

Abstract
Cellular factors tightly regulate the architecture of bundles of filamentous cytoskeletal proteins, giving rise to assemblies with distinct morphologies and physical properties, and a similar control of the supramolecular organization of nanotubes and nanorods in synthetic materials is highly desirable. However, it is unknown what principles determine how macromolecular interactions lead to assemblies with defined morphologies. In particular, electrostatic interactions between highly charged polyelectrolytes, which are ubiquitous in biological and synthetic self-assembled structures, are poorly understood. We have used a model system consisting of microtubules (MTs) and multivalent cations to examine how microscopic interactions can give rise to distinct bundle phases in biological polyelectrolytes. The structure of these supramolecular assemblies was elucidated on length scales from subnanometer to micrometer with synchrotron x-ray diffraction, transmission electron microscopy, and differential interference contrast microscopy. Tightly packed hexagonal bundles with controllable diameters were observed for large trivalent, tetravalent, and pentavalent counterions. Unexpectedly, in the presence of small divalent cations, we have discovered a living necklace bundle phase, comprised of 2D dynamic assemblies of MTs with linear, branched, and loop topologies. This new bundle phase is an experimental example of nematic membranes. The morphologically distinct MT assemblies give insight into general features of bundle formation and may be used as templates for miniaturized materials with applications in nanotechnology and biotechnology.
Keywords: cation, like-charge attraction, x-ray
In this article, we present our findings on the assembly behavior of microtubules (MTs), a model nanoscale tubule. MTs are hollow, cylindrical protein polymers, with inner and outer diameters of ≈15 and 25 nm, respectively, involved in a variety of cellular functions, including cell division, intracellular transport, and cell morphology. MTs often assemble into arrays and bundles as in axostyles in protozoa, the cortical array in plants, the mitotic spindle, and neuronal processes (1). MT-associated proteins (MAPs) regulate the interactions between MTs, giving rise to MT bundles with various degrees of order and different MT-MT spacings, both in native biological structures such as axons and dentrites and in in vivo overexpression experiments (2). In vitro experiments show that subtle mutations in MAPs can lead to bundles with radically different structures, converting hexagonally packed bundles into linear chains of MTs (3). However, it is unclear how MAP-controlled, MT-MT interactions lead to bundles with the observed architectures.
Understanding the fundamental mechanisms underlying the nature of the self-assembly of nanometer-scale tubules and rods is also important from a technological perspective. Nanotubes are currently being developed as miniaturized materials with applications as circuitry components, templates for nanosized wires and optical materials, enzyme encapsulation systems and biosensors, and vehicles for chemical, drug, and gene delivery (4, 5). The assembly properties of these nanotubes can have profound effects on their function. For example, the electrical and mechanical properties of carbon nanotubes and conducting polymers are strongly influenced by bundle morphology (6, 7). DNA bundled with cationic lipids are used in gene delivery, and the structure of the bundle phase is related to transfection efficiency (8). Thus, understanding how the morphology of bundles is determined is of both fundamental and practical importance.
Both biological and synthetic water-soluble nanotubes and nanorods are often highly charged. These rod-like polyelectrolytes, including DNA, filamentous actin, MTs, and filamentous viruses, can form bundles in the presence of multivalent cations (9-12). In these systems multivalent counterions condense onto the polyelectrolytes, in a process called Manning condensation, neutralizing most of their charge, which results in a much reduced repulsion (13). Attraction between the polyelectrolytes arises from correlations in condensed counterions on different rods (14), but, despite intensive experimental and theoretical effort, this phenomenon of like-charge attraction is poorly understood. The attraction may be caused by a long-range van der Waals-like interaction caused by transient correlated fluctuations in the condensed counterions (15), a short-range salt bridge-like cross-linking of the filaments by the condensed counterions (16) that may form a Wigner crystal, although that is not necessary (17), or a combination of these effects (18). It is also unknown how these interactions might lead to the observed bundle morphologies; in particular, it is unclear why these biopolymers self-assemble into finite size aggregates instead of macroscopically phase separating and if this is an equilibrium (M. Henle and P. Pincus, personal communication) or nonequilibrium effect (19).
In the presence of large, trivalent, tetravalent, and pentavalent cations, synchrotron small-angle x-ray diffraction (SAXRD) and transmission electron microscopy (TEM) show that taxol-stabilized MTs organize into ordered, hexagonally packed bundles of well defined size, with large wall-to-wall distances between MTs (Fig. 1 Left). These bundles are finite-size, hexagonal, columnar liquid crystals (20). Rare defects in the packing, visible on the nanoscale with TEM, cause the hexagonal bundles to bend and bifurcate on the mesoscale, as seen with differential interference contrast (DIC) microscopy. As the charge of the condensing agent is reduced from plus five, to plus four, to plus three, the bundles become less bent and forked and decrease in diameter, and the distance between MTs increases.
Fig. 1.

3D schematics of higher-order assembly of nanometer-scale MTs. Large trivalent, tetravalent, and pentavalent cations lead to the formation of hexagonal bundles (Left). Small divalent cations lead to the living necklace bundles with linear, branched, and loop morphologies (Right). The distinct bundle phases allow for tailored applications in miniaturized materials requiring high volume (hexagonal bundles) or high surface area (necklace bundles).
A radically different bundle structure, which we refer to as the living necklace bundle phase of MTs, is observed when the condensing ions are small, divalent cations. On the nanometer scale, TEM shows that these necklace bundles are highly flexible in cross section, giving rise to topologically distinct linear, branched, and loop morphologies (Fig. 1 Right). On the Å-length scale, SAXRD reveals the locally 2D and nematic nature of the bundles and the wall-to-wall MT distance. On the μm-length scale, DIC shows that the bundles are straight and rigid. The highly polymorphic nature of the bundles shows that they are living bundles that may dynamically change shape and topology because of thermal fluctuations. These living bundles are finite size, locally 2D membranes with nematic ordering, i.e., they consist of rod-like subunits (MTs) that break symmetry by spontaneously orienting but show only short-range positional ordering. These bundles are an experimental realization of nematic membranes, which have recently been predicted as a new universality class of membrane (21).
The control of bundle morphology demonstrated here should help provide fundamental insight into the nature of like-charge attraction and the general determinates of bundle structure. These higher-order assemblies of MTs are tubular analogs of “vesosomes” currently used in delivery applications (22). Furthermore, the MT bundles may be templated to form nanowires and sieves of controlled dimensions or as chemical and gene encapsulation systems. Varying the number of MTs per bundle, or using hexagonal bundles, which have a large internal volume, or living necklace bundles, which have a large surface area, allows the creation of bundles with physical properties tailored for different applications.
Materials and Methods
Materials. MT protein (MAPs plus tubulin) was purified from bovine brains by three cycles of polymerization/depolymerization and tubulin was purified from the MT protein mixture by phosphocellulose chromatography as described (23). MTs were polymerized from tubulin at ≈4 mg/ml in 50 mM Pipes (pH 6.8, adjusted with 80 mM NaOH), 1 mM MgCl2, 1 mM EGTA, 1 mM GTP, and 5% glycerol by incubating them in a 37°C water bath for 20 min and then stabilized by the addition of 40 μM taxol. MTs were sedimented through a sucrose cushion to remove unpolymerized tubulin dimers. Samples were made by diluting MTs with equal volume of cation solution, so final buffer and taxol concentrations were half those listed above.
X-Ray Scattering. Small-angle x-ray scattering experiments were performed at beamline 4-2 of the Stanford Synchrotron Radiation Laboratory. The scattering was done at 8.98 keV with a beam size of 0.2 × 0.2 mm and sample-to-detector distances of ≈2.2 m, determined by using silver behenate as a standard. A charge-coupled device-based area detector (MarCCD165, Mar USA, Evanston, IL) was used. The samples showed powder scattering, so images were averaged over 360° to obtain plots of scattering intensity vs. momentum transfer. Scans were performed for an average of 12 min, over which time no sample damage occurred, which was explicitly checked for by performing multiple short scans. Preliminary experiments were performed on a custom-built rotating anode small-angle x-ray scattering set-up. X-ray samples were centrifuged at 16,000 × g for 1 h and transferred to 1.5-mm quartz capillaries.
Optical Microscopy. A Nikon Diaphot 300 microscope with a Sutter Instruments (Novato, CA) Lambda LS xenon arc, an oil condenser, an oil, 1.4 numerical aperture, ×60 objective with an additional ×4 eye piece, and a Dage-MTI (Michigan City, IN) VE 1000 camera were used for high-resolution, video-enhanced DIC. Background subtraction, image enhancement, and two-frame averaging were performed with a Dage-MTI DSP-2000.
Electron Microscopy. TEM experiments were performed at 80 kV. Whole-mount samples were transferred to carbon-coated grids and stained with uranyl acetate. Samples for thin sections were centrifuged to a pellet at 16,000 × g for 1 h. Pellets were fixed with 2% glutaraldehyde and 0.2% tannic acid overnight, postfixed with 0.75% (wt/vol) OsO4 for 1 h, stained en bloc with 1% uranyl acetate for 1 h, dehydrated with acetone, embedded in spur plastic, and cut to ≈70 nm.
Results and Discussion
With low amounts of added cation the MTs remain unaggregated. MT bundles begin to form above a critical concentration (Cc) of cation, measured by DIC and SAXRD. Divalent cations, BaCl2 (Cc = 60 ± 10 mM), CaCl2 (Cc = 40 ± 10 mM), and SrCl2 (Cc = 60 ± 10 mM), trivalent cations, spermidine (Cc = 7.5 ± 1.5 mM) and oligolysine-three (Cc = 23 ± 0.75 mM), tetravalent cations, spermine (Cc = 1.5 ± 1 mM) and oligolysine-four (Cc = 2.5 ± 2 mM), and a pentavalent cation, oligolysine-five (Cc = 0.75 ± 0.25 mM) all were used to create MT bundles. No bundles were observed with monovalent salt (KCl and NaCl) as high as 500 mM or with the divalent ions MgCl2 and oligolysine-two. Taxol-stabilized MTs were unstable with low concentrations of CoCl2, MnCl2, ZnCl2, GdCl3, or cobalt hexamine. We have not observed a resolubilization of MTs at high concentrations of condensing cations, as has been seen with DNA (24), but at high salt concentrations all cations used caused the MTs to dissociate, with the tubulin subunits sometimes self-assembling into other highly-ordered structures (M.A.O.-L., D.J.N., U. Raviv, H.P.M., Y. Li, L.W., and C.R.S., unpublished results).
The mesoscopic structure of MT bundles is shown in video-enhanced DIC images (Fig. 2A) for large trivalent, tetravalent, and pentavalent condensing cations. Bundles formed with large cations bend and bifurcate on a length scale much smaller than the persistence length of MTs (≈1 mm). The bundles are rigid and do not fluctuate; the bends are static. Higher valent ions result in bundles that are more bent and these bundles appear thicker (they are brighter), but the widths of the bundles are below the optical resolution limit (≈250 μM). Individual MTs can easily be resolved by TEM, which clearly shows that the bundles are thick, with MTs tightly packed into a hexagonal array (Fig. 2B). Although the bundles are mostly well ordered, they do contain infrequent defects (Fig. 2B, arrow). The presence of defects provides a natural explanation for the quenched bending and forking of the MT bundles observed optically (Fig. 2 A), which may be analogous to the defect-mediated distortion of crystalline membranes (25) or the mechanical instabilities of the hexagonal phases of semiflexible chains (26).
Fig. 2.

Micron and nanometer scale images of the hexagonal bundle phase of microtubules. (A) DIC optical micrographs of hexagonal MT bundles with 3+ (20 mM spermidine), 4+ (5 mM spermine), and 5+ (2.5 mM oligolysine-five) counterions. (B) Plastic-embedded TEM cross section (Upper) and whole-mount TEM side view of hexagonal MT bundles (10 mM spermine) (Lower). A 3D schematic is shown in Fig. 1 Left.
In contrast, as shown in Fig. 3A, bundles created with small divalent cations are straight and unbranched on the μm-length scale and bend slightly in solution because of thermal fluctuations. TEM side views show that they are loosely packed sheets and dimers on the scale of tens of nm (Fig. 3A). Cross-sectional TEM images reveal that, on this length scale, the locally linear, raft-like nature of these bundles may result in a wide variety of higher-order structures with differing topology (Fig. 3 B-D). Some bundles, including MT dimers, are purely linear sheets with varying degrees of curvature (Fig. 3B). Many bundles display multiple branches in cross section, resulting in long necklace-like structures (Fig. 3C). The necklace bundles may close back on themselves to form loops, sometimes with multiple handles (Fig. 3D). These vesicular MT aggregates are reminiscent of the tubular phase of anisotropic membranes (27). The highly polymorphic nature of these bundles, caused by the observed structures having similar energies, indicates that they are very flexible in cross section. Thermal fluctuations will thus allow bundles with different morphologies to interconvert, showing that this is a phase of constantly changing bundles. No theory predicts the existence of these living necklace bundles of rod-like polyelectrolytes, which are very different from the larger, well-ordered, tight bundles seen with higher valent cations.
Fig. 3.

Micron and nanometer scale images of the necklace bundle phase of microtubules. (A) DIC (Right), whole-mount TEM side view (Upper Left), and plastic-embedded TEM side view (Lower Left) of MT necklace bundles with 100 mM BaCl2. (B-D) Plastic-embedded TEM cross sections of bundles with 100 mM BaCl2 showing linear (B), branched (C), and loop-like (D) morphologies. A 3D schematic is shown in Fig. 1 Right.
We have performed a series of synchrotron SAXRD experiments to gain further insight into the Å-scale structure of the MT bundles. Fig. 4A shows representative raw SAXRD scans that have been integrated 360° from a powder pattern on a 2D detector and are displayed as a function of the scattering vector, q. To quantitatively model these data, we have subtracted a background that consists of a polynomial that passes through the minimum of the scattering intensities (Fig. 4B). The MTs are modeled as hollow cylinders with an outer radius of 12.9 nm and a wall thickness of 3.2 nm (Fig. 4B, No Cation), in good agreement with the outer radius expected for MTs with 13 protofilaments (28).
Fig. 4.

Synchrotron x-ray scattering data of the angstrom and nanometer scale structure of the hexagonal bundle and necklace bundle phases of microtubules. (A) Raw SAXRD scattering data for MTs with no cation, 115 mM BaCl2 (2+), 15 mM spermidine (3+), 5 mM spermine (4+), or 5 mM oligolysine-five (5+) with hexagonal bundle peaks indexed. (B) Data in A after background subtraction (dots) with fitted model scattering curves (lines); see text. (C) Raw SAXRD data of MTs with 5 mM oligolysine-five, half the standard buffer concentration, and various amounts of added KCl. (D) Data in C after background subtraction (dots) with fitted model scattering curves (lines); see text. (E) Summary of SAXRD scattering fit parameters of MT bundles with CaCl2, SrCl2, BaCl2, oligoamines (spermidine and spermine), oligolysines, and oligolysine-five with half the standard buffer concentration and various amounts of added KCl. The MT wall-to-wall distance was determined by subtracting the MT diameter from the measured MT center-to-center distance. The number of MTs in cross section per bundle, for the hexagonal bundles, was computed by dividing the measured bundle cross-sectional area, L, (see text) by the cross-sectional area of a single MT.
The tight bundle phase, for ions with valence five to three, are modeled as a collection of hollow cylinders, with the dimensions given above, packed into a hexagonal lattice with a center-to-center distance of 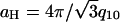 , which results in diffraction peaks at reciprocal lattice vectors G of q10,
, which results in diffraction peaks at reciprocal lattice vectors G of q10,  , q20 = 2q10,
, q20 = 2q10, 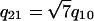 ,
,  , q30 = 3q10,
, q30 = 3q10, 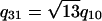 , and q40 = 4q10. The x-ray data could not be well fit by using Gaussian or Lorentzian structure factor lineshapes, but Lorentzian-squared peaks, ∝ 1/(w2 +|q - G|2)2, for the structure factor multiplied by the MT form factor and subsequently powder-averaged, gave an excellent fit to the data (Fig. 4B, 3+, 4+, 5+). The fitting parameters for each x-ray scan were the MT spacing, aH, one peak width for all of the peaks, w, and a separate amplitude for each peak. The fact that only one width was needed to fit all of the peaks in a given x-ray scan shows that the peak width is determined by the finite size of the bundles and that defects in the hexagonal packing are rare (29), as TEM also indicates. The average bundle thickness can be determined from w by using Warren's approximation (30), in which a finite lattice of linear dimension L very close to a reciprocal lattice vector G yields a structure factor
, and q40 = 4q10. The x-ray data could not be well fit by using Gaussian or Lorentzian structure factor lineshapes, but Lorentzian-squared peaks, ∝ 1/(w2 +|q - G|2)2, for the structure factor multiplied by the MT form factor and subsequently powder-averaged, gave an excellent fit to the data (Fig. 4B, 3+, 4+, 5+). The fitting parameters for each x-ray scan were the MT spacing, aH, one peak width for all of the peaks, w, and a separate amplitude for each peak. The fact that only one width was needed to fit all of the peaks in a given x-ray scan shows that the peak width is determined by the finite size of the bundles and that defects in the hexagonal packing are rare (29), as TEM also indicates. The average bundle thickness can be determined from w by using Warren's approximation (30), in which a finite lattice of linear dimension L very close to a reciprocal lattice vector G yields a structure factor 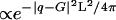 , giving
, giving 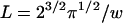 for Lorentzian squared peaks.
for Lorentzian squared peaks.
From the x-ray scans (Fig. 4 A and B) it is qualitatively clear that the peak width increases with decreasing valance, thus the bundle size decreases, and quantitative fits to the data bear this out (Fig. 4E). The bundles vary from ≈14 MTs in cross section for oligolysine-five to ≈8 MTs for oligolysine-three and spermidine (Fig. 4E Lower). The bundle size measured from SAXRD is consistent with TEM measurements (Fig. 2B). Within a specific family of cation (oligolysines or the oligoamines spermine and spermidine), the separation between MTs increases with decreasing cation charge, but mixtures with the oligoamines result in bundles with MT spacings that are consistently smaller than the spacing seen with oligolysines of the same charge (Fig. 4E Upper). The oligoamines are physically smaller than oligolysines of the same charge and have a lower Cc.
SAXRD scans of bundles assembled with divalent ions display very broad peaks (Fig. 4A, 2+), in contrast with the tight bundle phase. Indeed, instead of the hexagonal bundles observed with larger multivalent ions, detailed analysis shows that the SAXRD data can be quantitatively modeled as arising from a dimer of MTs. The only fit parameter is the MT-MT spacing (Fig. 4E Upper, Ca2+, Ba2+, and Sr2+). This finding is consistent with TEM results where cross sections show that there are only near-neighbor correlations between MTs (Fig. 3 B-D). The measured bundle size and MT wall-to-wall spacings appear to be equilibrium values as SAXRD reveals no differences in samples prepared by multiple pathways, starting from dilute (0.2 mg/ml) or concentrated (≈50 mg/ml) MT solutions, or samples aged for up to 1 week. The bundling is reversible; if MT bundles are centrifuged into a pellet and resuspended in buffer with no cation, the MTs become unbundled.
The structures of the cation-MT bundle phases are not sensitive to the concentration of condensing cation when all MTs are in the bundle phase, unlike what has been recently observed for DNA with low ionic strength (31). However, the addition of monovalent salt dramatically affects the MT bundles and causes the MT-MT spacing to swell (Fig. 4E Upper) and the number of MTs per bundle to decrease (Fig 4E Lower). The shift in sparing is seen in the SAXRD data in Fig. 4D where the diffraction peaks move to lower q, implying increased MT-MT spacing as KCl is increased [most easily observed by following the progression of the (2,1) peak, marked by the dashed line]. The MT bundles “melt” with 250 mM KCl added (Fig. 4D).
Conclusion
The structures of the MT bundle phases are stabilized by a balance of hydration and long-range electrostatic repulsive forces with attractive forces caused by van der Waals and ion correlations. The smallest wall-to-wall MT distances we measured were ≈3 nm, which is similar to the separation spontaneously assumed between lipid bilayers caused by a balance of van der Waals attraction and hydration repulsion (32). However, the maximum wall-to-wall distances we observed were >5.5 nm, and it is unclear what repulsive force could stabilize these large separations, because hydration repulsion is negligible at these distances and all theories of like-charge attraction predict very small mean separation (13-19). The stability of these large separations may be caused by the large radius of MTs, the presence of monovalent salt (in the buffer), the polyamphiphatic nature of MTs, or the nature of the distribution of charges on the tubulin surface (such as the unstructured, highly charged C terminus).
The bundles studied here have been created through nonspecific interactions so the living necklace bundle phase is likely to be a general feature of rod-like polyelectrolytes, as is the hexagonal bundle phase. These living necklace bundles are highly anisotropic self-assembled membranes of MTs with nematic in-plane ordering and varying topology. In addition to providing insight into the fundamental physics of rod-like polyelectrolytes, the control of bundle morphology demonstrated here, which depends on the charge and shape of the condensing ion, may help to assemble nanostructures for engineering and biomedical applications.
This work should motivate future theoretical models, which explore the possibility that the hexagonal and necklace bundles result from competing short-range attraction and long-range repulsive forces between MTs. For example, long-range repulsion between MTs (caused by electrostatics and osmotic pressures of the Na+ and Cl- ions present in experiments for charge neutrality of the macromolecules and the counterions) would favor the loosely bundled necklace phase, whereas short-range attraction (caused by counterions) favors the hexagonal bundle with more salt bridge-like contacts between an MT and its nearest neighbors. The model study presented in this article leading to distinct bundle phases qualitatively similar to those observed in vivo may be viewed as a step toward developing a better understanding of MAP-mediated MT assemblies in vivo.
This study demonstrates that the combination of quantitative reciprocal space synchrotron SAXRD measurements of statistically averaged structures and real-space imaging with electron and optical microscopy, can elucidate the noncrystalline structure of supramolecular assemblies of MTs in vitro. Thus, this work paves the way for future studies of noncrystalline structures of MT bundles and networks resulting from the interactions between MAPs and MTs under physiologically relevant conditions in vitro.
Acknowledgments
We thank M. Henle, P. Pincus, C. Santangelo, and Y. Rabin for insightful conversation regarding like-charged attraction and bundle structure. This work was supported by National Science Foundation Grant DMR 0203755 and National Institutes of Health Grants GM-59288 and NS-13560. Additional support was provided by National Science Foundation Grants CTS 0404444 and CTS 0103516 and the Department of Energy's Office of Basic Energy Sciences under Contract W-7405-ENG-36 with the University of California. U.R. acknowledges the support of the International Human Frontier Science Program Organization. The Materials Research Science and Engineering Center at the University of California, Santa Barbara, is supported by National Science Foundation Grant DMR-0080034. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Stanford Synchrotron Radiation Laboratory Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, National Institutes of Health, National Center for Research Resources, and Biomedical Technology Program.
Author contributions: D.J.N. and C.R.S. designed research; D.J.N., M.A.O.-L., and U.R. performed research; D.J.N. and C.R.S. analyzed data; H.P.M. and L.W. contributed new reagents/analytic tools; and D.J.N. and C.R.S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MT, microtubule; MAP, MT-associated protein; SAXRD, small-angle x-ray diffraction; TEM, transmission electron microscopy; DIC, differential interference contrast; Cc, critical concentration.
References
- 1.Bray, D. (2001) Cell Movements (Taylor & Francis, New York).
- 2.Chen, J., Kanai, Y., Cowan, N. J. & Hirokawa, N. (1992) Nature 360, 674-676. [DOI] [PubMed] [Google Scholar]
- 3.Lida, J., Itoh, T. J., Hotani, H., Nishiyama, K., Murofushi, H., Bulinski, J. C. & Hisanaga, S. (2002) J. Mol. Biol. 320, 97-106. [DOI] [PubMed] [Google Scholar]
- 4.White, G. M., Jr. & Love, J. C. (2001) Sci. Am. 285, 38-47. [Google Scholar]
- 5.Martin, C. R. (1994) Science 266, 1961-1966. [DOI] [PubMed] [Google Scholar]
- 6.Thess, A., Lee, R., Nikolaev, P., Dai, H. J., Petit, P., Robert, J., Xu, C. H., Lee, Y. H., Kim, S. G., Rinzler, A. G., et al. (1996) Science 273, 483-487. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser, A. B. (2001) Rep. Prog. Phys. 64, 1-49. [Google Scholar]
- 8.Koltover, I., Salditt, T., Radler, J. O. & Safinya, C. R. (1998) Science 281, 78-81. [DOI] [PubMed] [Google Scholar]
- 9.Tang, J., Wong, S., Tran, P. T. & Janmey, P. A. (1996) Ber. Bunsenges. Phys. Chem. 100, 796-806. [Google Scholar]
- 10.Vater, W., Bohm, K. J. & Unger, E. (1997) Cell Motil. Cytoskeleton 36, 76-83. [DOI] [PubMed] [Google Scholar]
- 11.Bloomfield, V. A. (1991) Biopolymers 31, 1471-1481. [DOI] [PubMed] [Google Scholar]
- 12.Wong, G. C. L., Lin, A., Tang, J. X., Li, Y., Janmey, P. A. & Safinya, C. R. (2003) Phys. Rev. Lett. 91, 018103-1-4. [DOI] [PubMed] [Google Scholar]
- 13.Levin, Y. (2002) Rep. Prog. Phys. 65, 1577-1632. [Google Scholar]
- 14.Grosberg, A. Y., Nguyen, T. T. & Shklovskii, B. I. (2002) Rev. Mod. Phys. 74, 329-345. [Google Scholar]
- 15.Oosawa, F. (1968) Biopolymers 6, 1633-1647. [Google Scholar]
- 16.Gronbech-Jensen, N., Mashl, R. J., Bruinsma, R. F. & Gelbart, W. M. (1997) Phys. Rev. Lett. 78, 2477-2480. [Google Scholar]
- 17.Netz, R. R. (2001) Eur. Phys. J. 5, 557-574. [Google Scholar]
- 18.Lau, A. W. C., Pincus, P., Levine, D. & Fertig, H. A. (2001) Phys. Rev. E 63, 051604. [DOI] [PubMed] [Google Scholar]
- 19.Ha, B. Y. & Liu, A. J. (1999) EuroPhys. Lett. 46, 624-630. [Google Scholar]
- 20.Selinger, J. V. & Bruinsma, R. F. (1991) Phys. Rev. A 43, 2910-2921. [DOI] [PubMed] [Google Scholar]
- 21.Xing, X., Mukhopadhyay, R., Lubensky, T. C. & Radzihovsky, L. (2003) Phys. Rev. E 68, 021108. [DOI] [PubMed] [Google Scholar]
- 22.Walker, S. A., Kennedy, M. T. & Zasadzinski, J. A. (1997) Nature 387, 61-64. [DOI] [PubMed] [Google Scholar]
- 23.Derry, W. B., Wilson, L. & Jordan, M. A. (1995) Biochemistry 34, 2203-2211. [DOI] [PubMed] [Google Scholar]
- 24.Raspaud, E., de la Cruz, M. O., Sikorav, J. L. & Livolant, F. (1998) Biophys. J. 74, 381-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson, D. R. (2001) Defects and Geometry in Condensed Matter Physics (Cambridge Univ. Press, Cambridge, U.K.).
- 26.Kleman, M. & Oswald, P. (1982) J. Phys. 43, 655-662. [Google Scholar]
- 27.Radzihovsky, L. & Toner, J. (1995) Phys. Rev. Lett. 75, 4752-4755. [DOI] [PubMed] [Google Scholar]
- 28.Li, H. L., DeRosier, D. J., Nicholson, W. V., Nogales, E. & Downing, K. H. (2002) Structure (London) 10, 1317-1328. [DOI] [PubMed] [Google Scholar]
- 29.Guiner, A. (1994) X-Ray Diffraction In Crystals, Imperfect Crystals, and Amorphous Bodies (Dover, New York).
- 30.Warren, B. E. (1941) Phys. Rev. 59, 693-698. [Google Scholar]
- 31.Conwell, C. C., Vilfan, I. D. & Hud, N. V. (2003) Proc. Natl. Acad. Sci. USA 100, 9296-9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lis, L. J., McAlster, M., Fuller, N., Rand, R. P. & Parsegian, V. A. (1982) Biophys. J. 37, 657-665. [PMC free article] [PubMed] [Google Scholar]