

Abstract
We have developed a versatile statistical analysis algorithm for the detection of genomic aberrations in human cancer cell lines. The algorithm analyzes genomic data obtained from a variety of array technologies, such as oligonucleotide array, bacterial artificial chromosome array, or array-based comparative genomic hybridization, that operate by hybridizing with genomic material obtained from cancer and normal cells and allow detection of regions of the genome with altered copy number. The number of probes (i.e., resolution), the amount of uncharacterized noise per probe, and the severity of chromosomal aberrations per chromosomal region may vary with the underlying technology, biological sample, and sample preparation. Constrained by these uncertainties, our algorithm aims at robustness by using a priorless maximum a posteriori estimator and at efficiency by a dynamic programming implementation. We illustrate these characteristics of our algorithm by applying it to data obtained from representational oligonucleotide microarray analysis and array-based comparative genomic hybridization technology as well as to synthetic data obtained from an artificial model whose properties can be varied computationally. The algorithm can combine data from multiple sources and thus facilitate the discovery of genes and markers important in cancer, as well as the discovery of loci important in inherited genetic disease.
Keywords: array-based comparative genomic hybridization, copy-number fluctuations, maximum a posteriori estimator
Genomes in a population are polymorphic, giving rise to diversity and variation. In cancer, even somatic cell genomes can rearrange themselves, often resulting in genomic deletion (hemi- or homozygous) and amplifications. Means for assessing these chromosomal aberrations quickly, inexpensively, and accurately have many potential scientific, clinical, and therapeutic implications (1, 2), particularly in the genomics of cancer and inherited diseases. Genome-based methods for studying cancer, in contrast to the gene expression-based methods, can exploit the stability of DNA (as a component of the cancerous cell, which does not vary as a function of the cell's physiological state). Karyotyping, determination of ploidy, and comparative genomic hybridization have been useful tools for this purpose even though they are crude and produce data that must be processed by sophisticated statistical algorithms to serve as useful guides to diagnosis and treatment.
Microarray methods are an important new technology that can be used to study variations between regular and cancer genomes. Imagine that one can sample the genome uniformly (independently and identically distributed) and reproducibly to create a large number of oligonucleotides (on the order of 100,000 probes) located every 30 kb or so. These oligonucleotides almost always come from regions of the genome that do not share homologous sequences elsewhere in the genome. These sequences (typically less than a few hundred base pairs long) occupy unique positions in the normal genome and have exactly two copies.
If one such oligonucleotide belongs to a region in a cancer genome that has an altered copy number, say, c (0 ≤ c ≠ 2), then when the cancer genome is sampled, this oligonucleotide will occur with a probability that is c/2 times that in the regular genome. The copy number can be computed by a ratiometric measurement of the abundance of an oligonucleotide in a cancer sample measured against that in the regular genome. This technique can be generalized to measure the copy number variations for many probes simultaneously with high-throughput microarray experiments. Even though the ratiometric measurements used and the associated regularizations tame the multiplicative noises in the system to some extent, there remains a large amount of uncharacterized noise (generally additive) that can render the data worthless unless a proper data-analysis algorithm is applied. Because the data may come from multiple sources collected with varying protocols, such an algorithm must be general and be based on a minimal set of prior assumptions about the methods. The algorithm we describe below reflects these desiderata.
Our Bayesian approach constructs a most plausible hypothesis concerning regional changes and the corresponding associated copy number. It can be viewed as an optimization process minimizing a score function that assigns penalties of different type for each kind of deviation from genomic normality (break-points, unexplainable probe values, noise, etc.); we discuss how these penalties are derived. We describe various algorithmic alternatives, their implementations, and the empirical results derived using real data (where the underlying facts are not directly verifiable) and simulated data (where the true facts are known).
Statistical Model
We start by describing a probabilistic generative model for observed copy number data. The model is Bayesian in spirit, in that we use parameterized prior distributions and use the posterior distribution function to estimate the underlying model. We use a maximum a posteriori (MAP) technique to estimate the underlying model. This idealized statistical model takes into account some major sources of copy number variation in an irregular genome and is described by two scalar parameters 0 ≤ pr, pb ≤ 1.
We assume that there is a copy-number distribution for probes at locations that have not been affected by the chromosomal aberrations associated with cancer. We call these probes regular probes. We also assume that the probability for a particular probe being regular is pr and that the associated regular copy-number distribution, after log transformation, is Gaussian, with mean μr and standard deviation σr. For the other probes, which we call deviated, the log-transformed copy-number distributions also are assumed to be Gaussian, with unknown mean and standard deviation, distinct from the regular distribution. There are usually many sets of probes drawn from different deviated distributions.
We also assume that there are locations in the genome that are particularly susceptible to amplification (also known as duplication) and deletion events. These aberrations change the copy numbers of probes locally. We model the number of such mutations as a Poisson process with parameter pbN, where N is the length of the genome (i.e., total number of probes).
We subdivide the probes along the genome into k nonoverlapping intervals. Probes belonging to a particular interval are assumed to have a similar evolutionary history of duplication and deletion events, and therefore have similar copy-number distributions. The number of intervals into which the probes can be separated represents the progressive degeneration of a cancer cell line. We do not model single nucleotide polymorphisms and other point-mutation events, and this undermodeling reappears as localized noise in our analyzed data.
In our picture, each interval in this subdivision has a “true” copy number. Our goal is to estimate the correct subdivision and the copy numbers associated with each subinterval. Despite its simplicity, our model can serve as the basis of a statistical algorithm to infer the aberrations without overfitting the data.
More formally, given a set of N probe copy-number values arranged on the genome, we assume that there is an unknown partition of this set into nonoverlapping subintervals. The probe copy number values in the jth interval are assumed to arise as independent samples from a Gaussian distribution N(μj, σj). The parameters relating to the jth interval can be represented as the tuple Ij = (μj, ij, σj), where μj and σj are the mean and standard deviation of the appropriate Gaussian distribution and ij is the position of the last probe in the interval. We call such a set of intervals I = {Ij|j = 1,..., k} an interval structure. When a particular interval in I is regular, its mean is the regular mean μr. If an interval Ij is deviated, then its population mean μj is unknown and is estimated by using the sample mean over the interval. In this work, we assume that all of the σj terms are equal to some common value σ, and we therefore omit them from the notation. We denote an interval structure IN with k intervals and whose intervals have associated means μ1,..., μk and endpoints i1,..., ik (necessarily ik = N) as 〈i1, μ1, i2, μ2,..., ik, μk〉.
Our goal is to estimate the unknown interval structure IN from an input sequence {vi, i = 1... N} of copy numbers of N successive probes.
The statistical model described thus far fits naturally into a Bayesian setting. We can start with a prior distribution on the set of interval structures depending only on the number of intervals and the number of regular probes with two scalar parameters pr and pb whose significance is described above.
This prior has two components, the first a Poisson distribution to model the number of intervals with Poisson parameter pbN. The second component is a sequence of Bernoulli trials, one for each probe with probability pr that a given probe is regular. Combining these factors, the prior distribution becomes
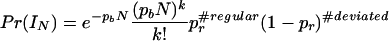 |
1 |
where #regular is the number of regular probes with the “regular” copy-number distribution and #deviated is the number of remaining probes in the interval structure IN. In each interval Ij, the data points are modeled by adding independent Gaussian noise to this prior structure and are drawn from the Gaussian distribution N(μj, σ).
The data likelihood function for the first n probes is given by the product of Gaussians:
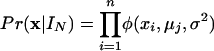 |
2 |
where the ith probe is covered by the jth interval of the interval structure IN and μj is the mean of the corresponding Gaussian distribution. φ denotes the density function of the Gaussian distribution. By multiplication, we obtain the posterior likelihood function:
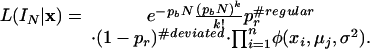 |
3 |
In the above expression for L, only the μ values of nonregular processes are unknown, and we estimate these values by using the sample mean for the interval. The MAP solution to the segmentation problem is obtained by finding the interval structure I* that maximizes this likelihood function or, equivalently, minimizes the negative log likelihood of L.
Algorithm and Implementation
A dynamic programming algorithm efficiently minimizes the negative posterior log likelihood function obtained above. Starting with an interval structure I = 〈i1, μ1,..., ik, μk〉, we can extend it to the interval structure I′ = 〈i1, μ1,..., ik+1, μk+1〉, where ik+1 > ik. The following formula computes the log likelihood for such an extension
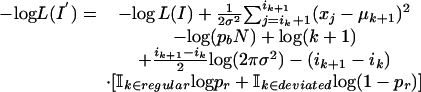 |
4 |
where the last term on the right side is chosen according to whether the last added interval (i.e., the one extending from ik + 1 to ik+1) is regular or not. 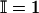 if the Boolean formula e is true, and 0 otherwise. We also point out that the MAP approach permits the estimation of the μ terms in a uniform manner. When the last added interval is regular, the value μk+1 is fixed at the global mean μ. When the last added interval is deviated, however, the MAP criterion automatically forces the choice of the sample mean of the data points covered by the last interval as the value for μk+1. One could build hierarchical models for the mean and use these “shrinkage-like” estimators as well (3), although we do not explore that approach here.
if the Boolean formula e is true, and 0 otherwise. We also point out that the MAP approach permits the estimation of the μ terms in a uniform manner. When the last added interval is regular, the value μk+1 is fixed at the global mean μ. When the last added interval is deviated, however, the MAP criterion automatically forces the choice of the sample mean of the data points covered by the last interval as the value for μk+1. One could build hierarchical models for the mean and use these “shrinkage-like” estimators as well (3), although we do not explore that approach here.
The negative log likelihood function satisfies an optimality condition that allows one to use a standard dynamic programming algorithm [of time-complexity O(N2)] in this setting.
Results
We evaluate the performance of this simple Bayesian scheme on three kinds of data. For each of these data sets, we will see that proper choice of the parameter values pr and pb leads to good segmentation. Indeed, coefficients chosen from within a fairly large region of the “pr-pb space” lead to a good segmentation because our procedure is stable over a large domain. The parameters pb, pr, μ, and σ play different roles: an increase in pb yields more intervals in the segmentation, and, as pr is increased, more probes come to be classified as regular, and therefore the number of different segments diminish.
The choice of μ is critical because it controls the bias in the resulting segmentation. The choice of σ is also important because increasing σ weakens the influence of the data on the segmentation obtained.
Representational Oligonucleotide Microarray Analysis (ROMA) Data from Breast Cancer Cell Lines. ROMA is a comparative genomic hybridization (CGH) technique developed by Wigler and colleagues (1) at Cold Spring Harbor Laboratory. It evolved from an earlier method, representational differential analysis, which was adapted for greatly increased volumes of data obtained by using an oligonucleotide microarray. ROMA uses a comparative “two-color” scheme to compare multiple genomes, each represented with reduced complexity by using a PCR-based method (4, 5). As in other array-based methods, ROMA performs simultaneous array hybridization to compare a normal genome at one fluorescent wavelength and a tumor genome at another. The DNA representations used by ROMA are based on amplification of short restriction endonuclease fragments and hence are predictable from the nucleotide sequence of the genome. We have tested our algorithm on the data sets from the Wigler laboratory obtained by ROMA from the genomes of breast cancer cell lines. The data set is based on 85,000 well characterized probes, each of length 70 bp, providing a resolution of a probe every 15-30 kb.
Figs. 1 and 2 show subsampled ROMA breast cancer data from chromosomes 2 and 8, respectively, overlaid with the segmentation found by our algorithm. The low-complexity DNA representation used in ROMA, together with a careful choice of probes, provides low-noise data that can be characterized accurately by the algorithm.
Fig. 1.

Segmented probes on chromosome 2, pr = 0.55, pb = 0.005, sampling rate 1 in 10.
Fig. 2.

Segmented probes on chromosome 8, pr = 0.55, pb = 0.01, sampling rate 1 in 10.
Array-Based CGH (arrayCGH) Data from Prostate Cancer Cell Lines. arrayCGH is a recently developed technique that maps duplicated or deleted chromosomal segments onto high-density arrays of well characterized bacterial artificial chromosomes (BACs), rather than onto metaphase chromosomes. This method has been used for precise mapping of duplications and deletions occurring in cancers and other human diseases, including birth defects and mental retardation (see ref. 6 for review and applications of this technique). Tumors that have been studied by using this method are breast, head and neck, Wilms, esophageal, pulmonary artery intimal, adrenocortical, renal, and prostate cancers and lymphomas. We have tested our algorithm on a data set obtained by high-resolution arrayCGH analysis of prostate cancer tissue. The data were supplied by a group at Nijmegen University Medical Center and obtained by hybridization on their custom array composed of ≈3,500 fluorescence in situ hybridization-verified clones selected to cover the genome with an average of one clone per megabase (7).
Fig. 3 shows the performance of our segmentation algorithm on data from prostate cancer cell lines obtained through arrayCGH experiments. We note that these data are noisier than the ROMA data considered previously. But, despite the increased noise, the segmentation algorithm is robust and yields reasonable segmentations.
Fig. 3.

Segmented probes on array-based CGH data. Chromosome 8, pr = 0.55, pb = 0.01, sampling rate 1 in 10.
Simulated Data. To further test our algorithm, we can use an artificial but biologically inspired model to generate synthetic data. To generate simulated copy-number data, we choose loci uniformly over a genome such that the probability of a duplication or deletion event taking place at that location on the genome is given by pb. At each of these points, we assign a new copy-number value that represents the mean for the new interval. The mean values are drawn from a power-transformed γ distribution to mimic the observed distribution of means in experimental data. The lengths of the intervals follow a geometric distribution such that the ratio of expected fragment length and the expected distance between the beginning of each interval is pr. Once the segmentation and the mean values are chosen, we generate the simulated data by adding random Gaussian noise. A typical simulated genome is shown in Fig. 4.
Fig. 4.

A simulated genome with μ = 0.0 and σ = 0.15 and the corresponding segmentation.
Effect of Noise on Performance. We investigate the effect of increasing the σ of the underlying model on the performance of the segmenter. Assuming the parameters of the model are correctly estimated, the segmenter can output the estimated mean value at every probe position. Using the known mean values, we can compare these two sequences of means. In our setting, a good measure of error is the number of misclassified probes, i.e., the number of probes that are known to be regular but were classified as amplified or deleted and vice versa. Fig. 5 shows the increase in the rate of misclassification as σ increases.
Fig. 5.

Average number of misclassified probes plotted against increasing σ on synthetic data. The average number of misclassified probes in >100 trials is normalized against the length of the simulated genome.
Prior Selection
Proper selection of a prior distribution has received extensive attention in the literature. Approaches include noninformative priors [Jeffreys (8)], reference priors [Bernardo (9); see also Berger and Bernardo (10) and Kass and Wasserman (11)], and conjugate priors [Raiffa and Schlaifer (12)] among others. Conjugate prior methods frequently arise in connection with exponential families of distributions [see Brown (13)]. Other approaches include using invariance properties to posit prior distributions with good performance. More recent and somewhat more data-dependent techniques include hierarchical and empirical Bayes techniques. Textbooks such as those by Bernardo and Smith (14), Berger (15), Carlin and Louis (16), Gelman et al. (17), and Robert (18) cover model selection as a part of Bayesian learning.
For the problem of estimating probe copy numbers, the prior distribution is specified by the two probability parameters, pr and pb. The other parameters (μ, the regular mean, and σ2, the regular variance) can be estimated by experiment. The problem of prior selection reduces to the problem of optimally selecting the values of pr and pb to prevent overfitting of the data.
Minimax approaches choose prior distributions that minimize the maximum value of the likelihood function (Eq. 4). This criterion is pessimistic, in that it chooses the prior that generates the worst likelihood value. See, for example, Berger (15), Brown (19-21), and Strawderman (22-24). In the nonparametric setting of function estimation, multiscale methods have been proven to be asymptotically minimax by Donoho and Johnstone (25-27).
We adapt an approach, based on statistical decision theory, that directly controls the level of overfitting without explicitly depending on the asymptotic performance guarantees of minimax approaches. We rely on the fact that, in any segmentation, each jump separates the probes in the two adjoining intervals. If a segmentation is overfitted, at least one of its jumps must be overfitted, too. We use Hotelling's t2 statistic [see Anderson (28) or Wilks (29)] at each jump to compute a measure of this overfitting.
We apply an F test to Hotelling's t2 statistic to test whether two sets of independent samples come from populations with the same mean. This F test is possible because we assume that the two sets of samples have the same (but still unknown) variance. Let x1, x2,..., xN1 and y1, y2,..., yN2 be the two sets of independent samples taken from successive intervals of size N1 and N2, respectively. Then, we define the statistic:
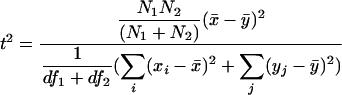 |
5 |
where x̄ and ȳ are the respective sample means, and df1 and df2 refer to the respective degrees of freedom of the two samples. Under the null hypothesis (of equal means), t2 follows an F distribution with 1, (df1 + df2) degrees of freedom. This leads to a one-tailed F test.
Intuitively, t2 needs to be large to avoid overfitting. The cumulative probability for the appropriate F distribution yields a score that determines the quality of the break. We also compute a score for the goodness of fit for the whole segmentation. This procedure yields a set of scores: one for each break and one for the goodness of fit. The minimum of these scores is used to evaluate the whole segmentation. We select the parameters pr and pb to maximize this score by searching at regular intervals over the parameter space. We can continue to refine the search in the neighborhood of the optimal values obtained. However, the algorithm is already extremely stable in a large region of the pr-pb space and yields, in practice, very good segmentations.
Discussion
The problem of detecting copy-number variations has assumed biological importance in recent years. Most extant algorithms use a global thresholding approach for this problem. This is the case, for example, in Vissers et al. (7) as well as many commercially available packages. These algorithms have the advantage of simplicity but perform poorly in the presence of noise and correlations. Other published approaches have used smoothing (30), hidden Markov models (HMMs) (31, 32), and mixtures of Gaussians, as well as approaches that try to estimate the correlations between probes (33, 34). Although smoothing certainly improves the performance of threshold-based approaches, the specifics remain somewhat ad hoc, and the method requires tuning dependent on the source and resolution of the data.
HMMs have the advantage of having a general (although slow) learning algorithm; however, their performance is very sensitive to the topology of the HMM. For this reason, researchers tend to analyze very narrow classes of data with a particular HMM, e.g., a prostate cancer cell line. Very rarely are normal-normal data so analyzed, because this analysis usually necessitates the construction of an HMM with a different topology, leading to questions about the comparative power of such analyses. The main problem with both this approach and others based on assuming a distributional form for cancerous data is that the cancerous insertion-deletion polymorphisms are characterized by being nonnormal, rather than belonging to a specific distributional form. Therefore, fitting cancer data leads to the construction of a specific HMM topology that might depend on the specific cancer as well as the goodness of fit desired by the statistician.
Olshen and Venkatraman (35) have advocated another approach based on recursive change-point detection in the copy number data. The existence of a large literature on change-point analysis makes this approach attractive. Conversely, an efficient implementation of this algorithm is difficult. The specific statistic chosen for change-point detection in this and other work of the group perform poorly on normal-normal data due to overly pessimistic criteria. In some sense, our approach of putting a Bayesian prior on the number of change points enables us to be aggressive about detecting change points.
We have devised a versatile MAP estimator algorithm to analyze arrayCGH data. This algorithm uses a model that captures the genomic amplification-deletion processes but is relatively insensitive to additive noise in the data. When the algorithm was tested on a wide variety of data from ROMA- and arrayCGH-based methods, this particular feature of the algorithm provided strength and robustness. We note that the correct choice of pr and pb is critical in the segmentation algorithm. High values of pb tend to yield overfitted solutions, whereas high values of pr drive us toward biased solutions that mark all segments as regular. The advantage of having an algorithm with only two numerical parameters is that a simple and natural statistical criterion enables the proper choice of these parameters in all cases.
We parenthetically note that our approach extends to multidimensional data mutatis mutandis. The relevant likelihood function needs to be changed to the following
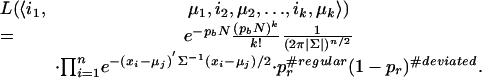 |
6 |
The t2 statistic can be modified similarly.
Prior work by Donoho and colleagues (36-38) on detecting geometrical features in point clouds by using multiresolution methods relates to the ideas presented here. These papers focus on the use of multiresolution approaches for efficiency and statistical stability. There is also prior work by Kolaczyk (see ref. 39, for example) that gives a unified Bayesian treatment to multiresolution analysis and covers large classes of both continuous as well as discrete processes. Our approach leads to an efficient algorithm for sequence-like data, which can be used in a multiscale setting if desired. Furthermore, in our approach, the probabilistic generative model directly leads to the cost function; thus, other generative models, e.g., poisson models, can be easily considered in this setting. It should be noted that the Hotelling's t2 statistic cannot be easily generalized to this setting.
Acknowledgments
We thank Yi Zhou (New York University Bioinformatics Group) and two anonymous reviewers for many helpful discussions, suggestions, and relevant references to statistical literature. We also thank Lakshmi Muthuswami (Cold Spring Harbor Laboratory), and Eric Schoenmakers and Joris Veltman (Nijmegen University Medical Center, Nijmegen, The Netherlands) for providing the data used here and for explaining their biological significance. The work reported in this paper was supported by grants from the National Science Foundation (NSF) Qubic Program, the NSF Information Technology Research Program, the Defense Advanced Research Projects Agency, a Howard Hughes Medical Institute Biomedical Support Research Grant, the U.S. Department of Energy, the U.S. Air Force (Air Force Research Laboratory), the National Institutes of Health, and the New York State Office of Science, Technology and Academic Research.
Author contributions: B.M. designed research; R.-S.D., A.R., and B.M. performed research; R.-S.D., A.R., and B.M. contributed new reagents/analytical tools; R.-S.D., A.R., H.O., M.H.W., and B.M. analyzed data; R.-S.D., A.R., and B.M. wrote the paper; and R.L. and M.H.W. contributed data.
Abbreviations: MAP, maximum a posteriori; ROMA, representational oligonucleotide microarray analysis; CGH, comparative genomic hybridization; arrayCGH, array-based CGH; HMM, hidden Markov model.
References
- 1.Lucito, R., West, J., Reiner, A., Alexander, J., Esposito, D., Mishra, B., Powers, S., Norton, L. & Wigler, M. (2000) Genome Res. 10, 1726-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mishra, B. (2002) Comput. Sci. Eng. 4, 42-49. [Google Scholar]
- 3.Daniels, M. J. & Kass, R. E. (1999) J. Am. Stat. Assoc. 94, 1254-1263. [Google Scholar]
- 4.Lisitsyn, N., Lisitsyn, N. & Wigler, M. (1993) Science, 258, 946-951. [DOI] [PubMed] [Google Scholar]
- 5.Lucito, R., Nakimura, M., West, J. A., Han, Y., Chin, K., Jensen, K., McCombie, R., Gray, J. W. & Wigler, M. (1998) Proc. Natl. Acad. Sci. USA 95, 4487-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albertson, D. G. & Pinkel, D. (2003) Hum. Mol. Genet, 12, Suppl. 2, R145-R152. [DOI] [PubMed] [Google Scholar]
- 7.Vissers, L. E. L. M., de Vries, B. B. A., Osoegawa, K., Janssen, I. M., Feuth, T., Choy, C. O., Straatman, H., van der Vliet, W., Huys, E. H. L. P. G., van Rijk, A., et al. (2003) Am. J. Hum. Genet. 73, 1261-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeffreys, H. (1946) Proc. R. Soc. London Ser. A 186, 453-461. [DOI] [PubMed] [Google Scholar]
- 9.Bernardo, J. M. (1979) J. R. Stat. Soc. Ser. B 41, 113-147. [Google Scholar]
- 10.Berger, J. O. & Bernardo, J. M. (1992) in Bayesian Statistics 4, eds. Berger, J. O., Bernardo, J. M., Dawid, A. P. & Smith, A. F. M. (Oxford Univ. Press, Oxford), pp. 35-60
- 11.Kass, R. E. & Wasserman, L. A. (1996) J. Am. Stat. Assoc. 91, 1343-1370. [Google Scholar]
- 12.Raiffa, H. & Schlaifer, R. (1961) Applied Statistical Decision Theory (Wiley, New York).
- 13.Brown, L. D. (1986) Foundations of Exponential Families (Institute of Mathematical Statistics, Hayward, CA), Monograph Series 6.
- 14.Bernardo, J. M. & Smith, A. F. M. (1994) Bayesian Theory (Wiley, New York).
- 15.Berger, J. O. (1985) Statistical Decision Theory and Bayesian Analysis (Springer, New York), 2nd Ed.
- 16.Carlin, B. P. & Louis, T. A. (1996) Bayes and Empirical Bayes Methods for Data Analysis (Chapman & Hall, London).
- 17.Gelman, A., Carlin, J. B., Stern, H. S. & Rubin, D. D. (1995) Bayesian Data Analysis (Chapman & Hall, London).
- 18.Robert, C. P. (2001) The Bayesian Choice (Springer, New York).
- 19.Brown, L. D. (1971) Ann. Math. Stat. 42, 855-903. [Google Scholar]
- 20.Brown, L. D. (1993) in Statistical Decision Theory and Related Topics 5, eds. Gupta, S. S. & Berger, J. O. (Springer, New York), pp. 1-18.
- 21.Brown, L. D. (2000) J. Am. Stat. Assoc. 95, 1277-1282. [Google Scholar]
- 22.Strawderman, W. E. (1971) Ann. Math. Stat. 42, 385-388. [Google Scholar]
- 23.Strawderman, W. E. (1974) J. Multivariate Anal. 4, 255-263. [Google Scholar]
- 24.Strawderman, W. E. (2000) J. Am. Stat. Assoc. 95, 1364-1368. [Google Scholar]
- 25.Donoho, D. & Johnstone, I. M. (1998) Ann. Stat. 26, 879-921. [Google Scholar]
- 26.Donoho, D. & Johnstone, I. M. (1999) Stat. Sinica, 9, 1-32. [Google Scholar]
- 27.Donoho, D. L. (1999) Ann. Stat. 27, 859-897. [Google Scholar]
- 28.Anderson, T. W. (1958) An Introduction to Multivariate Statistical Analysis (Wiley, New York).
- 29.Wilks, S. S. (1962) Mathematical Statistics (Wiley, New York).
- 30.Jong, K., Marchiori, E., Meijer, G., van der Vaart, A. & Ylstra, B. (June 16, 2004) Bioinformatics, 10.1093/bioinformatics/bth355. [DOI] [PubMed]
- 31.Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P., Maner, S., Massa, H., Walker, M., Chi, M., et al. (2004) Science 305, 525-528. [DOI] [PubMed] [Google Scholar]
- 32.Fridlyand, J., Snijders, A. M., Pinkel, D., Albertson, D. G. & Jain, A. N. (2004) J. Multivariate Anal. 90, 132-153. [Google Scholar]
- 33.Wang, J., Meza-Zepeda, L. A., Kresse, S. H. & Myklebost, O. (2004) BMC Bioinformatics 5, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, Y. & Guo, S. W. (2004) Front. Biosci. 9, 540-549. [DOI] [PubMed] [Google Scholar]
- 35.Olshen, A. B. & Venkatraman, E. S. (2002) in American Statistical Association Proceedings of the Joint Statistical Meetings (American Statistical Association, Alexandria, VA) pp. 2530-2535.
- 36.Arias-Castro, E., Donoho, D. & Huo, X. (2003) Technical Report 2003-22 (Department of Statistics, Stanford University, Stanford, CA).
- 37.Arias-Castro, E., Donoho, D. & Huo, X. (2003) Technical Report 2003-17 (Department of Statistics, Stanford University, Stanford, CA).
- 38.Donoho, D. & Huo, X. (2001) in Multiscale and Multiresolution Methods, Springer Lecture Notes in Computational Science and Engineering, eds. Barth, T. J., Chan, T. & Haimes, R. (Springer, New York), Vol. 20, pp. 149-196. [Google Scholar]
- 39.Kolaczyk, E. D. (1999) J. Am. Stat. Assoc. 94, 920-933. [Google Scholar]