Google Scholar]
- 52.Huang, S., Shu, L., Dilling, M. B., Easton, J., Harwood, F. C., Ichijo, H. & Houghton, P. J. (2003) Mol. Cell 11, 1491-1501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}