

Abstract
One of the major successes in computational biology has been the unification, by using the graphical model formalism, of a multitude of algorithms for annotating and comparing biological sequences. Graphical models that have been applied to these problems include hidden Markov models for annotation, tree models for phylogenetics, and pair hidden Markov models for alignment. A single algorithm, the sum-product algorithm, solves many of the inference problems that are associated with different statistical models. This article introduces the polytope propagation algorithm for computing the Newton polytope of an observation from a graphical model. This algorithm is a geometric version of the sum-product algorithm and is used to analyze the parametric behavior of maximum a posteriori inference calculations for graphical models.
This article develops an algorithm for graphical models based on the mathematical foundation for statistical models proposed in ref. 1 and applies it to biological sequence-analysis problems. Its relevance for computational biology can be summarized in the following theses:
Graphical models are a unifying statistical framework for biological sequence analysis.
Parametric inference is important for obtaining biologically meaningful results.
The polytope propagation algorithm solves the parametric inference problem.
1. Inference with Graphical Models for Biological Sequence Analysis
Thesis i states that graphical models are good models for biological sequences. This point of view is based on the emerging understanding and practical success of probabilistic algorithms in computational biology and also on the observation that inference algorithms for graphical models subsume many apparently nonstatistical methods. A noteworthy example of the latter interpretation is the explanation of classic alignment algorithms, such as Needleman-Wunsch and Smith-Waterman, in terms of the Viterbi algorithm for pair hidden Markov models (HMMs) (2). Graphical models are now used for many problems, including motif detection, gene-finding, alignment, phylogeny reconstruction, and protein-structure prediction. For example, most gene prediction methods are now based on HMMs, and previously nonprobabilistic methods now have HMM-based reimplementations.
In typical applications, biological sequences are modeled as observed random variables Y1,..., Yn in a graphical model. The observed random variables may correspond to sequence elements such as nucleotides or amino acids. Hidden random variables X1,..., Xm encode information of interest that is unknown but that one would like to infer. For example, the information could be an annotation, alignment, or ancestral sequence in a phylogenetic tree. One of the strengths of graphical models is that, by virtue of being probabilistic, they can be combined into powerful models in which the hidden variables are more complex. For example, HMMs can be combined with pair HMMs to align and annotate sequences simultaneously (3). One of the drawbacks of such approaches is that the models have more parameters, and as a result, inferences could be less robust.
For a fixed observed sequence σ1 σ2... σn and fixed parameters, two types of problems from statistical learning theory are as follows.
- The calculation of marginal probabilities:
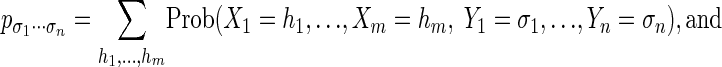
- The calculation of maximum a posteriori (MAP) log probabilities:
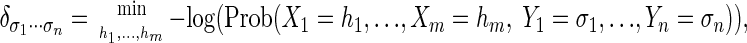
where the hi range over all the possible assignments for the hidden random variables Xi. In practice, the solution to problem 2 is of interest because it is the one that solves the problem of finding the genes in a genome or the “best” alignment for a pair of sequences. A shortcoming of this approach is that the solution ĥ = (ĥ1,..., ĥm may vary considerably with a change in parameters.
Thesis ii suggests that a parametric solution to the inference problem can help in ascertaining the reliability, robustness, and biological meaning of an inference result. By parametric inference, we mean the solution of problem 2 for all model parameters simultaneously. In this way, we can decide whether a solution obtained for particular parameters is an artifact or if it is largely independent of the chosen parameters. This approach has been applied successfully to the problem of pairwise sequence alignment in which parameter choices are known to be crucial in obtaining good alignments (4-6). Our aim is to develop this approach for arbitrary graphical models. In thesis iii, we claim that the polytope propagation algorithm is efficient for solving the parametric inference problem for a small number of parameters. In certain cases, it is not much slower than solving problem 2 for fixed parameters. The algorithm is a geometric version of the sum-product algorithm, which is the standard tool for inference with graphical models. Although it is exponential in the number of parameters, it is polynomial in the size of the graphical model.
The mathematical setting for understanding the polytope propagation algorithm is tropical geometry. The connection between tropical geometry and parametric inference in statistical models is developed in the companion to this article (1). Here, we describe the details of the polytope propagation algorithm (section 3) in the following two familiar settings: the HMM for annotation (section 2) and the pair HMM for alignment (section 4). In section 5, we discuss some practical aspects of parametric inference, such as specializing parameters, the construction of single cones (which eliminates the need for identifying all possible MAP explanations), and the relevance of our findings to Bayesian computations.
2. Parametric Inference with HMMs
HMMs play a central role in sequence analysis, in which they are used widely to annotate DNA sequences (7). A simple example is the CpG island annotation problem (ref. 8, section 3). The computational identification of CpG islands is important because they are associated with promoter regions of genes and are known to be involved in gene silencing.
Unfortunately, there is no sequence characterization of CpG islands. A generally accepted definition due to Gardiner-Garden and Frommer (9) is that a CpG island is a region of DNA at least 200 bp in length with a G+C content of at least 50% and a ratio of observed to expected CpG sites of at least 0.6. This arbitrary definition has since been refined (e.g., ref. 10); however, even analysis of the complete sequence of the human genome (11) has failed to reveal precise criteria for what constitutes a CpG island. HMMs can be used to predict CpG islands (ref. 8, section 3). We have selected this application of HMMs to illustrate our approach to parametric inference in a mathematically simple setting.
The CpG island HMM that we consider has n hidden binary random variables Xi and n observed random variables Yi that take on the values {A, C, G, T} (see figure 1 in ref. 1). In general, an HMM can be characterized by the following conditional independence statements for i = 1,..., n:
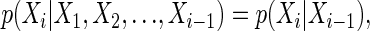 |
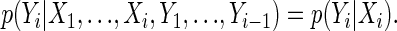 |
The CpG island HMM has 12 model parameters, namely, the entries of the following transition matrices:
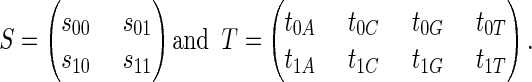 |
Here, the hidden-state space has just two states (non-CpG = 0, and CpG = 1) with transitions allowed between them, but in more complicated applications such as gene finding, the state space is used to model numerous gene components (such as introns and exons), and the sparsity pattern of the matrix S is crucial. In its algebraic representation (ref. 1, section 2), the HMM is given as the image of the polynomial map as follows:
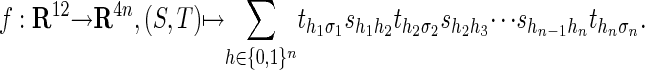 |
1 |
The inference problem 1 asks for an evaluation of one coordinate polynomial fσ of the map f. This evaluation can be done in linear time (in n) by using the forward algorithm (12), which recursively evaluates the following formula:
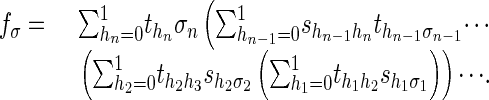 |
2 |
Problem 2 is to identify the largest term in the expansion of fσ. Equivalently, if we write uij = -log(sij) and vij = -log(tij), then problem 2 is to evaluate the following piecewise-linear function:
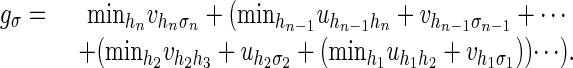 |
3 |
This formula can be evaluated efficiently by recursively computing the parenthesized expressions. This evaluation is known as the Viterbi algorithm in the HMM literature. The Viterbi and forward algorithms are instances of the more general sum-product algorithm (13).
We are proposing, in this article, the computation of the collection of cones in R12 on which the piecewise-linear function gσ is linear. This computation may be feasible because the number of cones grows polynomially in n. Each cone is indexed by a binary sequence h ∈ {0, 1}n, which represents the CpG islands found for any system of parameters (uij, vij) in that cone. A binary sequence that arises in this manner is an explanation for σ in the sense described in section 4 of ref. 1. Our results in ref. 1 imply that the number of explanations scales polynomially with n.
Theorem 1. For any given DNA sequence σ of length n, the number of bit strings ĥ ∈ {0, 1}n (which are explanations for the sequence σ in the CpG island HMM) is bounded above by a constant times n5.25.
Proof: There are a total of 2·4 + 4 = 12 parameters, which is the dimension of the ambient space. However, note that, for a fixed observed sequence, the number of times that the observation A is made is fixed, and a similar case is true for C, G, and T. Furthermore, the total number of transitions in the hidden states must equal n. Together, these constraints remove five degrees of freedom. Thus, we can apply theorem 7 from ref. 1 with d = 12 - 5 = 7. We conclude that the total number of vertices of the Newton polytope of fσ is O(n(7·6/8)) = O(n5.25).
We explain the biological meaning of our parametric analysis with a very small example. Let us consider the following special case of the CpG island HMM. First, assume that tiA = tiT and that tiC = tiG (i.e., the output probability depends only on whether the nucleotide is a purine or pyrimidine). Furthermore, assume that t0A = t0G, which means that the probability of emitting a purine or a pyrimidine in the non-CpG island state is equal (i.e., the base composition is uniform in non-CpG islands).
Suppose that the observed sequence is σ = AATAGCGG. We ask for all the possible explanations for σ (that is, for all possible MAP CpG island annotations for all parameters). A priori, the number of explanations is bounded by 28 = 256, which is the total number of binary strings of length 8. However, of the 256 binary strings, only 25 are explanations. Fig. 1 is a geometric representation of the solution to this problem: the Newton polytope of fσ is a four-dimensional polytope with 25 vertices. Fig. 1 is a Schlegel diagram of this polytope. It was created with polymake software. The 25 vertices in Fig. 1 correspond to the 25 annotations that are the explanations for σ as the parameters vary. Two annotations are connected by an edge if and only if their parameter cones share a wall. From this geometric representation, we can determine all parameters that result in the same MAP prediction.
Fig. 1.

The Schlegel diagram of the Newton polytope of an observation in the CpG island HMM.
3. Polytope Propagation
The evaluation of gσ for fixed parameters by using the formulation in Eq. 3 is known as the Viterbi algorithm in the HMM literature. We begin by reinterpreting this algorithm as a convex optimization problem.
Definition 2. Given a polynomial,
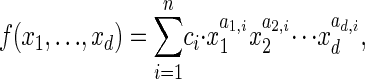 |
the Newton polytope of f is defined as follows to be the convex hull of the lattice points in Rd corresponding to the monomials in f:
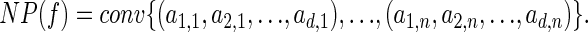 |
Recall that for a fixed observation there are natural polynomials associated with a graphical model, which we have been denoting by fσ. In the CpG island example from section 2, these polynomials are the coordinates fσ of the polynomial map f in Eq. 1. Each coordinate polynomial fσ is the sum of 2n monomials, where n =|σ|. The crucial observation is that even though the number of monomials grows exponentially with n, the number of vertices of the Newton polytope NP(fσ) is much smaller. The Newton polytope is important for us because its vertices represent the solutions to the inference problem 2.
Proposition 3. The MAP log probabilities δσ in problem 2 can be determined by minimizing a linear functional over the Newton polytope of fσ.
Proof: This assertion is nothing but a restatement of the fact that when passing to logarithms, monomials in the parameters become linear functions in the logarithms of the parameters.
Theorem 4 (Polytope Propagation). Let fσ be the polynomial associated to a fixed observation σ from a graphical model. The list of all vertices of the Newton polytope of fσ can be computed efficiently by recursive convex hull and Minkowski sum computations on unions of polytopes.
Proof: Observe that if f1, f2 are polynomials, then NP(f1·f2) = NP(f1) + NP(f2), where the + denotes the Minkowski sum of the two polytopes. Similarly, NP(f1 + f2) = conv(NP(f1) ∪ NP(f2)) if f1 and f2 are polynomials with positive coefficients. The recursive description of fσ given in Eq. 2 can be used to evaluate the Newton polytope efficiently. The necessary geometric primitives are precisely Minkowski sum and convex hull of unions of convex polytopes. These primitives run in polynomial time because the dimension of the polytopes is fixed. This is the case in our situation because we consider graphical models with a fixed number of parameters. Hence, we can run the sum-product algorithm efficiently in the semiring known as the polytope algebra. The size of the output scales polynomially (ref. 1, theorem 7).
Fig. 2 shows an example of the polytope propagation algorithm for a HMM with all random variables binary and with the following transition and output matrices:
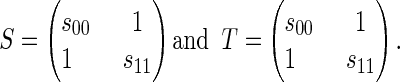 |
Fig. 2.

Graphical representation of the polytope propagation algorithm for an HMM. For a particular pair of parameters (shown as a vector in the circle), there is one optimal Viterbi path (shown as large vertices on the polytopes).
Here, we specialized to only two parameters in order to simplify the diagram. When we run polytope propagation for long enough DNA sequences σ in the CpG island HMM of section 2 with all 12 free parameters, we get a diagram just like Fig. 2, but with each polygon replaced by a seven-dimensional polytope.
It is useful to note that, for HMMs, the Minkowski sum operations are simply shifts of the polytopes, and therefore, the only nontrivial geometric operations that are required are the convex hulls of unions of polytopes. The polytope in Fig. 1 was computed by using polytope propagation. This polytope has dimension 4 (rather than 7) because the sequence σ = AATAGCGG is so short. We emphasize that the small size of our examples is only for clarity; there is no practical or theoretical barrier to computing much larger instances.
For general graphical models, the running time of the Minkowski sum and convex hull computations depends on the number of parameters, and the number of vertices in each computation. These are clearly bounded by the total number of vertices of NP(fσ), which are bounded above as follows (ref. 1, theorem 7):
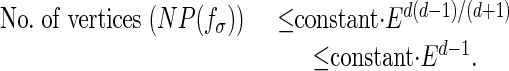 |
Here, E is the number of edges in the graphical model (often linear in the number of vertices of the model). The dimension d of the Newton polytope NP(fσ) is fixed because it is bounded above by the number of model parameters. The total running time of the polytope propagation algorithm can then be estimated by multiplying the running time for the geometric operations of Minkowski sum and convex hull with the running time of the sum-product algorithm. It follows that, for directed acyclic graphical models, the running time scales polynomially in E.
We have shown (ref. 1, section 4) that the vertices of NP(fσ) correspond to explanations for the observation σ. In parametric inference, we are interested in identifying the parameter regions that lead to the same explanations. Because parameters can be identified with linear functionals, it is the case that the set of parameters that lead to the same explanation (i.e., a vertex v) are the linear functionals that minimize on v. The set of these linear functionals is the normal cone of NP(fσ) at v. The collection of all normal cones at the various vertices v forms the normal fan of the polytope. By using Proposition 3, we obtain the following:
Proposition 5. The normal fan of the Newton polytope of fσ solves the parametric inference problem for an observation σ in a graphical model. It is computed by using the polytope propagation algorithm.
4. Parametric Sequence Alignment
The sequence alignment problem is concerned with finding the best alignment between two sequences that have evolved from a common ancestor by means of a series of mutations, insertions, and deletions. Formally, given two sequences 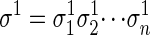 and
and 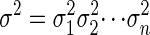 over the alphabet {0, 1,..., l - 1}, an alignment is a string over the alphabet {M, I, D} such that #M + #D = n and #M + #I = m. Here, #M, #I, and #D denote the number of characters M, I, and D in the word, respectively. An alignment records the “edit steps” from the sequence σ1 to the sequence σ2, where edit operations consist of changing characters, preserving them, or inserting/deleting them. An I in the alignment string corresponds to an insertion in the first sequence, a D is a deletion in the first sequence, and an M is either a character change or lack thereof. We write
over the alphabet {0, 1,..., l - 1}, an alignment is a string over the alphabet {M, I, D} such that #M + #D = n and #M + #I = m. Here, #M, #I, and #D denote the number of characters M, I, and D in the word, respectively. An alignment records the “edit steps” from the sequence σ1 to the sequence σ2, where edit operations consist of changing characters, preserving them, or inserting/deleting them. An I in the alignment string corresponds to an insertion in the first sequence, a D is a deletion in the first sequence, and an M is either a character change or lack thereof. We write  for the set of all alignments. For a given
for the set of all alignments. For a given  , we denote the jth character in h by hj, we use h to indicate #M + #I in the prefix h1h2... hi, and we use h(j) to indicate #M + #D in the prefix h1h2... hj. The cardinality of the set
, we denote the jth character in h by hj, we use h to indicate #M + #I in the prefix h1h2... hi, and we use h(j) to indicate #M + #D in the prefix h1h2... hj. The cardinality of the set 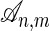 of all alignments can be computed as the coefficient of xmyn in the generating function 1/(1 - x - y - xy). These coefficients are known as Delannoy numbers in combinatorics (ref. 14, section 6.3).
of all alignments can be computed as the coefficient of xmyn in the generating function 1/(1 - x - y - xy). These coefficients are known as Delannoy numbers in combinatorics (ref. 14, section 6.3).
Bayesian multinets were introduced in ref. 15 and are extensions of graphical models by the introduction of class nodes and a set of local networks corresponding to values of the class nodes. In other words, the value of a random variable can change the structure of the graph underlying the graphical model. The pair HMM (see Fig. 3) is an instance of a Bayesian multinet. In this model, the hidden states (unshaded nodes forming the chain) take on one of the values M, I, or D. Depending on the value at a hidden node, either one or two characters are generated; this feature is encoded by plates (squares around the observed states) and class nodes (unshaded nodes in the plates). The class nodes take on the value 0 or 1 corresponding to whether or not a character is generated. Therefore, pair HMMs are probabilistic models of alignments, in which the structure of the model depends on the assignments to the hidden states.
Fig. 3.

A pair HMM for sequence alignment.
Our next result gives the precise description of the pair HMM for sequence alignment in the language of algebraic statistics; namely, we represent this model by means of a polynomial map f. Let σ1 and σ2 be the output strings from a pair HMM (of lengths n and m, respectively). Then,
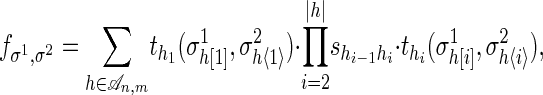 |
4 |
where shi-1hi is the transition probability from state hi-1 to hi, and 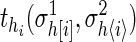 are the output probabilities for a given state hi and the corresponding output characters on the strings σ1 and σ2.
are the output probabilities for a given state hi and the corresponding output characters on the strings σ1 and σ2.
Proposition 6. The pair HMM for sequence alignment is the image of a polynomial map f: R9+2l+l2 → Rln+m. The coordinates of f are polynomials of degree n + m + 1 in Eq. 4.
We need to explain why the number of parameters is 9 + 2l + l2. First, there are the following nine parameters:
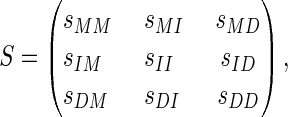 |
which play the same role as in section 2 (namely, they represent transition probabilities in the Markov chain). There are l2 parameters tM(a, b) =: tMab for the probability that letter a in σ1 is matched with letter b in σ2. The insertion parameters tI(a, b) depend only on the letter b, and the deletion parameters tD(a, b) depend only on the letter a, so there are only 2l of these parameters. In the following example, which explains the algebraic representation of Proposition 6, we use t1b and tDa to denote these parameters.
Consider two sequences σ1 = ij and σ2 = klm of length n = 2 and m = 3 over any alphabet. The number of alignments is 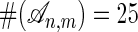 , and they are given in Table 1. The polynomial fσ1,σ2 is the sum of the 25 monomials (of degrees 9, 7, and 5) in the right column of Table 1. For example, if we consider strings over the binary alphabet {0, 1}, then there are 17 parameters (9 s and 8 t parameters), and the pair HMM for alignment is the image of a map f: R17 → R32. The coordinate of f that is indexed by (i, j, k, l, m) ∈ {0, 1}5 equals the 25-term polynomial gotten by summing the right column of Table 1.
, and they are given in Table 1. The polynomial fσ1,σ2 is the sum of the 25 monomials (of degrees 9, 7, and 5) in the right column of Table 1. For example, if we consider strings over the binary alphabet {0, 1}, then there are 17 parameters (9 s and 8 t parameters), and the pair HMM for alignment is the image of a map f: R17 → R32. The coordinate of f that is indexed by (i, j, k, l, m) ∈ {0, 1}5 equals the 25-term polynomial gotten by summing the right column of Table 1.
Table 1. Alignments for a pair of sequences of lenghts 2 and 3.
| Alignment | Gap representation | Monomial |
|---|---|---|
| IIIDD | (···ij,klm··) | tlkSlltllSlltlmSlDtDiSDDtDj |
| IIDID | (··i·j,kl·m·) | tlkSlltllSlDtDiSDltlmSIDtDj |
| IIDDI | (··ij·,kl··m) | tlkSlltllSlDtDiSDDtDjSDltlm |
| IDIID | (·i··j,k·lm·) | tlkSlDtDiSDltllSlltlmSlDtDj |
| IDIDI | (·i·j·,k·l·m) | tlkSlDtDiSDltllSlDtDjSDitlm |
| IDDII | (·ij··,k··lm) | tlkSlDtDiSDDtDjSDltllSlltlm |
| DIIID | (i···j,·klm·) | tDiSDltlkSlltllSlltlmSlDtDj |
| DIIDI | (i··j·,·kl·m) | tDiSDltlkSlltllSlDtDjSDltlm |
| DIDII | (i·j··,·k·lm) | tDiSDltlkSlDtDjSDltllSlltlm |
| DDIII | (ij···,··klm) | tDiSDDtDjSDltlkSlltllSlltlm |
| MIID | (i··j, klm·) | tMikSMltllSlltlmSlDtDj |
| MIDI | (i·j·, kl·m) | tMikSMltllSlDtDjSDltlm |
| MDII | (ij··, k·lm) | tMikSMDtDjSDltllSlltlm |
| IMID | (·i·j, klm·) | tlkSlMtMilSMltlmSlDtDj |
| IMDI | (·ij·, kl·m) | tlkSlMtMilSMDtDjSDltlm |
| IIMD | (··ij, klm·) | tlkSlltllSlMtMimSMDtDj |
| IIDM | (··ij, kl·m) | tlkSlltllSlDtDiSDMtMjm |
| IDMI | (·ij·, k·lm) | tlkSlDtDiSDMtMjlSMltlm |
| IDIM | (·i·j, k·lm) | tlkSlDtDiSDltllSlMtMjm |
| DMII | (ij··, ·klm) | tDiSDMtMjkSMltllSlltlm |
| DIMI | (i·j·, ·klm) | tDiSDltlkSlMtMjlSMltlm |
| DIIM | (i··j, ·klm) | tDiSDltlkSlltllSlMtMjm |
| MMI | (ij·, klm) | tMikSMMtMjlSMltlm |
| MIM | (ij·, klm) | tMikSMltllSlMtMjm |
| IMM | (·ij, klm) | tlkSlMtMilSMMtMjm |
The parametric inference problem for sequence alignment is solved by computing the Newton polytopes NP(fσ1,σ2) with the polytope propagation algorithm. In the terminology introduced in section 4 of ref. 1, an observation σ in the pair HMM is the pair of sequences (σ1, σ2), and the possible explanations are the optimal alignments of these sequences with respect to the various choices of parameters. In summary, the vertices of the Newton polytope NP(fσ1,σ2) correspond to the optimal alignments. If the observed sequences σ1, σ2 are not fixed, then we are in the situation described in proposition 6 of ref. 1. Each parameter choice defines a function from pairs of sequences to alignments:
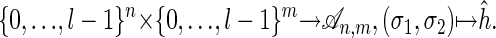 |
The number of such functions grows double-exponentially in n and m, but only a tiny fraction of them are inference functions, which means that they correspond to the vertices of the Newton polytope of the map f. It is an interesting combinatorial problem to characterize the inference functions for sequence alignment.
An important observation is that our formulation in problem 2 is equivalent to combinatorial “scoring schemes” or “generalized edit distances,” which can be used to assign weights to alignments (2). For example, the simplest scoring scheme consists of the following two parameters: a mismatch score mis and an insertion/deletion (indel) score gap (4, 6, 16). The weight of an alignment is the sum of the scores for all positions in the alignment, where a match is assigned a score of 1. This scheme is equivalent to specializing the logarithmic parameters U = -log(S) and V = -log(T) of the pair HMM as follows:
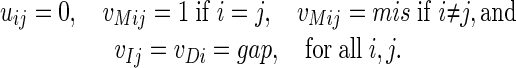 |
5 |
This specialization of the parameters corresponds to intersecting the normal fan of the Newton polytope with a two-dimensional affine subspace (whose coordinates are called mis and gap).
Efficient software for parametrically aligning the sequences with two free parameters exists (xparal; ref. 5). Consider the example of the following two sequences: σ1 = AGGACCGATTACAGTTCAA and σ2 = TTCCTAGGTTAAACCTCATGCA. xparal will return four cones; however, a computation of the Newton polytope reveals seven vertices (three of which correspond to positive mis or gap values). The polytope propagation algorithm has the same running time as xparal: for two sequences of length n and m, the method requires O(nm) two-dimensional convex hull computations. The number of points in each computation is bounded by the total number of points in the final convex hull (or equivalently, the number, K, of explanations). Therefore, each convex hull computation requires at most O(Klog(K)) operations, thus giving an O(nmKlog(K)) algorithm for solving the parametric alignment problem. However, this running time can be improved by observing that the convex hull computations that need to be carried out have a very special form; namely, in each step of the algorithm, we need to compute the convex hull of two superimposed convex polygons. This procedure is a primitive of the divide-and-conquer approach to convex hull computation, and there is a well known O(K) algorithm for solving it (ref. 17, section 3.3.5). Therefore, for two parameters, our recursive approach to solving the parametric problem yields an O(Kmn) algorithm, matching the running time of xparal and the conjecture of Waterman et al. (6).
To demonstrate the practicality of our approach for higher-dimensional problems, we implemented a four-parameter recursive parametric alignment solver. The more general alignment model includes different transition/transversion parameters (instead of just one mismatch parameter) and separate parameters for opening gaps and extending gaps. A transition is mutation from one purine (A or G) to another, or from one pyrimidine (C or T) to another, and a transversion is a mutation from a purine to a pyrimidine or vice versa. More precisely, if we let Pu = {A, G} and Py = {C, T}, the model is as follows:
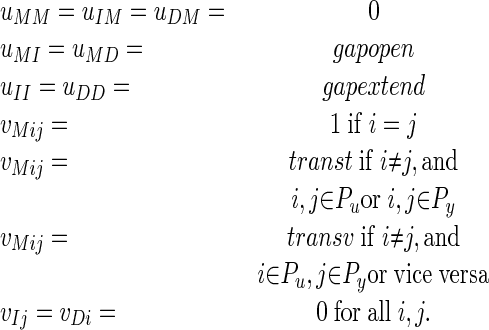 |
For the two sequences σ1 and σ2 in the example above, the number of vertices of the four-dimensional Newton polytope is 224 (compared with 7 for the two-parameter case).
5. Practical Aspects of Parametric Inference
We begin by pointing out that parametric inference is useful for Bayesian computations. Consider the problem in which we have a prior distribution π(s) on our parameters s = (s1,..., sd) and in which we would like to compute the posterior probability of a MAP explanation ĥ:
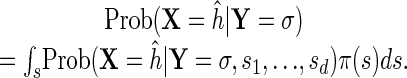 |
6 |
This problem is important because it can give a quantitative assessment of the validity of ĥ in a setting in which we have prior, but not certain, information about the parameters, and also because we may want to sample ĥ according to its posterior distribution (for an example of how this can be applied in computational biology, see ref. 18). Unfortunately, these integrals may be difficult to compute. We propose the following simple Monte Carlo algorithm for computing a numerical approximation to the integral [6].
Proposition 7. Select N parameter vectors s(1),..., s(N) according to the distribution π(s), where N is much larger than the number of vertices of the Newton polytope NP(fσ). Let K be the number of s(i) such that -log[s(i)] lies in the normal cone of NP(fσ) indexed by the explanation ĥ. Then, K/N approximates Eq. 6.
Proof: The expression Prob(X = ĥ|Y = σ, s1,..., sd) is zero or one depending on whether the vector -log(s) = (-log(s1),..., -log(sd)) lies in the normal cone of NP(fσ) indexed by ĥ. This membership test can be done without ever running the sum-product algorithm if we precompute an inequality representation of the normal cones.
The bound on the number of vertices of the Newton polytope in section 4 of ref. 1 provides a valuable tool for estimating the quality of this Monte Carlo approximation. We believe that the tropical geometry developed in ref. 1 will also be useful for more refined analytical approaches to Bayesian integrals. The study of Newton polytopes can also complement the algebraic geometry approach to model selection proposed in ref. 19.
Another application of parametric inference is to problems in which the number of parameters may be very large, but in which we want to fix a large subset of parameters, thereby reducing the dimensions of the polytopes. Gene-finding models, for example, may have up to thousands of parameters and input sequences can be millions of base pairs long however, we are usually only interested in studying the dependence of inference on a select few. Although specializing parameters reduces the dimension of the parameter space, the explanations correspond to vertices of a regular subdivision of the Newton polytope, rather than just to the vertices of the polytope itself. This identification is explained below (see ref. 1 for more background).
Consider a graphical model with parameters s1,..., sd of which the parameters s1,..., sr are free but sr+1 = Sr+1,..., sd = Sd, where the Si are fixed nonnegative numbers. Then, the coordinate polynomials fσ of our model specialize to polynomials in r unknowns whose coefficients ca are nonnegative numbers:
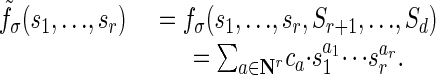 |
The support of this polynomial is the finite set 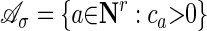 . The convex hull of
. The convex hull of 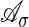 in Rr is the Newton polytope of the polynomial f̃σ = f̃σ(s1,..., sr). For example, in the case of the HMM with output parameters specialized, the Newton polytope of f̃σ is the polytope associated with a Markov chain. Kuo (20) shows that the size of these polytopes does not depend on the length of the chain.
in Rr is the Newton polytope of the polynomial f̃σ = f̃σ(s1,..., sr). For example, in the case of the HMM with output parameters specialized, the Newton polytope of f̃σ is the polytope associated with a Markov chain. Kuo (20) shows that the size of these polytopes does not depend on the length of the chain.
Let h be any explanation for σ in the original model and let (u1,..., ur, ur+1,..., un) be the vertex of the Newton polytope of fσ corresponding to that explanation. We abbreviate ah = (u1,..., ur) and 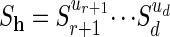 . The assignment h ↦ ah defines a map from the set of explanations of σ to the support
. The assignment h ↦ ah defines a map from the set of explanations of σ to the support 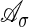 . The convex hull of the image coincides with the Newton polytope of f̃σ. We define the following:
. The convex hull of the image coincides with the Newton polytope of f̃σ. We define the following:
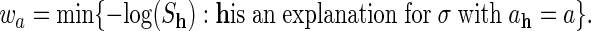 |
7 |
If the specialization is sufficiently generic, then this maximum is attained uniquely, and for simplicity, we will assume that this is the case. If a point 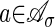 is not the image of any explanation h, then we set wa = ∞. The assignment a ↦ wa is a real valued function on the support of our polynomial f̃σ, and it defines a regular polyhedral subdivision Δw of the Newton polytope NP(f̃σ). Namely, Δw is the polyhedral complex consisting of all lower faces of the polytope gotten by taking the convex hull of the points (a, wa) in Rr+1. See ref. 21 for details on regular triangulations and regular polyhedral subdivisions.
is not the image of any explanation h, then we set wa = ∞. The assignment a ↦ wa is a real valued function on the support of our polynomial f̃σ, and it defines a regular polyhedral subdivision Δw of the Newton polytope NP(f̃σ). Namely, Δw is the polyhedral complex consisting of all lower faces of the polytope gotten by taking the convex hull of the points (a, wa) in Rr+1. See ref. 21 for details on regular triangulations and regular polyhedral subdivisions.
Theorem 8. The explanations for the observation σ in the specialized model are in bijection with the vertices of the regular polyhedral subdivision Δw of the Newton polytope of the specialized polynomial f̃σ.
Proof: The point (a, wa) is a vertex of Δw if and only if the following open polyhedron is nonempty:
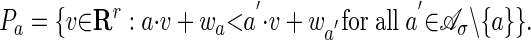 |
If v is a point in Pa, then we set si = exp(-vi) for i = 1,..., r, and we consider the explanation h which attains the minimum in Eq. 7. All parameters have been specialized, and h is the solution to problem 2. This argument is reversible: any explanation for σ in the specialized model arises from one of the nonempty polyhedra Pa. Note that the collection of polyhedra Pa defines a polyhedral subdivision of Rr, which is geometrically dual to the subdivision Δw of the Newton polytope of f̃σ.
In practical applications of parametric inference, it may be of interest to compute only one normal cone of the Newton polytope (for example, the cone containing some fixed parameters). We conclude this section by observing that the polytope propagation algorithm is suitable for this computation as well.
Proposition 9. Let v be a vertex of a d-dimensional Newton polytope of an HMM. Then, the normal cone containing v can be computed by using a polytope propagation algorithm in dimension d - 1.
Proof: We run the standard polytope propagation algorithm described in section 4, but at each step, we record only the minimizing vertex in the direction of the log parameters, together with its neighboring vertices in the edge graph of the Newton polytope. It follows by induction that given this information at the nth step, we can use it to find the minimizing vertices and related neighbors in the (n + 1)st step.
6. Summary
We envision a number of biological applications for the polytope propagation algorithm, including the following:
Full parametric inference using the normal fan of the Newton polytope of an observation when the graphical model under consideration has only few model parameters.
Use of the edge graph of the polytope to identify stable parts of alignments and annotations.
Construction of the normal cone containing a specific parameter vector when computation of the full Newton polytope is infeasible.
Computation of the posterior probability (in the sense of Bayesian statistics) of an alignment or annotation. The regions for the relevant integrations are the normal cones of the Newton polytope.
As we have shown, the computation of Newton polytopes for (interesting) graphical models is certainly feasible for a few free parameters, and we expect that further analysis of the computational geometry should yield efficient algorithms in higher dimensions. For example, the key operation, computation of convex hulls of unions of convex polytopes, is likely to be considerably easier than general convex hull computations even in high dimensions. Fukuda et al. (22) give polynomial time algorithms for computing convex hulls of unions of convex polytopes that are in general position.
If computation of the Newton polytope is impractical, it is still possible to identify the cone containing a specific parameter, and this cone can be used to measure quantitatively the robustness of the inference. Parameters near a boundary are unlikely to lead to biologically meaningful results. Furthermore, the edge graph can be used to identify common regions in the explanations corresponding to adjacent vertices. In the case of alignment, biologists might see a collection of alignments rather than just one optimal one, with common subalignments highlighted. This output is different from returning the k best alignments because suboptimal alignments may not be vertices of the Newton polytope. The solution that we propose explicitly identifies all suboptimal alignments that can result from similar parameter choices.
Acknowledgments
L.P. was supported in part by National Institutes of Health Grant R01-HG02362-02. B.S. was supported by a Hewlett Packard Visiting Research Professorship 2003/2004 at the Mathematical Sciences Research Institute (MSRI) at the University of California, Berkeley, and in part by National Science Foundation Grant DMS-0200729.
Abbreviations: MAP, maximum a posteriori; HMM, hidden Markov model.
References
- 1.Pachter, L. & Sturmfels, B. (2004) Proc. Natl. Acad. Sci. USA 101 16132-16137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bucher, P. & Hofmann, K. (1996) in Proceedings of the Conference on Intelligent Systems for Molecular Biology, eds. States, D. J., Agarwal, P., Gaasterland, T., Hunter, L. & Smith, R. (AAAI, Menlo Park, CA), pp. 44-51.
- 3.Alexandersson, M., Cawley, S. & Pachter, L. (2003) Genome Res. 13 496-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernández-Baca, D., Seppäläinen, T. & Slutzki, G. (2000) in Combinatorial Pattern Matching, Lecture Notes in Computer Science, eds. Giancarlo, R. & Sankoff, D. (Springer, Berlin), Vol. 1848, pp. 68-82. [Google Scholar]
- 5.Gusfield, D. & Stelling, P. (1996) Methods Enzymol. 266 481-494. [DOI] [PubMed] [Google Scholar]
- 6.Waterman, M., Eggert, M. & Lander, E. (1992) Proc. Natl. Acad. Sci. USA 89 6090-6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldi, P. & Brunak, S. (1998) Bioinformatics. The Machine Learning Approach. (MIT Press, Cambridge, MA).
- 8.Durbin, R., Eddy, S., Krogh, A. & Mitchison, G. (1998) Biological Sequence Analysis (Probabilistic Models of Proteins and Nucleic Acids) (Cambridge Univ. Press, Cambridge, U.K.).
- 9.Gardiner-Garden, M. & Frommer, M. (1987) J. Mol. Biol. 196 261-282. [DOI] [PubMed] [Google Scholar]
- 10.Takai, D. & Jones, P. A. (2002) Proc. Natl. Acad. Sci. USA 99 3740-3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., et al. (2001) Nature 409 860-921, and errata [DOI] [PubMed] [Google Scholar]; (2001), 411 720, and; (2001) 412 565.
- 12.Jordan, M. I. & Weiss, Y. (2002) in Handbook of Brain Theory and Neural Networks, ed. Arbib, M. (MIT Press, Cambridge, MA), 2nd Ed.
- 13.Kschischang, F., Frey, B. & Loeliger, H. A. (2001) IEEE Trans. Inform. Theory 47 498-519. [Google Scholar]
- 14.Stanley, R. (1999) Enumerative Combinatorics (Cambridge Univ. Press, Cambridge, MA), Vol. 2.
- 15.Friedman, N., Geiger, D. & Goldszmidt, M. (1997) Machine Learning 29 131-161. [Google Scholar]
- 16.Gusfield, D., Balasubramanian, K. & Naor, D. (1994) Algorithmica 12 312-326. [Google Scholar]
- 17.Preparata, F. P. & Shamos, M. I. (1985) Computational Geometry: An Introduction (Springer, Berlin).
- 18.Liu, J. (1994) J. Am. Stat. Assoc. 89 958-966. [Google Scholar]
- 19.Rusakov, D. & Geiger, D. (2002) Proceedings of the 18th Conference in Uncertainty in Artificial Intelligence, eds. Darwiche, A. & Friedman, N. (Morgan Kaufmann, San Francisco), pp. 438-445.
- 20.Kuo, E. (2004) arXiv: math.CO/0401342.
- 21.Sturmfels, B. (1996) Gröbner Bases and Convex Polytopes (Am. Math. Soc., Providence, RI), Vol. 8.
- 22.Fukuda, K., Liebling, T. H. & Lütlof, C. (2001) Comput. Geom. 20 13-23. [Google Scholar]