

Abstract
Kawasaki disease (KD) is the most common acquired pediatric heart disease. We analyzed Whole Genome Sequences (WGS) from a 6-member African American family in which KD affected two of four children. We sought rare, potentially causative genotypes by sequentially applying the following WGS filters: sequence quality scores, inheritance model (recessive homozygous and compound heterozygous), predicted deleteriousness, allele frequency, genes in KD-associated pathways or with significant associations in published KD genome-wide association studies (GWAS), and with differential expression in KD blood transcriptomes. Biologically plausible genotypes were identified in twelve variants in six genes in the two affected children. The affected siblings were compound heterozygous for the rare variants p.Leu194Pro and p.Arg247Lys in Toll-like receptor 6 (TLR6), which affect TLR6 signaling. The affected children were also homozygous for three common, linked (r2 = 1) intronic single nucleotide variants (SNVs) in TLR6 (rs56245262, rs56083757 and rs7669329), that have previously shown association with KD in cohorts of European descent. Using transcriptome data from pre-treatment whole blood of KD subjects (n = 146), expression quantitative trait loci (eQTL) analyses were performed. Subjects homozygous for the intronic risk allele (A allele of TLR6 rs56245262) had differential expression of Interleukin-6 (IL-6) as a function of genotype (p = 0.0007) and a higher erythrocyte sedimentation rate at diagnosis. TLR6 plays an important role in pathogen-associated molecular pattern recognition, and sequence variations may affect binding affinities that in turn influence KD susceptibility. This integrative genomic approach illustrates how the analysis of WGS in multiplex families with a complex genetic disease allows examination of both the common disease–common variant and common disease–rare variant hypotheses.
Introduction
Although the ability to generate whole genome sequences (WGS) from individual subjects has existed for over a decade, the use of such methods to discover novel disease-causing variants in multiplexed families affected by complex genetic disease has been limited [1]. However, robust methods have been developed to identify rare, disease causative variants in WGS of families with monogenic diseases [2, 3]. Furthermore, such methods have proven useful in identifying individual patients with common diseases that are caused by rare, highly penetrant variants [4]. Individuals with rare Mendelian forms of common complex diseases are often distinguished by extreme phenotypes–such as very early onset, or disease refractory to usual treatments–and by multiple affected members in single families [5]. Here we sought to test this hypothesis in a multiplexed family with Kawasaki disease (KD).
Susceptibility to KD, the most common cause of acquired heart disease in children, is postulated to result from a complex set of genetic variants of which only a limited number have been validated to date [6]. This self-limited illness of unknown etiology presents with the sudden onset of fever and mucocutaneous signs and is associated with coronary artery vasculitis. Inflammation in the arterial wall can compromise the structural integrity, which leads to aneurysm formation in 25% of untreated children [7]. The major sequelae of aneurysms include thrombosis, scarring with stenosis, myocardial ischemia, infarction, and death [8–11]. KD is over-represented among children of Asian descent. In Japan, the country of highest incidence (306/100,000 children <5 years; one in every 60 male and 75 female children affected, respectively), there are more than 14,000 new cases each year and rates continue to rise [12]. In the United States, system dynamic models suggest that by 2030, one in every 1600 adults in the U.S. will have suffered from KD [13]. Data from limited patient series suggests that African American is disproportionately affected by KD [14–16]. Despite their apparent increased susceptibility, children of African American descent has been excluded from previous KD genetic analyses. As with other complex disorders, elucidation of the genetic determinants of KD has hitherto relied on candidate gene and genome-wide association studies (GWAS) using matched population controls and family linkage studies [17–27]. Thus far, many of the associated genetic variants have been located in introns with no associated molecular function identified [6]. Only the C allele of SNV rs28493229 in the inositol 1,4,5-trisphosphate 3-kinase C (ITPKC) gene on chromosome 19q13.2 has been shown to affect gene transcription and impact intracellular calcium signaling and inflammasome activation [22, 28].
Here, we examined the common disease–rare variant hypothesis in KD in an African Americans family with two affected and two unaffected siblings and their unaffected, biologic parents. We report rare, likely pathogenic genotypes in biologically plausible genes that co-segregated with disease in whole genome sequences.
Results
The genomes of all six family members were sequenced with paired, short reads to an aligned mean read depth of 33.2-fold. Unique nucleotide variants (8,018,553) were identified in the family, of which 7,592,729 were of high quality (Fig 1). We created three filter pipelines for further analysis. First, we applied the following filters: recessive homozygous only in the affected children, potentially deleterious (located in an exon, promoter region, splice site, or 3’UTR), and rare (allele frequency <1% or not available in 1,000 Genomes database). To these 34 variants in 31 genes, we applied two additional filters: gene found in KD pathway (defined by Ingenuity Pathway Analysis) and differentially expressed (p<0.05) in our KD transcriptome database [29]. This identified a CAG repeat variant (CAG10 homozygosity in the two affected siblings) in myocyte enhancer factor 2A (MEF2A). The reference allele for this variant is CAG11 and the variant was predicted to be deleterious. The deletion was confirmed by resequencing all six family members (S1 Table).
Fig 1. Workflow of variant discovery and validation.

The diagram illustrates a discovery and validation workflow starting from called variants in an African American family of six members with two KD-affected children as a discovery analysis on WGS followed by knowledge-based filtering derived from published GWAS, disease pathogenesis, and gene-level validation using differential transcript abundance. Confidence filter: a call quality at least 20 and read depth at least 10. Differentially expressed: ≥ 1.2-fold change between acute and convalescent whole blood transcripts. Abbrv.: AF: allele frequency; NA: not available, GWAS: genome-wide association study; KD: Kawasaki disease; TLR6: Toll-like receptor 6; MEF2A: myocyte enhancer factor 2A; ARRDC4:arrestin domain containing 4; SLK: STE20 like kinase; TACSTD2: tumor-associated calcium signal transducer 2.
An alternative set of filters was applied to the 7,592,729 high quality variants: compound heterozygous in two affected siblings only, allele frequency <3% in 1000 Genomes database, found in KD pathway, and differentially expressed in the KD transcriptome database. This resulted in only two rare variants (p.Leu194Pro and p.Arg247Lys) in a single gene (TLR6) that were compound heterozygous only in the affected siblings (Table 1). The MEF2A and TLR6 rare and potentially pathogenic variants that were found in KD pathways were grouped together in Tier 1 (Fig 1 and Table 1). Both TLR6 and MEF2A were differentially expressed in acute and convalescent whole blood RNA samples, with the highest expression levels in the acute phase of KD (Fig 2a and 2b).
Table 1. Genetic variants found in Tiers 1 and 2.
| Tier | Mode of inherit | Gene Symbol | Chr. | rs ID | Gene Region | Risk allele | Allelefreq.* | GWAS p-value |
|---|---|---|---|---|---|---|---|---|
| 1 | CH | TLR6 | 4 | 35220466 | Exonic | C | 0.6 | NA |
| 1 | CH | TLR6 | 4 | 5743809 | Exonic | A | 1.5 | NA |
| 1 | HR | MEF2A | 15 | 373652230 | Exonic | CAG deletion | NA | NA |
| 2 | HR | TACSTD2 | 1 | 14008 | Exonic | T | 15.7 | 0.01 |
| 2 | HR | TLR6 | 4 | 12650224 | 3'UTR | A | 21.3 | 0.002 |
| 2 | HR | TLR6 | 4 | 6822503 | 3'UTR | A | 63.3 | 0.008 |
| 2 | HR | TLR6 | 4 | 12645200 | 3'UTR | T | 63.3 | 0.008 |
| 2 | HR | TLR6 | 4 | 5743826 | 3'UTR | T | 4.2 | 0.003 |
| 2 | HR | TLR6 | 4 | 6837101 | Promoter | A | 38.4 | 0.03 |
| 2 | HR | SLK | 10 | 6584583 | Promoter | C | 83.1 | 0.01 |
| 2 | HR | SLK | 10 | 10786779 | Promoter | G | 79.3 | 0.02 |
| 2 | HR | ARRDC4 | 15 | 1552673 | Promoter | C | 76.2 | 0.03 |
RH = Homozygous Recessive, CH = Compound Heterozygous, TLR6: Toll-like receptor 6, MEF2A: Myocyte Enhancer Factor 2A, TACSTD2: tumor-associated calcium signal transducer 2, SLK: STE20 like kinase, ARRDC4: arrestin domain containing 4,
* 1000 genome allele frequencies for All, GWAS: European descent GWAS [19], NA: not available
Fig 2. Paired acute and convalescent whole blood gene expression levels for 131 KD subjects for the six genes implicated in KD susceptibility.

P values are uncorrected. Transcript levels from microarray database as previously described, not corrected for cell number [29]. Abbrv.: TLR6: Toll-like receptor 6; MEF2A: myocyte enhancer factor 2A; ARRDC4:arrestin domain containing 4; SLK: STE20 like kinase; TACSTD2: tumor-associated calcium signal transducer 2.
To explore whether common, deleterious variants were also preferentially associated with KD, we analyzed 302 recessive homozygous and deleterious variants in 180 genes. Of these, nine variants in four genes were significantly associated with KD in an imputed European descent, KD GWAS dataset (nominal p <0.05), and were differentially expressed in whole blood during acute versus convalescent KD [19, 29]. We used our European descent GWAS because no published association studies of African Americans KD subjects were available. These nine common variants were grouped in Tier 2 (Fig 1). Interestingly, TLR6 was one of the four Tier 2 genes, as well as arrestin domain containing 4 (ARRDC4), STE20 like kinase (SLK), and tumor-associated calcium signal transducer 2 (TACSTD2). The Tier 2 TLR6 variants were located either in the promoter or 3’UTR and had population allele frequencies greater than 3% in the 1,000 Genomes database (risk allele frequency 4.2–63.3%) (Table 1).
eQTL analysis of common TLR6 variants in KD
Among the six genes in the two tiers, TLR6 had multiple variants in both tiers, suggesting that TLR6 may contribute both to common, sporadic KD as well as uncommon, familial KD. To explore if there were any additional TLR6 variants for which only the affected children were homozygous, we re-analyzed the original unfiltered TLR6 WGS from all six family members. We found 34 additional variants for which only the affected children were homozygous (2 synonymous exonic SNVs and 32 intronic SNVs)(S2 Table). All were common variants with allele frequencies of 0.32–0.75 in African datasets in the 1,000 Genomes database. Next, we looked for association of these 34 variants with KD susceptibility in the imputed European descent GWAS dataset [19]. There were three intronic SNVs (rs56245262, rs56083757 and rs7669329) associated with KD susceptibility with p = 6.9x10-6 (S2 Table and Fig 3). These three intronic SNVs and the seven SNVs in TLR6 from Tiers 1 and 2 are shown in Table 2. The two SNVs in the 3’UTR (rs6822503 and rs12645200) were in linkage disequilibrium (LD, with r2 = 0.97 in Africans, r2 = 1 in European descendants) in the imputed European descent GWAS dataset, as were the three intronic SNVs (rs56245262, rs56083757 and rs7669329), with r2 = 1 in both African and European descent cohorts. We chose rs6822503 and rs56245262 (earlier chromosome position) as representative SNVs for additional analysis.
Fig 3. Location of SNVs associated with KD susceptibility in TLR6.

Association results using the imputed GWAS database were plotted against chromosome location and gene structure of TLR6. Red dots show the SNVs for which only the affected children were homozygous recessive. SNVs above the blue line were associated with a P-value <0.05. TLR6 is encoded on the negative strand so the gene structure is shown 3’ to 5’.
Table 2. TLR6 variants in African American family with two affected siblings.
| rs ID | Gene Region | Ethnicity | Risk allele | Family members | WGS | GWAS p-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA* | HIS** | ASN | CEU | KD 1 | KD 2 | Non-KD 1 | Non-KD 2 | M | F | |||||
| 12650224 | 3'UTR | 0.23 | 0.17 | 0.28 | 0.03 | A | Hom | Hom | Het | - | Het | Het | Tier2 | 0.002 |
| 6822503 | 3'UTR | 0.77 | 0.46 | 0.76 | 0.52 | A | Hom | Hom | Het | - | Het | Het | Tier2 | 0.008 |
| 12645200 | 3'UTR | 0.77 | 0.45 | 0.76 | 0.52 | T | Hom | Hom | Het | - | Het | Het | Tier2 | 0.008 |
| 5743826 | 3'UTR | 0.08 | 0.02 | 0 | 0.01 | T | Hom | Hom | Het | - | Het | Het | Tier2 | 0.003 |
| 35220466 | Exon (p.Arg247Lys) | 0.01 | 0 | 0 | 0 | T | Het | Het | Het | - | - | Het | Tier 1 | ND |
| 5743809 | Exon (p.Leu194Pro) | 0.08 | 0 | 0 | 0 | G | Het | Het | - | - | Het | - | Tier 1 | ND |
| 56245262 | Intronic | 0.42 | 0.37 | 0.62 | 0.23 | A | Hom | Hom | Het | - | Het | Het | NA | 6.9E-06 |
| 56083757 | Intronic | 0.42 | 0.37 | 0.62 | 0.23 | G | Hom | Hom | Het | - | Het | Het | NA | 6.9E-06 |
| 7669329 | Intronic | 0.42 | 0.37 | 0.62 | 0.23 | C | Hom | Hom | Het | - | Het | Het | NA | 6.9E-06 |
| 6837101 | Promotor | 0.45 | 0.34 | 0.35 | 0.29 | A | Hom | Hom | Het | - | Het | Het | Tier2 | 0.03 |
AA: African American, HIS: Hispanic, ASN: Asian, CEU: European descent, KD: Kawasaki disease affected child, Non-KD: unaffected child, M: mother, F: father, GWAS: European descent GWAS [19], Hom: homozygous of risk allele, -: homozygous of non-risk allele, NA: not applicable,
* 1000 Genomes African Ancestry in Southwest US,
** 1000 Genomes:Phase_3:AMR
To understand the possible effects of common variants in TLR6 in KD pathogenesis, we performed an eQTL analysis for the two representative SNVs (rs56245262 and rs6822503) and one SNV (rs6837101) in the promoter region of TLR6. Since TLR2/6 activates the transcription factors, NFKB and AP1, we focused the eQTL analysis using only the 415 genes targeted by these transcription factors. For the intronic variant (rs56245262), only one gene (IL6) among 415 NFKB and/or AP1 targets showed a significant difference in acute whole blood transcript levels as a function of genotype (nominal p< 0.001). IL6 transcript levels were lower in subjects homozygous for the risk allele (p = 0.0007 vs. non-risk allele homozygotes, and p = 0.007 vs. heterozygotes) (Fig 4). For variants in the 3’UTR (rs6822503), no gene showed significantly different transcript levels as a function of genotype with a p< 0.001. No genes were regulated as a function of the genotype of the promoter SNV (rs6837101).
Fig 4. Transcript levels for IL6 in whole blood samples from KD subjects as a function of genotype for rs56245262 in TLR6.

TLR6 rs56245262 risk allele (A) is shown in red. Transcriptome data from a. Acute, pre-treatment (n = 146: T/T n = 54, T/A n = 69, A/A n = 23), b. Paired acute, pre-treatment and convalescent (n = 131: T/T n = 53, T/A n = 58, A/A n = 20). P-values were calculated using Mann Whitney test for (a) and Wilcoxon matched-paired test for (b).
Analysis of patient characteristics as a function of genotype
Since the TLR6 intronic SNVs were associated with differential expression of IL6, we reasoned that patient clinical characteristics related to inflammation might also vary as a function of genotype. We used our published dataset of 161 subjects [30] with detailed demographic and clinical information and 7,602,343 imputed genotypes (from the Illumina HumanOmni1-Quad® chip) to analyze differences in clinical parameters as a function of genotype for the intronic variant (rs56245262)(Table 3). Homozygosity for the risk allele was more common among self-declared Asians (10 of 24 (44%) A/A genotype). This result was consistent with the observation that the A allele frequency is higher in Asian populations (A allele frequencies in 1000 Genome database: Asian 0.62, A/A 0.42, Hispanic 0.37, European descent 0.23). Of interest, the pre-treatment erythrocyte sedimentation rate (ESR) was higher in subjects homozygous for the risk allele (A/A) (median ESR 80 mm/h for A/A vs. 59 mm/h for T/T, p = 0.01).
Table 3. Characteristics of subjects (n = 161) by genotype of TLR6 rs56245262.
| TLR6 rs56245262 | p* | |||
|---|---|---|---|---|
| A/A n = 24 |
A/T n = 78 |
T/T n = 59 |
||
| Male, n (%) | 15 (63) | 48 (62) | 33 (56) | NS |
| Age, years median (IQR) | 2.7 (0.9–3.8) | 2.6 (1.4–4.1) | 2.2 (1.2–4.4) | NS |
| Illness Day, median (range)** | 6 (3–9) | 6 (3–10) | 6 (2–10) | NS |
| Ethnicity, n (%) | ||||
| Asian | 10 (44) | 13 (17) | 5 (8) | 0.002*** |
| African-American | 1 (0) | 5 (6) | 0 (0) | NS |
| Caucasian | 3 (12) | 18 (23) | 21 (36) | NS |
| Hispanic | 3 (24) | 22 (28) | 17 (29) | NS |
| More than race | 7 (20) | 20 (26) | 16 (27) | NS |
| Coronary artery status | ||||
| Aneurysms, n (%) | 3 (13) | 7 (9) | 8 (14) | NS |
| Dilated, n (%) | 4 (16) | 19 (24) | 13 (22) | |
| Normal, n (%) | 17 (71) | 52 (67) | 38 (64) | |
| Z-worst, median (IQR) | 1.5 (1.0–2.6) | 1.7 (1.1–2.9) | 1.9 (1.3–2.9) | NS |
| IVIG resistant, n (%) | 4 (17) | 19 (25) | 12 (20) | NS |
| Lab data, median (IQR) | ||||
| WBC, ×103/mm3 | 16.3 (11.6–19.4) | 13.2 (10.5–18) | 13.9 (11.7–18.7) | NS |
| Absolute neutrophil, ×103/mm3 | 10.7 (7.4–15.2) | 9.0 (6.7–12.5) | 8.4 (6.4–11.6) | NS |
| Platelet count, ×103/mm3 | 401 (334–462) | 400 (292–500) | 392 (312–472) | NS |
| ESR, mm/h | 80 (57–105) | 59 (41–80) | 59 (44–78) | 0.01**** |
| CRP, mg/dl | 7.9 (4.3–13.6) | 8.4 (4.7–16.8) | 8.2 (5.2–16.1) | NS |
| GGT | 34 (25–96) | 48 (18–134) | 41 (22–101) | NS |
* p-values were calculated by Mann–Whitney U test for continuous variables and chi test for categorical variables except ethnicity comparison.
** Illness day 1: first calendar day of fever.
*** Fisher-Freeman-Halton test. Asian comparing to all other ethnicities combined.
**** Mann-Whitney test for the levels of A/A vs. T/T genotype. IVIG: Intra venous immunoglobulin G therapy CRP: C-reactive protein, ESR: Erythrocyte sedimentation rate, WBC: White blood count
Discussion
Analysis of WGS of an African Americans family with two affected and two unaffected siblings and their unaffected, biologic parents highlighted genetic variation in TLR6 in KD susceptibility. These TLR6 variants included both compound heterozygosity for two rare, likely deleterious SNVs and homozygosity for common KD risk SNVs. Subsequently, using an acute KD whole blood transcriptome data set, eQTL analysis of the common SNVs suggested decreased transcript levels of IL6 and higher ESR at diagnosis in individuals homozygous for the risk allele. This integrative genomic approach illustrates how WGS in families with multiple members affected with a complex genetic disease can yield insights into both the common disease–common variant and common disease–rare variant hypotheses.
TLR6- NFKB signaling pathway
TLRs recognize pathogen-associated molecular patterns (PAMPS), lead to activation of the transcription factors, NFKB and AP1, and transcription of genes that control inflammation. TLR6 forms a heterodimer with TLR2 that recognizes peptidoglycan, diacyl lipoproteins, and zymosan derived from Gram positive bacteria, mycoplasmas, and fungi, respectively [31]. TLR6 is constitutively expressed in humans on myeloid dendritic cells, monocytes, B cells, coronary artery endothelial cells (EC), and coronary artery vascular smooth muscle cells [32, 33]. In contrast, TLR2 is widely expressed on immune cells but only expressed on human EC and vascular smooth muscle cells following induction by pro-inflammatory cytokines. Population differences in downstream cytokine production have been observed following TLR stimulation. In a study of African children, higher levels of TNFα were produced following in vitro stimulation with a specific TLR2/6 ligand in a whole blood assay when compared to children of European descent [34].
We found 41 TLR6 variants located in the promoter, intron 1, exon 2, and the 3’UTR associated with KD susceptibility. The three common intronic SNVs (rs56245262, rs56083757 and rs7669329) influenced the transcription of IL6 and were associated with KD susceptibility both in the European descent GWAS and the African Americans family. IL6, a key cytokine controlled by the transcription factors NFKB and AP1 among others, is reported to be high in the serum of acute KD patients [35]. Our transcriptome data show that IL6 transcripts are low in risk allele carriers during the acute disease (Fig 4) [29]. The variants were located in a 1.5kb region in the middle of the single 28kb intron, 11kb from the splice donor site and 17kb from the splice acceptor site. Five transcripts of TLR6 have been predicted to result from alternative splicing (https://www.ncbi.nlm.nih.gov/nuccore?LinkName=gene_nuccore_refseqrna&from_uid=10333) and it is possible that the intronic SNVs could influence splicing efficiency of these variants (S1 Fig). The eQTL database, GTEx (http://www.gtexportal.org/home/), showed that there were tissue-specific effects of these intronic SNVs with the risk allele decreasing TLR6 transcripts in transformed fibroblasts but increasing TLR6 transcripts in a transformed B-cell line (S2 Fig). No data were available for other tissues of potential interest in KD including endothelial cells, vascular smooth muscle cells, and cardiomyocytes.
Two of the four TLR6 3’UTR SNVs (rs12645200 and rs6822503) were in LD. We failed to find any microRNAs that were predicted to bind these four loci according to the miRdSNV data base (http://mirdSNV.ccr.buffalo.edu/index.php [36] and miRNASNV V2 (http://bioinfo.life.hust.edu.cn/miRNASNV2/index.php) [37, 38]. For one of the 3’UTR variants, rs6822503, the risk allele was in weak LD (r2 = 0.58, D’ = 0.94) with the C allele of the exonic variant c.G1083C (rs3821985) in Africans, and the two affected siblings were homozygous for the C allele at this locus (S1 Table). Shey et al stimulated whole blood from 70 healthy Africans with the diacylated lipopeptides, FSL-1 and PAM2 (TLR6/2 ligands), and found reduced IL6 levels in cells from subjects homozygous for the C allele of rs3821985 compared to G homozygotes [39]. This finding suggests that our KD-affected siblings might produce lower levels of IL6 upon TLR2/6 stimulation.
The two affected children were compound heterozygotes for the two SNVs in TLR6 exon 2 (p.Leu194Pro and p.Arg247Lys) having inherited the former from the mother and the latter from the father. These SNVs change amino acids located on the extracellular surface of TLR6 in a region predicted to be involved with ligand binding, which could influence ligand-binding affinity (Fig 5). 3D proteomic structure modeling of the two non-synonymous mutations was computationally predicted (SNPs3D: http://www.snps3d.org/modules.php?name=SnpAnalysis&locus_ac=10333). p.Leu194Pro is predicted to be deleterious due to loss of an intramolecular hydrogen bond and p.Arg247Lys is a variant classified as non-deleterious but on the protein ectodomain. The functional impact of these TLR6 variants was tested using an NF-κB luciferase reporter assay in human embryonic kidney 293T cells expressing the TLR6 variants and stimulated with TLR agonists [40]. p.Arg247Lys showed a 15.6% decrease in ability to respond to the TLR2/6 agonist, PAM2CSK4. Cells transfected with p.Leu194Pro constructs showed a more marked decrease in NF-kB activation (25.4%). Thus, compound heterozygosity for these SNVs in the affected children is expected to reduce NF-kB activation.
Fig 5. TLR6 exonic SNV positions.

Positions of the exonic SNVs rs5743809 and rs3522046 on the TLR6 protein diagrammed using NCBI Molecular Modeling database (MMDB). The replaced amino acids (p.Leu194Pro and p.Arg247Lys) are shown in green. Variants are located in the extra-cellular domain in the predicted ligand-binding region and may alter hydrogen binding.
Role of NFKB and IL6 in the modulation of immune responses
A self-limited inflammatory disease like KD must activate potent anti-inflammatory pathways that ultimately lead to the resolution of inflammation. TLR2/6 stimulation initiates inflammation but also stimulates the regulatory compartment of the immune response. In mice, stimulation of TLR2/TLR6 expressed on dendritic cells leads to their differentiation into tolerogenic dendritic cells secreting IL-10 and promotes T cell differentiation into a regulatory (Treg) phenotype [41]. Work by Franco et al. has highlighted the importance of IL-10 secretion by natural regulatory T cells and tolerogenic myeloid dendritic cells in the resolution of inflammation in KD patients [42, 43]. Downstream of TLR2/6, both NFKB and IL6 have complex immune functions [44, 45]. In the carrageenan-induced rat pleurisy model, blocking NFKB led to protracted inflammation with reduced leukocyte apoptosis and decreased release of the anti-inflammatory molecule, TGFβ1, thus highlighting the important role of NFKB in the resolution of inflammation [44].
IL6 functions by binding the IL6 receptor (R) and gp130, a transmembrane signal transduction protein. The IL6R is expressed only on hepatocytes and a subset of inflammatory cells including macrophages, neutrophils, and naïve T cells. However, gp130 is ubiquitously expressed [45]. During acute inflammation, neutrophils infiltrate tissues and undergo apoptosis with shedding of IL6-sIL6R. This complex binds gp130 on endothelial cells leading to signal transduction that results in monocyte/macrophage recruitment and removal of apoptotic neutrophils. Thus, reduced levels of IL6 might be expected to allow persistence of neutrophils in the arterial wall and in the circulation. Although IL6 levels in the serum are high in acute KD, this may represent synthesis of IL6 by hepatocytes as part of the acute phase response rather than synthesis by circulating immune cells as eQTL are often tissue-specific [35]. This could have important implications for persistence of the acute inflammatory state in KD (Fig 6). Of interest, a pilot study of tocilizumab (monoclonal antibody against human IL6R) plus IVIG for treatment of acute KD was terminated for safety concerns when the first three patients enrolled developed CAA (Prof. Emeritus Shumpei Yokota, Yokohama City University, Japan, personal communication).
Fig 6. Hypothetical role of decreased IL6 signaling in the persistence of vascular inflammation in acute KD.
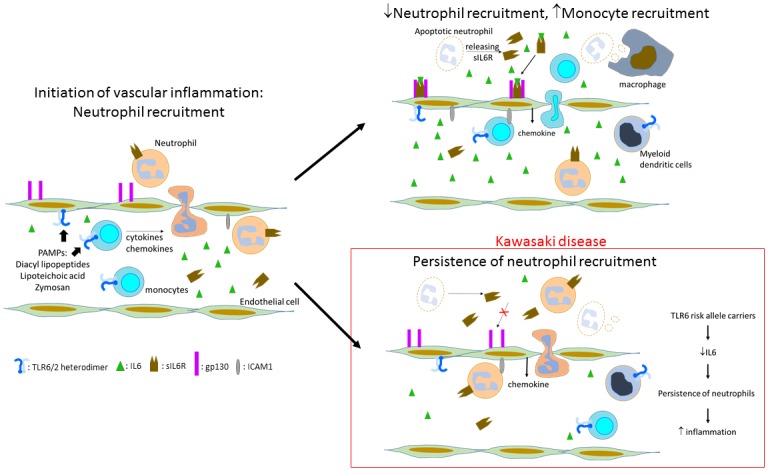
The TLR6 variants associated with KD are predicted to yield lower levels of NFKB upon stimulation, thus resulting in lower levels of IL6. During vascular inflammation, neutrophil apoptosis is associated with shedding of the soluble IL6R-IL6 complex, which binds to gp130 on endothelial cells. This stimulates a signaling pathway that switches the adhesion molecule and chemokine profile to one that favors attraction of monocytes to the vessel wall with subsequent downregulation of vascular inflammation. In KD, lower levels of IL6 may lead to persistence of neutrophil recruitment to the vessel wall and prolonged inflammation.
Tier 1 genes
TLR6 and the transcription factor, MEF2A, were the two Tier 1 genes. They were found in KD pathways and were differentially expressed in the KD transcriptome database with significantly higher levels in the acute phase. MEF2A plays a critical role in transcriptional activation of IL-2 during T cell activation [46]. Our associated SNV, rs373652230, was a (CAG) deletion at amino acid 420 in a poly-lysine tract and multiple studies have reported (CAG)n variants associated with coronary artery disease, although the mechanism underlying this association has not been elucidated [47]. Although the Ingenuity Variant Analysis identified the CAG10 variant as rare (allele frequency <1%), studies in European descent, Asian, and Turkish populations found allele frequencies from 12.7–21.9%. We were unable to find data regarding the allele frequency of the (CAG) repeats in African Americans populations. Of interest, the intronic TLR6 SNV, rs56083757 and the promoter SNV rs6837101, were also predicted to influence the DNA binding of MEF2A (HaploReg: http://www.broadinstitute.org/mammals/haploreg/detail_v4.1.php?query=&id=rs56083757). No other functional significance could be assigned to the other SNVs.
The nature of the PAMP that might stimulate TLR6/TLR2 in KD is unknown although a hypothesis has linked KD to inhalation of Candida antigens carried on aerosols arising from agricultural areas in China [48]. Of speculative interest, the glucans from the Candida cell wall are potent ligands for TLR2/6. In addition, C57BL/6 mice are homozygous for the p.Leu194Pro variant in TLR6 and two murine models of coronary artery vasculitis in this species use intraperitoneal injection of either Candida albicans or Lactobacillus casei cell wall extracts, both of which are potent ligands for TLR2/6[49, 50].
Tier 2 genes
Six genes harboring nine variants met the criteria of homozygous recessive, predicted deleterious, significantly associated in the imputed GWAS, and differentially expressed in acute versus convalescent KD (Fig 2). The TLR6 promoter and 3’UTR SNVs were discussed above.
The Tier 2 gene, TACSTD2, is involved in calcium signaling and may be linked in this way to KD pathogenesis. Validated calcium signaling genes linked to KD pathogenesis currently include ITPKC, ORAI1, and SLC8A1 [22, 30, 51]. TACSTD2 encodes for Trop2, a membrane-spanning protein that transduces an intracellular calcium signal. The protein is over-expressed in many epithelial cancers and is thought to be involved in metastasis and malignant cell invasion [52]. Its transcription is regulated by a number of transcription factors including NFKB and its expression was high in acute versus convalescent KD.
The serine/threonine Ste-like protein kinase, SLK, belongs to the family of germinal center kinases, is ubitquitously expressed, and is involved with stress-induced apoptosis, cytoskeletal remodeling, and cell motility [53]. ARRDC4 is a member of the α-arrestin family, which, as a class, is involved with fine-tuning cellular responses to cell surface signals [54]. Both SLK and ARRDC4 were transcriptionally upregulated in acute KD but the mechanism through which these genes may participate in KD susceptibility is unclear.
Strengths and limitations
This is the first analysis of WGS in an African Americans family and provides a database that can be mined for future studies of genetic structure in this population. Many different filters can be applied for subsequent analyses that may uncover additional variants that influence susceptibility to KD. Neither affected child developed coronary artery aneurysms, so only variants affecting susceptibility can be analyzed from this dataset. We recognize that using a whole blood transcriptomic database from a mixed ethnic population could miss transcripts that are only differentially expressed in relevant cardiovascular tissues and among African Americans. This analysis underscores the need to focus genetic and genomic studies on minority populations such as African Americans who are disproportionately affected by KD compared to children of European descent.
Conclusion
The first analysis of WGS from an African Americans family with two siblings affected with KD revealed genetic variation in TLR6 that may be linked to the pro-inflammatory state during acute KD. Previous GWAS and linkage association studies had not identified this gene as influencing susceptibility to KD. Another variant, in TACSTD2, has an intriguing link to calcium signaling that will need to be pursued in future studies. The analytic approach presented here is a novel method for finding potentially relevant variants in WGS in families with individuals affected by complex genetic diseases.
Materials and methods
Subjects
Members of the African American family selected for whole genome sequencing included two affected sons, an older unaffected son and daughter, and unrelated/unaffected father and mother, all of whom provided written informed consent for study participation. The study was approved by the Institutional Review Board at the University of California San Diego. The two affected subjects were both diagnosed by one of the co-authors (JCB). Neither developed coronary artery abnormalities by echocardiogram and both responded to a single dose of IVIG with defervescence and resolution of inflammation.
Whole genome sequencing
Whole blood samples or Scope® mouthwash rinses were obtained from family members and 10 μg of genomic DNA was extracted and submitted to Illumina Clinical Services Laboratory in San Diego, CA, USA for sequencing using HiSeq 2000. DNA was fragmented and attached to the surface of glass microscope slides. Fluorescently labeled nucleotides were used to sequence the fragments. Laser excitation of the nucleotides was followed by digital imaging. The sequence fragments were assessed for quality scores.
Sequence processing and variant calling
Raw images were processed by Illumina’s CASAVA version 1.8 pipeline to generate six sample FASTQ files for a downstream analysis. The FASTQ files were aligned to a human reference genome (hg19), PCR duplicates marked and variants called using Edico Genome’s DRAGEN pipeline [55], using default parameters.
Variant prioritization
VCF files were uploaded to Ingenuity Variant Analysis™ (QIAGEN Redwood City, CA) for tertiary analysis. Variant prioritization was performed by sequentially applying filters in 7 steps of confidence, genetics (recessive homozygous and compound heterozygous), predicted deleterious, rare, found in KD pathway, significant in published KD GWAS, and differentially expressed in KD transcriptome database (Fig 1). The Confidence filter retained variants with a call quality at least 20, read depth at least 10, and eliminated the top 5% of the most exonically variable genes or 100-bp regions in 1000 Genomes to remove false positive variants such as mucin and olfactory receptor genes. The annotated variants were imported into an in-house MySQL database to perform genetic analysis of two inheritance types, recessive homozygous and compound heterozygous. The Homozygous filter required homozygous variants to be present exclusively in the two KD affected children but not in any of the four unaffected individuals. The Compound heterozygous filter was constructed based on the following rules [56]:
A variant is in a heterozygous state in both of KD affected children.
A variant must not occur in a homozygous state in any of the four unaffected individuals, i.e. two siblings and two parents.
A variant that is in a heterozygous state in an affected child must be heterozygous in exactly one of the parents but not both.
The Predicted deleterious filter followed the guideline classification of American College of Medical Genetics (ACMG) and loss-of-function in terms of frameshift, in-frame in/del, start/stop codon change, missense, or splice site loss all implemented in IVA. The Rare filter retained variants whose allele frequency was less than 1% (for recessive homozygosity) or 3% (for compound heterozygosity) in any of three public datasets; 1000 genomes, Exome Aggregation Consortium (ExAC), and NHLBI ESP Exomes. The Pathway filter generated by Ingenuity program retained genes implicated in pathways relevant to KD pathogenesis. These were defined using the Ingenuity system with the key terms “susceptibility to KD”, “calcium signaling pathway, coronary artery aneurysm”, and “coronary artery abnormalities”. The GWAS filter retained variants that were significantly (nominal P-value < 0.05) associated in 405 KD subjects versus 6,252 normal controls in our previously published KD GWAS dataset genotyped on the Illumina HumMap 550 SNV array [19]. Since GWAS targets common SNVs with an allele frequency > 5%, the GWAS filter was applied to variants surviving the Deleterious filter in Fig 1. Quality control was performed with missing rate, minor allele frequency (MAF) and Hardy-Weinberg Equilibrium (HWE) cutoff values. The original dataset was expanded through imputation using SHAPEIT2 for phasing and IMPUTE2 for imputation with the most recent versions of 1000 Genomes version 3 and HapMap3 CEU panels (hg18) as reference data. LiftOver tool from UCSC Genome Browser (Kent et al. Genome Res 2002 PMID:12045153) was used to convert genomes coordinates from assembly hg18 to hg19. The new imputation with updated reference panels increased the total number of imputed SNVs to 7,602,343 from 4,545,265. The Transcriptome filter set gene-level prioritization by retaining differentially expressed genes between 131 paired acute and convalescent whole blood RNA samples (PAXgene).[29] Transcript levels were measured using the Illumina HumanRef-12 V4 BeadChip with 47,000 probes and quality control and analysis were as described. The cutoff for differential expression was an adjusted P-value < 0.05.
Validation genotyping
MEF2A
The six family members were re-sequenced for the MEF2A (CAG)n variant.
Primers were designed using Primer 3: forward primer (caagtccgaaccgatttcac) and reverse primers (gccaagcacaattggagaat) (product size 247 bp). Fifty nanograms of DNA from each sample was amplified using high-fidelity Taq polymerase (Platinum® Taq DNA Polymerase High Fidelity, Life Technologies) for 35 cycles following the manufacturer’s instructions. PCR products were resolved on 2% agarose gels, excised, and purified (QIAquick Gel Extraction Kit, Qiagen). PCR products were sequenced using forward and reverse primers (Eaton Bioscience Inc., San Diego).
eQTL analysis
Detailed methods were described previously [30]. A total of 673 probes from 415 genes from the database of literature-curated human TF-target interactions for NFKB and AP1 were used for eQTL analysis [57].
Data sharing
Data is available upon request. Users wishing to access our data should contact us.
Supporting Information
(TIF)
Genotype-Tissue Expression (GTEx: http://www.gtexportal.org/home/).
(TIF)
(PDF)
(PDF)
Acknowledgments
We thank the family who donated their whole genome sequences. We also thank Francesca Marrassi PhD. for helpful discussion of the TLR6 exonic variants and the International Kawasaki Disease Genetics consortium for the original GWAS dataset. This work was funded by the NIH Roadmap for Medical Research, Grant U54HL108460, a grant from the Gordon and Marilyn Macklin Foundation and by Rady Children’s Institute for Genomic Medicine, San Diego.
Data Availability
The IRB at UCSD has reviewed the consent forms signed by the family for the collection and analysis of their DNA. It has been determined that the consents cover the sharing of data from the DNA analysis for the purpose of future research by qualified investigators. However, the consent form does not cover the depositing of genetic data in a national database. Data access may be granted by contacting the Principal Investigator, Dr. Jane C. Burns by email at jcburns@ucsd.edu.
Funding Statement
National Institute of Health grant U54HL108460 to LOM; Gordon and Marilyn Macklin Foundation's private grant to JCB and AHT; Rady Children’s Institute for Genomic Medicine, San Diego. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Although Dr. Flatley was CEO of Illumina at the time that this research was initiated, Illumina did not play a role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript and only provided financial support for the whole genome sequencing. Dr. Flatley’s role was as a contributor to the interpretation of data and review of the manuscript.
References
- 1.Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464(7293):1351–6. 10.1038/nature08990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3(5):377–87. 10.1016/S2213-2600(15)00139-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stittrich AB, Ashworth J, Shi M, Robinson M, Mauldin D, Brunkow ME, et al. Genomic architecture of inflammatory bowel disease in five families with multiple affected individuals. Hum Genome Var. 2016;3:15060 10.1038/hgv.2015.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dinwiddie DL, Bracken JM, Bass JA, Christenson K, Soden SE, Saunders CJ, et al. Molecular diagnosis of infantile onset inflammatory bowel disease by exome sequencing. Genomics. 2013;102(5–6):442–7. 10.1016/j.ygeno.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onouchi Y. Genetics of Kawasaki disease: what we know and don't know. Circ J. 2012;76(7):1581–6. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki A, Kamiya T, Kuwahara N, Ono Y, Kohata T, Takahashi O, et al. Coronary arterial lesions of Kawasaki disease: cardiac catheterization findings of 1100 cases. Pediatr Cardiol. 1986;7(1):3–9. 10.1007/BF02315475 [DOI] [PubMed] [Google Scholar]
- 8.Daniels LB, Tjajadi MS, Walford HH, Jimenez-Fernandez S, Trofimenko V, Fick DB Jr., et al. Prevalence of Kawasaki disease in young adults with suspected myocardial ischemia. Circulation. 2012;125(20):2447–53. Epub 2012/05/19. 10.1161/CIRCULATIONAHA.111.082107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato H, Sugimura T, Akagi T, Sato N, Hashino K, Maeno Y, et al. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation. 1996;94(6):1379–85. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura Y, Aso E, Yashiro M, Tsuboi S, Kojo T, Aoyama Y, et al. Mortality among Japanese with a history of Kawasaki disease: results at the end of 2009. J Epidemiol. 2013;23(6):429–34. Epub 2013/09/18. 10.2188/jea.JE20130048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon JB, Daniels LB, Kahn AM, Jimenez-Fernandez S, Vejar M, Numano F, et al. The Spectrum of Cardiovascular Lesions Requiring Intervention in Adults After Kawasaki Disease. Jacc. 2016;9(7):687–96. 10.1016/j.jcin.2015.12.011 [DOI] [PubMed] [Google Scholar]
- 12.Nakamura Y. Lessones from Epidemiologic Studies of Kawasaki Disease in Japan. Circulation. 2015;131(Supplement 2):12. [Google Scholar]
- 13.Huang SK, Lin MT, Chen HC, Huang SC, Wu MH. Epidemiology of Kawasaki disease: prevalence from national database and future trends projection by system dynamics modeling. J Pediatr. 2013;163(1):126–31 e1. Epub 2013/01/15. 10.1016/j.jpeds.2012.12.011 [DOI] [PubMed] [Google Scholar]
- 14.Gibbons RV, Parashar UD, Holman RC, Belay ED, Maddox RA, Powell KE, et al. An evaluation of hospitalizations for Kawasaki syndrome in Georgia. Arch Pediatr Adolesc Med. 2002;156(5):492–6. [DOI] [PubMed] [Google Scholar]
- 15.Abuhammour WM, Hasan RA, Eljamal A, Asmar B. Kawasaki disease hospitalizations in a predominantly African-American population. Clin Pediatr (Phila). 2005;44(8):721–5. [DOI] [PubMed] [Google Scholar]
- 16.Holman RC, Curns AT, Belay ED, Steiner CA, Schonberger LB. Kawasaki syndrome hospitalizations in the United States, 1997 and 2000. Pediatrics. 2003;112(3 Pt 1):495–501. Epub 2003/09/02. [DOI] [PubMed] [Google Scholar]
- 17.Chang CJ, Kuo HC, Chang JS, Lee JK, Tsai FJ, Khor CC, et al. Replication and meta-analysis of GWAS identified susceptibility loci in Kawasaki disease confirm the importance of B lymphoid tyrosine kinase (BLK) in disease susceptibility. PLoS One. 2013;8(8):e72037 10.1371/journal.pone.0072037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. 2012;44(5):517–21. Epub 2012/03/27. 10.1038/ng.2220 [DOI] [PubMed] [Google Scholar]
- 19.Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. 2011;43(12):1241–6. Epub 2011/11/15. 10.1038/ng.981 [DOI] [PubMed] [Google Scholar]
- 20.Burgner D, Davila S, Breunis WB, Ng SB, Li Y, Bonnard C, et al. A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. 2009;5(1):e1000319 Epub 2009/01/10. 10.1371/journal.pgen.1000319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khor CC, Davila S, Shimizu C, Sheng S, Matsubara T, Suzuki Y, et al. Genome-wide linkage and association mapping identify susceptibility alleles in ABCC4 for Kawasaki disease. J Med Genet. 2011;48(7):467–72. Epub 2011/05/17. 10.1136/jmg.2010.086611 [DOI] [PubMed] [Google Scholar]
- 22.Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nature genetics. 2008;40(1):35–42. 10.1038/ng.2007.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan Y, Ma Y, Liu Y, Hu H, Shen Y, Zhang S, et al. Combined analysis of genome-wide-linked susceptibility loci to Kawasaki disease in Han Chinese. Human genetics. 2013;132(6):669–80. Epub 2013/03/05. 10.1007/s00439-013-1279-2 [DOI] [PubMed] [Google Scholar]
- 24.Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nature genetics. 2012;44(5):522–5. Epub 2012/03/27. 10.1038/ng.2227 [DOI] [PubMed] [Google Scholar]
- 25.Kim JJ, Hong YM, Sohn S, Jang GY, Ha KS, Yun SW, et al. A genome-wide association analysis reveals 1p31 and 2p13.3 as susceptibility loci for Kawasaki disease. Hum Genet. 2011;129(5):487–95. Epub 2011/01/12. 10.1007/s00439-010-0937-x [DOI] [PubMed] [Google Scholar]
- 26.Tsai FJ, Lee YC, Chang JS, Huang LM, Huang FY, Chiu NC, et al. Identification of novel susceptibility Loci for kawasaki disease in a Han chinese population by a genome-wide association study. PLoS One. 2011;6(2):e16853 10.1371/journal.pone.0016853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shrestha S, Wiener HW, Aissani B, Shendre A, Tang J, Portman MA. Imputation of class I and II HLA loci using high-density SNPs from ImmunoChip and their associations with Kawasaki disease in family-based study. Int J Immunogenet. 2015;42(3):140–6. 10.1111/iji.12190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alphonse MP, Duong TT, Shumitzu C, Hoang TL, McCrindle BW, Franco A, et al. Inositol-Triphosphate 3-Kinase C Mediates Inflammasome Activation and Treatment Response in Kawasaki Disease. J Immunol. 2016. [DOI] [PubMed] [Google Scholar]
- 29.Hoang LT, Shimizu C, Ling L, Naim AN, Khor CC, Tremoulet AH, et al. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. 2014;6(11):541 10.1186/s13073-014-0102-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimizu C, Eleftherohorinou H, Wright VJ, Kim J, Alphonse MP, Perry JC, et al. Genetic variation in the SLC8A1 calcium signaling pathway is associated with susceptibility to Kawasaki disease and coronary artery abnormalities. Circulation Cardovascular Genetics. (In Press). [DOI] [PubMed] [Google Scholar]
- 31.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- 32.Satta N, Kruithof EK, Reber G, de Moerloose P. Induction of TLR2 expression by inflammatory stimuli is required for endothelial cell responses to lipopeptides. Mol Immunol. 2008;46(1):145–57. 10.1016/j.molimm.2008.07.017 [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Murthy V, Schultz K, Tatro JB, Fitzgerald KA, Beasley D. Toll-like receptor 3 signaling evokes a proinflammatory and proliferative phenotype in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;291(5):H2334–43. 10.1152/ajpheart.00252.2006 [DOI] [PubMed] [Google Scholar]
- 34.Labuda LA, de Jong SE, Meurs L, Amoah AS, Mbow M, Ateba-Ngoa U, et al. Differences in innate cytokine responses between European and African children. PLoS One. 2014;9(4):e95241 10.1371/journal.pone.0095241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furukawa S, Matsubara T, Yone K, Hirano Y, Okumura K, Yabuta K. Kawasaki disease differs from anaphylactoid purpura and measles with regard to tumour necrosis factor-alpha and interleukin 6 in serum. Eur J Pediatr. 1992;151(1):44–7. [DOI] [PubMed] [Google Scholar]
- 36.Bruno AE, Li L, Kalabus JL, Pan Y, Yu A, Hu Z. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3'UTRs of human genes. BMC Genomics. 2012;13:44 10.1186/1471-2164-13-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gong J, Liu C, Liu W, Wu Y, Ma Z, Chen H, et al. An update of miRNASNP database for better SNP selection by GWAS data, miRNA expression and online tools. Database (Oxford). 2015;2015:bav029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong J, Tong Y, Zhang HM, Wang K, Hu T, Shan G, et al. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum Mutat. 2012;33(1):254–63. 10.1002/humu.21641 [DOI] [PubMed] [Google Scholar]
- 39.Shey MS, Randhawa AK, Bowmaker M, Smith E, Scriba TJ, de Kock M, et al. Single nucleotide polymorphisms in toll-like receptor 6 are associated with altered lipopeptide- and mycobacteria-induced interleukin-6 secretion. Genes Immun. 2010;11(7):561–72. 10.1038/gene.2010.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Ali M, Corre B, Manry J, Barreiro LB, Quach H, Boniotto M, et al. Functional characterization of naturally occurring genetic variants in the human TLR1-2-6 gene family. Hum Mutat. 2011;32(6):643–52. 10.1002/humu.21486 [DOI] [PubMed] [Google Scholar]
- 41.Depaolo RW, Tang F, Kim I, Han M, Levin N, Ciletti N, et al. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe. 2008;4(4):350–61. 10.1016/j.chom.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franco A, Touma R, Song Y, Shimizu C, Tremoulet A, Kanegaye J, et al. Specificity of regulatory T cells that modulate vascular inflammation. Autoimmunity. 2014;47:95–104. 10.3109/08916934.2013.860524 [DOI] [PubMed] [Google Scholar]
- 43.Burns JC, Franco A. The immunomodulatory effects of intravenous immunoglobulin therapy in Kawasaki disease. Expert Rev Clin Immunol. 2015;11(7):819–25. 10.1586/1744666X.2015.1044980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001;7(12):1291–7. 10.1038/nm1201-1291 [DOI] [PubMed] [Google Scholar]
- 45.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–88. 10.1016/j.bbamcr.2011.01.034 [DOI] [PubMed] [Google Scholar]
- 46.Pan F, Ye Z, Cheng L, Liu JO. Myocyte enhancer factor 2 mediates calcium-dependent transcription of the interleukin-2 gene in T lymphocytes: a calcium signaling module that is distinct from but collaborates with the nuclear factor of activated T cells (NFAT). J Biol Chem. 2004;279(15):14477–80. 10.1074/jbc.C300487200 [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Niu W, Wu Z, Su X, Chen Q, Lu L, et al. Variants in exon 11 of MEF2A gene and coronary artery disease: evidence from a case-control study, systematic review, and meta-analysis. PLoS One. 2012;7(2):e31406 10.1371/journal.pone.0031406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodo X, Curcoll R, Robinson M, Ballester J, Burns JC, Cayan DR, et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc Natl Acad Sci U S A. 2014;111(22):7952–7. 10.1073/pnas.1400380111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lehman TJ, Warren R, Gietl D, Mahnovski V, Prescott M. Variable expression of Lactobacillus casei cell wall-induced coronary arteritis: an animal model of Kawasaki's disease in selected inbred mouse strains. Clin Immunol Immunopathol. 1988;48(1):108–18. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi K, Oharaseki T, Wakayama M, Yokouchi Y, Naoe S, Murata H. Histopathological features of murine systemic vasculitis caused by Candida albicans extract—an animal model of Kawasaki disease. Inflamm Res. 2004;53(2):72–7. 10.1007/s00011-003-1225-1 [DOI] [PubMed] [Google Scholar]
- 51.Onouchi Y, Fukazawa R, Yamamura K, Suzuki H, Kakimoto N, Suenaga T, et al. Variations in ORAI1 Gene Associated with Kawasaki Disease. PLoS ONE. 2016;11(1):e0145486 10.1371/journal.pone.0145486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer. 2015;6(3–4):84–105. 10.18632/genesandcancer.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cybulsky AV, Guillemette J, Papillon J. Ste20-like kinase, SLK, activates the heat shock factor 1—Hsp70 pathway. Biochim Biophys Acta. 2016;1863(9):2147–55. 10.1016/j.bbamcr.2016.05.015 [DOI] [PubMed] [Google Scholar]
- 54.Shea FF, Rowell JL, Li Y, Chang TH, Alvarez CE. Mammalian alpha arrestins link activated seven transmembrane receptors to Nedd4 family e3 ubiquitin ligases and interact with beta arrestins. PLoS ONE. 2012;7(12):e50557 10.1371/journal.pone.0050557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller NA, Farrow EG, Gibson M, Willig LK, Twist G, Yoo B, et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 2015;7:100 10.1186/s13073-015-0221-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamphans T, Sabri P, Zhu N, Heinrich V, Mundlos S, Robinson PN, et al. Filtering for compound heterozygous sequence variants in non-consanguineous pedigrees. PLoS One. 2013;8(8):e70151 10.1371/journal.pone.0070151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han H, Shim H, Shin D, Shim JE, Ko Y, Shin J, et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci Rep. 2015;5:11432 10.1038/srep11432 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
Genotype-Tissue Expression (GTEx: http://www.gtexportal.org/home/).
(TIF)
(PDF)
(PDF)
Data Availability Statement
The IRB at UCSD has reviewed the consent forms signed by the family for the collection and analysis of their DNA. It has been determined that the consents cover the sharing of data from the DNA analysis for the purpose of future research by qualified investigators. However, the consent form does not cover the depositing of genetic data in a national database. Data access may be granted by contacting the Principal Investigator, Dr. Jane C. Burns by email at jcburns@ucsd.edu.