

Abstract
Bacterial sepsis is a major killer in hospitalized patients. Coagulase-negative staphylococci (CNS) with the leading species Staphylococcus epidermidis are the most frequent causes of nosocomial sepsis, with most infectious isolates being methicillin-resistant. However, which bacterial factors underlie the pathogenesis of CNS sepsis is unknown. While it has been commonly believed that invariant structures on the surface of CNS trigger sepsis by causing an over-reaction of the immune system, we show here that sepsis caused by methicillin-resistant S. epidermidis is to a large extent mediated by the methicillin resistance island-encoded peptide toxin, PSM-mec. PSM-mec contributed to bacterial survival in whole human blood and resistance to neutrophil-mediated killing, and caused significantly increased mortality and cytokine expression in a mouse sepsis model. Furthermore, we show that the PSM-mec peptide itself, rather than the regulatory RNA in which its gene is embedded, is responsible for the observed virulence phenotype. This finding is of particular importance given the contrasting roles of the psm-mec locus that have been reported in S. aureus strains, inasmuch as our findings suggest that the psm-mec locus may exert effects in the background of S. aureus strains that differ from its original role in the CNS environment due to originally “unintended” interferences. Notably, while toxins have never been clearly implied in CNS infections, our tissue culture and mouse infection model data indicate that an important type of infection caused by the predominant CNS species is mediated to a large extent by a toxin. These findings suggest that CNS infections may be amenable to virulence-targeted drug development approaches.
Author Summary
Coagulase-negative staphylococci (CNS) are the leading cause of sepsis in hospitalized patients, causing a significant number of deaths. This situation is further worsened by a limitation of therapeutic options due to the fact that most CNS infectious isolates are resistant to methicillin. CNS sepsis has been assumed to be due to on over-reacting immune response triggered by invariant bacterial surface structures. By using tissue culture and animal infection model-based evidence, we here show that in contrast to that notion, the PSM-mec toxin produced by methicillin-resistant strains of the leading CNS species Staphylococcus epidermidis has a strong impact on the severity of sepsis and its outcome. This is the first report to link a toxin to the pathogenesis of the most frequent bacterial cause of sepsis. Notably, these findings pave the way for anti-virulence strategies against this widespread and deadly type of infection.
Introduction
Bacterial sepsis is a frequent cause of death in hospitalized patients. Coagulase-negative staphylococci (CNS) are the leading cause of nosocomial sepsis, especially in neonates [1–3]. CNS sepsis most often originates from the infection of indwelling medical devices, such as in catheter-related bloodstream infections (CRBSIs) or central line-associated blood stream infections (CLABSIs) [4]. Most prominent among CNS infections are those due to the skin commensal Staphylococcus epidermidis [5]. However, the bacterial factors contributing to the development of sepsis, in particular in CNS, are poorly understood.
Given that toxins have long been assumed to be widely absent from CNS [6], sepsis caused by S. epidermidis and other CNS, similar to other Gram-positive bacteria, has so far been believed to be due predominantly to an overwhelming immune reaction directed against invariable, pro-inflammatory cell surface molecules, such as teichoic acids and lipopeptides [7]. Recently, the notion that CNS do not commonly produce toxins had to be revised with the discovery of the pro-inflammatory and cytolytic phenol-soluble modulin (PSM) staphylococcal toxin family [8]. However, due to the difficulties associated with genetic manipulation of S. epidermidis and other CNS, the roles of PSMs in CNS infections, including most notably sepsis, have hitherto remained unexplored.
Most S. epidermidis blood infections are caused by methicillin-resistant strains (MRSE), with methicillin resistance rates even exceeding those found among S. aureus [9]. Methicillin resistance is encoded on so-called staphylococcal chromosome cassette (SCC) mec mobile genetic elements, which are believed to have originated from CNS, from where they were transferred to S. aureus [10]. While other PSMs are core-genome encoded [8], one PSM toxin, called PSM-mec, is encoded within SCCmec elements of subtypes II, III, and VIII [11, 12]. The psm-mec gene is embedded in a short regulatory (sr) RNA, which in S. aureus has been reported to down-regulate the production of other PSMs and thereby decrease virulence [13, 14]. While this effect has been claimed to generally explain lower virulence of hospital-associated as compared to community-associated MRSA strains [13], it is quite moderate and extensively strain-dependent [11, 13]. Recently, the psm-mec locus has been introduced on a plasmid into some CNS that naturally lack psm-mec, and was reported to trigger gene regulatory changes [15]; but the roles that the PSM-mec peptide or the psm-mec srRNA naturally play in CNS including S. epidermidis are unknown.
Here we analyzed the role of the psm-mec locus in S. epidermidis sepsis by using tissue culture and animal infection models. Our findings show for the first time that a toxin can have a strong impact on CNS sepsis, setting the stage for anti-virulence strategies directed against this frequent and deadly infection.
Results and Discussion
To analyze the impact of the psm-mec locus on S. epidermidis sepsis, we produced isogenic psm-mec deletion mutants (Δpsm-mec) in two MRSE strains, a clinical isolate (SE620) and the genome-sequenced strain RP62A. PSM-mec production in these strains is representative of clinical PSM-mec-positive MRSE (S1 Fig), which we determined in a clinical S. epidermidis strain collection from Norway to occur in ~ 2/3 (59/91) of the ~ 50% (91/180) methicillin-resistant S. epidermidis. We also introduced a point mutation in the start codon of the psm-mec gene in the genome of strain SE620 to differentiate between effects mediated by the PSM-mec peptide versus those due to the psm-mec srRNA (psm-mec*). Notably, the stability of the psm-mec RNA was not significantly altered by introduction of the 1-basepair start codon mutation (S2 Fig).
We first analyzed those mutants in a murine sepsis model. Mortality was significantly reduced in the Δpsm-mec mutants of both strains (Fig 1A and 1B). There was no significant difference between the Δpsm-mec mutant and the psm-mec* start codon mutant (Fig 1A). Furthermore, CFU in the blood and the kidneys were strongly reduced in the Δpsm-mec mutants of both strains and the psm-mec* start codon mutant (Fig 1C–1F). These results demonstrate a strong contribution of the PSM-mec toxin to bacteremia and mortality due to S. epidermidis sepsis, while the psm-mec srRNA did not show any impact.
Fig 1. Mouse sepsis model.

Female, 6–10 weeks old, C57BL/6NCRl mice (n = 5 for all groups except SE620Δpsm-mec and SE620psm-mec*, n = 10) were injected via the tail vein with 5 x 108 CFU of the indicated bacterial strains and monitored for disease development every 8 h for up to 120 h. (A,B) Survival curves; (C,D) CFU in blood at 12 h; (E,F) CFU in kidneys at 12 h. Statistical analysis is by Log-rank (Mantel-Cox) tests for survival curves, otherwise using 1-way ANOVA with Bonferroni post-tests. Error bars show ±SEM. N.S., not significant. Δpsm-mec, isogenic psm-mec deletion mutant; psm-mec*, psm-mec gene start codon mutant.
We showed previously that synthetic PSM-mec peptide is strongly pro-inflammatory and has moderate to strong cytolytic capacity [12]. To analyze the contribution that the psm-mec locus has to pro-inflammatory and cytolytic capacity in the S. epidermidis background, we measured cytokine concentrations during experimental murine sepsis and determined cytolytic capacity of the bacterial strains toward human neutrophils in vitro. Cytokine concentrations during sepsis are the result of a systemic reaction due to several immune cell types, and are thus best determined in vivo, while cytolytic capacity can be most accurately measured in vitro.
The PSM-mec peptide, but not the psm-mec srRNA, had a strong and significant impact on the production of cytokines during murine sepsis (Fig 2). At 12 h after infection, the mouse IL-8 homologue CXCL1 was significantly reduced when mice were infected with the Δpsm-mec or psm-mec* start codon mutant of strain SE620, to about half the concentration measured in mice infected with the wild-type strain (Fig 2A). Concentrations of IL-1β and TNF-α were even more strongly reduced to levels not significantly different from those measured in mock (PBS) infected animals (Fig 2B and 2C). In the RP62A background, the phenotypes were similar, with differences being more pronounced during earlier stages of the infection (measured at 2 versus 12 h) (Fig 3). These results showed that the cytokine storm that commonly accompanies bacterial sepsis is strongly dependent on the PSM-mec toxin in S. epidermidis.
Fig 2. Mouse sepsis model, cytokine concentrations, strain SE620.

Cytokine concentrations (A, CXCL1, B, IL-1β, C, TNF-α) were determined at 12 h post infection in the mouse sepsis model using commercial ELISA kits. Statistical analysis is by 1-way ANOVA with Bonferroni post-tests. Error bars show ±SEM. ****, P<0.0001. Δpsm-mec, isogenic psm-mec deletion mutant; psm-mec*, psm-mec gene start codon mutant.
Fig 3. Mouse sepsis model, cytokine concentrations, strain RP62A.
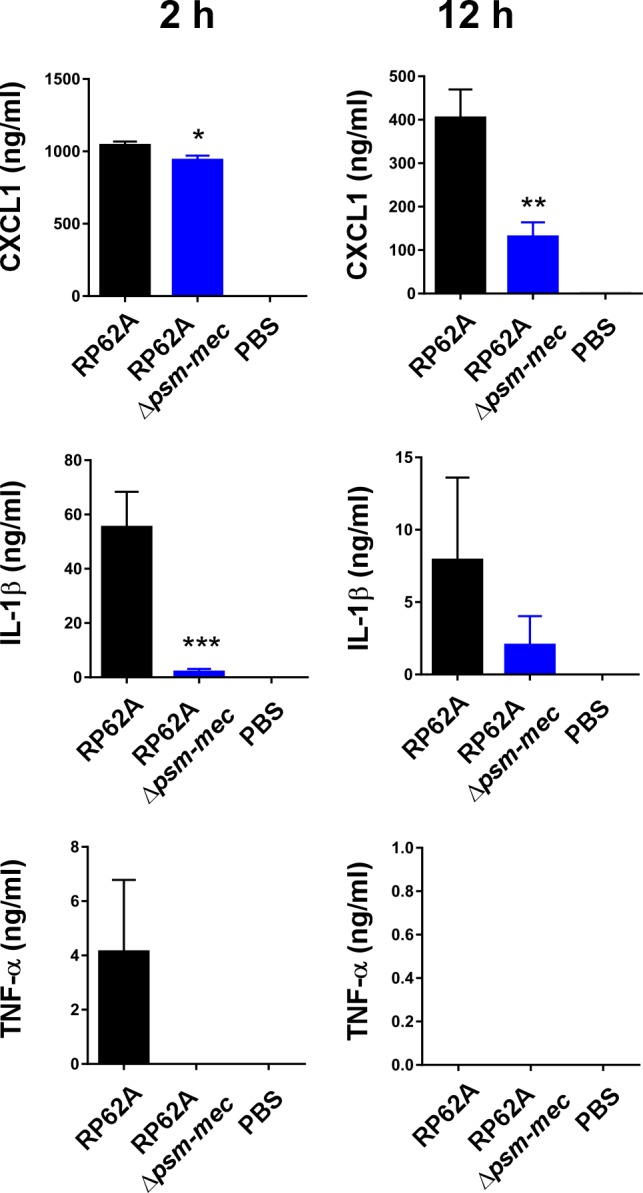
Cytokine concentrations in the blood of mice infected with RP62A or its psm-mec deletion mutant at 2 and 12 h after infection. Statistical analysis is by 1-way ANOVA; multiple comparisons using Bonferroni post-tests. Error bars show ±SEM. *, P<0.05; **, P<0.01; ***, P<0.001. Statistical results are only shown for the RP62A versus psm-mec mutant comparison. Note TNF-α concentrations were below the detection limit at 12 h. Δpsm-mec, isogenic psm-mec deletion mutant; psm-mec*, psm-mec gene start codon mutant.
In addition to being pro-inflammatory, the PSM-mec toxin has pronounced cytolytic capacity [12]. Cytolysis by PSMs is believed to be most important for infection when bacteria are engulfed in the phagosome of neutrophils and other phagocytes [16, 17]. Survival of bacteria when incubated with human neutrophils and survival in whole human blood was significantly higher with the S. epidermidis wild-type strain than with Δpsm-mec or psm-mec* start codon mutants, as was killing of neutrophils when incubated with whole bacteria (Fig 4), emphasizing the role of the PSM-mec toxin in evasion of neutrophil killing and resistance to the strong bactericidal capacities of immune defense mechanisms in human blood. Together, these results indicate that the both the pro-inflammatory and cytolytic capacities of the PSM-mec peptide contribute to the development of S. epidermidis sepsis.
Fig 4. Bacterial survival during incubation with human neutrophils and in whole human blood.

(A,B) Survival in whole, heparinized human blood. (C,D) Survival during incubation with human neutrophils (MOI 10:1). (E,F) Killing of human neutrophils (MOI 100:1). Error bars show ±SEM. *, P <0.05, **, P <0.01. ***, P <0.001. ****, P <0.0001 (unpaired t-tests for RP62A; 1-way ANOVA with Bonferroni post-tests for SE620). Δpsm-mec, isogenic psm-mec deletion mutant; psm-mec*, psm-mec gene start codon mutant. In (A) and (C), no comparisons between SE620Δpsm-mec and SE620psm-mec* were statistically significant.
In S. aureus, the psm-mec locus has also been implicated in biofilm-forming capacity, although effects were generally minor and highly strain-dependent [12]. Similar to S. aureus, biofilm formation in S. epidermidis was affected only slightly by the psm-mec locus, and as this was seen only in one strain, similarly strain-dependent (Fig 5). In that strain, SE620, the effect was due to the PSM-mec peptide, not the psm-mec srRNA. These findings indicate that during indwelling medical device-associated blood stream infections by S. epidermidis, the impact of PSM-mec generally is by contributing to the development of sepsis, as we have shown here, rather than by promoting biofilm formation on the device itself.
Fig 5. Impact of psm-mec on biofilm formation in S. epidermidis.
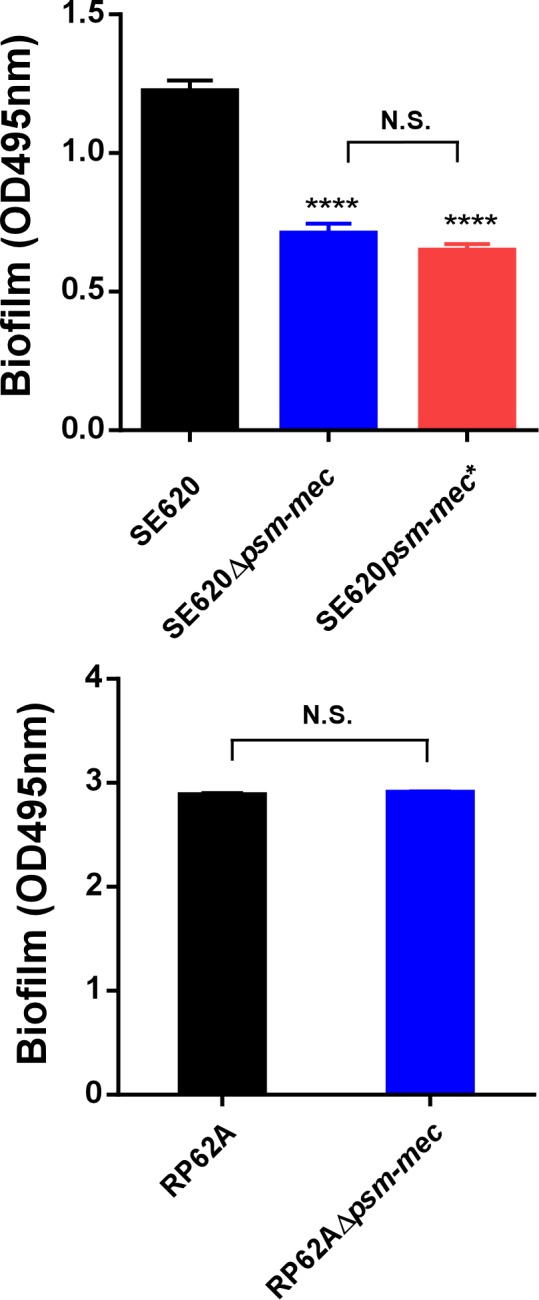
Biofilm formation by S. epidermidis strains and isogenic psm-mec deletion and psm-mec start codon mutants. Biofilm formation was measured using a semi-quantitative microtiter plate assay. 24 wells per group were measured. ****, P<0.0001 (1-way ANOVA, Bonferroni post tests vs. wild-type strain); N.S., not significant. Error bars show ±SEM. Δpsm-mec, isogenic psm-mec deletion mutant; psm-mec*, psm-mec gene start codon mutant.
Our results showed that the psm-mec srRNA is not involved with sepsis or other relevant virulence phenotypes in S. epidermidis. As a previous study suggested that the psm-mec srRNA leads to gene regulatory changes in S. epidermidis [15], based on the introduction of a psm-mec expressing plasmid into S. epidermidis, we also directly investigated whether the psm-mec locus has a gene regulatory impact in S. epidermidis. The most important regulatory effect of the psm-mec locus in S. aureus, by which the sometimes negative impact of the psm-mec locus on virulence in S. aureus was explained, has been reported to consist in the alteration of the expression of other, core genome-encoded PSMs [18]. PSM expression was altered only to a very low extent in the psm-mec-negative as compared to the wild-type S. epidermidis strains, with changes only significant for some PSMs and never exceeding a factor of ~ 1.5 (Fig 6). This demonstrates that there is only a very minor effect of the psm-mec srRNA on PSM expression when analyzed directly in the S. epidermidis background. Furthermore, we analyzed genome-wide gene expression in the psm-mec mutants of both strains by microarray analysis (Tables 1 and 2). For microarray analysis, strains were grown to the maximum of PSM-mec expression as determined by qRT-PCR (10 h) (Fig 7). While we observed gene regulatory changes that were due to the psm-mec srRNA, they mostly comprised metabolic (e.g., riboflavin and purin/pyrimidine synthesis) rather than virulence genes, and were inconsistent between the two strains. Notably, the results of the previously claimed impact of the psm-mec locus on virulence would be negative [13, 15, 18], contrasting the positive effect we observed in the mouse sepsis model. Such a gene regulatory mechanism can thus be ruled out as underlying psm-mec-mediated development of S. epidermidis sepsis.
Fig 6. PSM production in S. epidermidis strains and isogenic psm-mec deletion and psm-mec start codon mutants.

PSMs were measured in stationary phase (16 h) cultures using RP-HPLC/ESI-MS in triplicate. Error bars show ±SEM. $, Residual PSM-mec production in the SE620 psm-mec start codon mutant is likely due to strong gene expression and usage of a non-canonical start codon, as we previously found also in S. aureus psm-mec start codon mutants [14].
Table 1. Microarray results, RP62A1.
| RP62A | |||
|---|---|---|---|
| Gene | Number2 | Function | RP62AΔpsm-mec vs. RP62A3 |
| pyrB | SERP0766 | aspartate carbamoyltransferase | 18.14 |
| ribE | SERP1327 | riboflavin synthase subunit alpha | 16.04 |
| ribBA | SERP1326 | 3,4-dihydroxy-2-butanone-4-phosphate synthase | 15.16 |
| purM | SERP0656 | phosphoribosylaminoimidazole synthetase | 14.94 |
| purF | SERP0655 | amidophosphoribosyltransferase | 14.77 |
| purN | SERP0657 | phosphoribosylglycinamide formyltransferase | 14.72 |
| serS | SERP2545 | seryl-tRNA synthetase | 13.55 |
| purH | SERP0658 | bifunctional phosphoribosylaminoimidazolecarboxamide formyltransferase/IMP cyclohydrolase | 13.45 |
| purD | SERP0659 | phosphoribosylamine—glycine ligase | 11.92 |
| arcB-2 | SERP2351 | ornithine carbamoyltransferase | 11.33 |
| ribD | SERP1328 | riboflavin biosynthesis protein RibD | 10.99 |
| - | SERP2546 | hypothetical protein | 9.88 |
| ribH | SERP1325 | 6,7-dimethyl-8-ribityllumazine synthase | 9.50 |
| arcC | SERP2352 | carbamate kinase | 9.07 |
| purL | SERP0654 | phosphoribosylformylglycinamidine synthase II | 7.33 |
| - | SERP2381 | NADH:flavin oxidoreductase/fumarate reductase, flavoprotein subunit | 7.27 |
| #N/A | SE2245 | 7.00 | |
| - | SERP2279 | hypothetical protein | 6.65 |
| - | SERP2278 | hypothetical protein | 6.50 |
| - | SERP2380 | drug transporter | 6.26 |
| cysH | SERP2192 | phosophoadenylyl-sulfate reductase | -95.16 |
| sat | SERP2186 | sulfate adenylyltransferase | -18.63 |
| - | SERP0094 | cysteine synthase/cystathionine beta-synthase family protein | -13.93 |
| cysK | SERP0152 | cysteine synthase | -9.84 |
| - | SERP0095 | trans-sulfuration enzyme family protein | -8.77 |
| - | SERP2196 | MarR family transcriptional regulator | -8.64 |
| - | SERP1478 | GntR family transcriptional regulator | -8.26 |
| - | SERP2003 | amino acid ABC transporter ATP-binding protein | -7.99 |
| - | SERP2187 | hypothetical protein | -7.65 |
| - | SERP2195 | alpha keto acid dehydrogenase complex, E3 component, lipoamide dehydrogenase | -7.46 |
| cysC | SERP2185 | adenylylsulfate kinase | -7.41 |
| - | SERP2004 | amino acid ABC transporter permease | -6.80 |
| lacE | SERP1790 | PTS system, lactose-specific IIBC components | -6.80 |
| - | SERP1980 | nitrite extrusion protein | -5.78 |
| cysI | SERP2190 | sulfite reductase subunit beta | -5.74 |
| lacG | SERP1789 | 6-phospho-beta-galactosidase | -5.70 |
| lacF | SERP1791 | PTS system, lactose-specific IIA component | -5.07 |
| - | SERP0056 | hypothetical protein | -4.91 |
| - | SERP2005 | amino acid ABC transporter amino acid-binding protein | -4.86 |
| lacA | SERP1795 | galactose-6-phosphate isomerase subunit LacA | -4.81 |
1 The top 20 down- and up-regulated genes are shown.
2 Gene numbers are for strain RP62A, unless a specific gene is not annotated or exists in that strain, in which case the number for strain ATCC12228 is shown.
3Up-regulation is shown by positive numbers, down-regulation by negative numbers.
Table 2. Microarray results, SE6201.
| SE620 | |||||
|---|---|---|---|---|---|
| Gene | Number2 | Function | SE620Δpsm-mec vs. SE620 | SE620psm-mec* vs. SE620 | SE620Δpsm-mec vs.SE620psm-mec* |
| - | SERP2245 | tributyrin esterase EstA | 4.25 | 1.24 | 3.42 |
| ribE | SERP1327 | riboflavin synthase subunit alpha | 4.18 | 16.69 | -3.99 |
| ribBA | SERP1326 | 3,4-dihydroxy-2-butanone-4-phosphate synthase | 3.62 | 12.67 | -3.50 |
| - | SERP2546 | hypothetical protein | 3.41 | 1.41 | 2.42 |
| ribD | SERP1328 | riboflavin biosynthesis protein RibD | 3.06 | 10.33 | -3.38 |
| - | SERP0664 | hypothetical protein | 3.00 | 1.89 | 1.59 |
| - | SERP2321 | hypothetical protein | 2.92 | 5.19 | -1.77 |
| rbsK | SERP2100 | Ribokinase | 2.82 | 2.29 | 1.23 |
| ribH | SERP1325 | 6,7-dimethyl-8-ribityllumazine synthase | 2.79 | 8.78 | -3.15 |
| purS | SERP0652 | phosphoribosylformylglycinamidine synthase, PurS protein | 2.76 | 2.95 | -1.07 |
| - | SERP1933 | hypothetical protein | 2.71 | 1.65 | 1.64 |
| - | SERP2364 | succinyl-diaminopimelate desuccinylase | 2.66 | 3.38 | -1.27 |
| - | SERP2354 | tributyrin esterase EstA | 2.64 | 1.88 | 1.40 |
| - | SERP1498 | ammonium transporter | 2.58 | 4.47 | -1.74 |
| - | SERP1933 | hypothetical protein | 2.53 | 1.63 | 1.55 |
| mraY | SERP0747 | phospho-N-acetylmuramoyl-pentapeptide-transferase | 2.49 | 1.27 | 1.96 |
| purQ | SERP0653 | phosphoribosylformylglycinamidine synthase I | 2.40 | 2.48 | -1.03 |
| - | SERP2357 | amino acid ABC transporter permease | 2.39 | 2.17 | 1.10 |
| - | SERP1529 | hypothetical protein | 2.34 | -1.90 | 4.46 |
| purF | SERP0655 | amidophosphoribosyltransferase | 2.32 | 2.85 | -1.23 |
| - | SERP0473 | hypothetical protein | -6.11 | -1.43 | -4.28 |
| - | SERP0473 | hypothetical protein | -5.46 | -1.31 | -4.17 |
| - | SERP1474 | hypothetical protein | -5.27 | -11.93 | 2.26 |
| - | SERP1478 | GntR family transcriptional regulator | -4.32 | -11.37 | 2.63 |
| - | SERP2158 | amino acid permease | -3.75 | -3.27 | -1.14 |
| - | SERP0944 | ThiJ/PfpI family protein | -3.71 | -1.71 | -2.16 |
| trpG | SERP0938 | anthranilate synthase component II | -3.65 | -1.87 | -1.96 |
| - | SERP1475 | ABC transporter ATP-binding protein | -3.46 | -8.38 | 2.42 |
| czrA | SERP1755 | CzrA family transcriptional regulator | -3.30 | -2.32 | -1.42 |
| - | SERP0273 | alpha/beta hydrolase | -3.08 | -1.73 | -1.78 |
| - | SERP0507 | CBS domain-containing protein | -3.06 | -1.48 | -2.07 |
| - | SERP2091 | hypothetical protein | -2.94 | -1.32 | -2.22 |
| - | SE0735 | -2.91 | -1.12 | -2.60 | |
| - | SERP1476 | hypothetical protein | -2.91 | -9.68 | 3.33 |
| - | SERP1477 | ABC transporter ATP-binding protein | -2.89 | -9.52 | 3.30 |
| - | SERP0620 | hypothetical protein | -2.80 | -1.04 | -2.69 |
| - | SERP2129 | short chain dehydrogenase/reductase family oxidoreductase | -2.78 | -1.28 | -2.17 |
| - | SE0082 | -2.71 | -1.12 | -2.42 | |
| - | SERP0385 | ABC transporter ATP-binding protein | -2.69 | -1.56 | -1.73 |
| - | SERP1479 | hypothetical protein | -2.66 | -2.94 | 1.11 |
1 The top 20 down- and up-regulated genes are shown. Sorting was by the SE620Δpsm-mec vs. SE620 comparison and the gene expression changes for the same genes for the other comparisons are shown.
2 Gene numbers are for strain RP62A, unless a specific gene is not annotated or exists in that strain, in which case the number for strain ATCC12228 is shown.
3Up-regulation is shown by positive numbers, down-regulation by negative numbers.
Fig 7. Time-dependent expression of psm-mec.
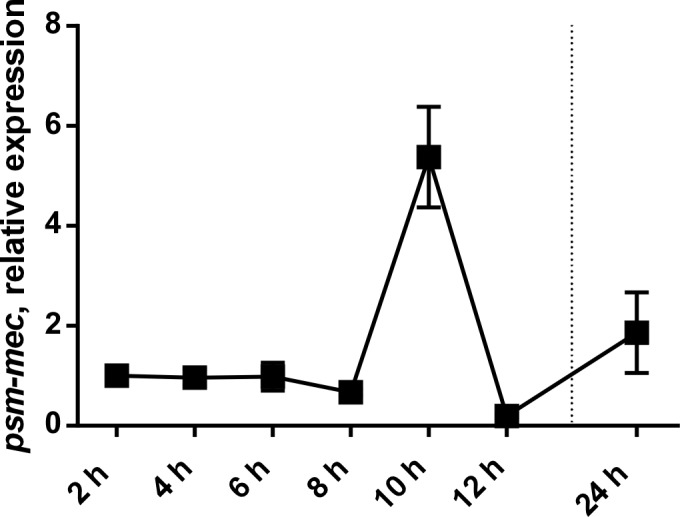
Expression of the psm-mec RNA relative to that of 16S RNA in S. epidermidis RP62A during growth in TSB. The experiment was performed in triplicate. Error bars show ±SEM.
Our results may explain the highly inconsistent phenotypes that have been attributed to psm-mec in S. aureus [11–13], inasmuch as the psm-mec locus may exert effects in the background of S. aureus strains that differ from its original role in the CNS environment. One such possibility that remains to be investigated is that the highly expressed psm-mec mRNA interferes with other DNA or RNA sequences in S. aureus. Furthermore, the psm-mec srRNA barely exceeds the limits of the psm-mec gene [14], which contrasts the only other case of an srRNA with an embedded peptide toxin in staphylococci, namely the well-described regulatory RNAIII of the staphylococcal accessory gene regulator (Agr) system. RNAIII significantly exceeds the boundaries of the embedded PSM peptide gene, hld [19]. Together, these observations suggest that the psm-mec srRNA does not serve a well-defined general purpose in virulence gene regulation.
In conclusion, our study reveals that sepsis due to MRSE is mediated to a large extent by the PSM-mec peptide toxin, representing the first example of a toxin being made responsible for the development of CNS sepsis. Our study was largely based on the investigation of isogenic psm-mec mutants in clinical strains of S. epidermidis, using tissue culture and animal infection models. Future clinical work is needed to assess whether PSM-mec and/or other toxins contribute to sepsis in humans. Importantly, our results suggest that CNS sepsis may be amenable to virulence-targeted therapeutic approaches, such as those targeting the quorum-sensing system Agr [20], which strictly regulates PSM expression [21], or monoclonal antibody-based therapy directed against the toxin.
Methods
Bacterial strains and growth conditions
Strain RP62A is a genome-sequenced clinical MRSE isolate [22]. Strain SE620 is an MRSE clinical isolate from Norway [23]. Isogenic Δpsm-mec deletion mutants and the psm-mec* start codon mutant were produced with the constructs previously used for S. aureus [12, 14], using a strategy with the allelic exchange vector pKOR1 [24]. The psm-mec locus and adjacent DNA do not differ between S. aureus and S. epidermidis [25]. For construction of the psm-mec* mutant, the start codon mutation was created by introducing a ClaI restriction site (introducing ATC instead of the ATG start codon) using primer PSMEClarev GAGGGTATGCATATCGATTTCACTGGTGTTATTACAAGC and primer PSMECladir (reverse complement of PSMEClarev). Two PCR fragments were amplified using those primers and primers psmEatt1 and psmEatt2, respectively [12], cut with ClaI, ligated, and cloned into pKOR1. The resulting plasmid was used for allelic replacement as described [24]. Growth patterns of the mutants were indistinguishable from those of the wild-type (S3 Fig). Strains were grown in tryptic soy broth (TSB), unless otherwise noted.
Mouse sepsis model
Female, 6–10 weeks old, C57BL/6NCRl (Charles River) mice were used. The mice were injected via the tail vein with 5 x 108 CFU in 100 μl phosphate-buffered saline (PBS) of the indicated bacterial strains grown to mid-exponential growth phase and monitored for disease development every 8 h for up to 120 h. This dosis was determined to be minimally necessary to achieve mortality and production of inflammatory cytokines (S4 Fig). Animals were euthanized immediately if showing signs of respiratory distress, mobility loss, or inability to eat and drink. Cytokine concentrations were measured at 2 and/or 12 h, as indicated, using commercially available ELISA kits (IL-1β, TNF-α, BD BioSciences; CXCL1, R&D Systems).
Interaction of bacteria with human neutrophils and bacterial survival in human blood
For survival in whole blood experiments, about 108 bacteria in 100 μl Dulbecco’s PBS from mid-exponential growth phase were added to 500 μl heparinized human blood and mixtures were incubated for 6 h. Aliquots were taken at 2-h intervals, and CFU were determined by plating and incubating plates overnight at 37°C.
For neutrophil interaction experiments, neutrophils were isolated from the venous blood of human volunteers as described [26]. Bacteria from mid-exponential growth phase were mixed with neutrophils at an MOI (bacteria/neutrophils) of 10:1. Bacteria/neutrophil mixtures were incubated at 37°C, 5% CO2, 90% humidity for 6 h. At 2-h intervals, 50 μl of Triton X-100 was added to the 200-μl bacteria/neutrophil suspensions, aliquots were plated, and plates incubated at 37°C overnight for CFU counting. Alternatively, the rate of neutrophil lysis promoted by the bacteria was determined after 4–h incubation using a lactate dehydrogenase (LDH) assay at an MOI of 100:1.
Biofilm formation
Biofilm formation was assessed in a semi-quantitative 96-well microtiter plate assay as previously described [27], using TSB + 0.5% glucose.
PSM measurement
Relative PSM concentrations in culture filtrates were determined as described using reversed-phase high-pressure liquid chromatography/electrospray mass spectrometry (RP-HPLC/ESI-MS) [28].
Quantitative real-time (RT)-PCR and microarray analysis
Quantitative RT-PCR was performed as previously described [29] with the following oligonucleotides: psm-mecF, TGCATATGGATTTCACTGGTGTTA, psm-mecR, CGTTGAATATTTCCTCTGTTTTTTAGTTG, psm-mec probe, ATTTAATCAAGACTTGCATTCAG. Expression was measured relative to that of 16S RNA. Cultures were grown to the maximum of psm-mec expression as determined by qRT-PCR (10 h). Total RNA and cDNA were prepared as described [30]. Biotinylated S. aureus cDNA was hybridized to custom Affymetrix GeneChips (RMLChip 3) with 100% coverage of chromosomal genes from strains S. epidermidis RP62A and scanned according to standard GeneChip protocols (Affymetrix). Each experiment was replicated 3 times. Affymetrix GeneChip Operating Software was used to perform the preliminary analysis of the custom GeneChips at the probe-set level. Subsequent data analysis was performed as described [30]. The complete set of microarray data was deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE85265.
Analysis of mRNA stability
To determine psm-mec mRNA stability in strains S. epidermidis SE620 and the psm-mec* start codon mutant, bacteria were cultured at 37°C for 10 hours. At time t = 0 min, rifampicin (50 mg/ml stock in DMSO) was added to the cultures to a final concentration of 100 μg/ml. One-ml aliquots were taken and immediately centrifuged at 4°C to pellet cells, which were then frozen at -70°C. Remaining cultures were further incubated at 37°C with shaking; one-ml aliquots were taken at the indicated times and RNA was subsequently isolated from all cell pellets as described [29]. Samples were analyzed by qRT-PCR using primers psm-mecR and psm-mecF with a SuperScript III Platinum SYBR Green One-Step qRT-PCR kit (Invitrogen) according to the manufacturer’s instructions. Expression was measured relative to that of 16S RNA.
Statistics
Statistical analysis was performed using GraphPad Prism Version 6.0. Comparisons were by 1-way or 2-way ANOVA for comparisons of three and more, and by unpaired t-tests for comparisons of 2 groups. Error bars show ±SEM.
Ethics statement
The animal protocol (LB1E) was reviewed and approved by the Animal Care and Use Committee at the NIAID, NIH, according to the animal welfare act of the United States (7 U.S.C. 2131 et. seq.). All mouse experiments were performed at the animal care facility of the NIAID, Building 50, in accordance with approved guidelines. All animals were euthanized by CO2 at the end of the studies. Human neutrophils were isolated from blood obtained under approved protocols at the NIH Blood Bank or with a protocol (633/2012BO2) approved by the Institutional Review Board for Human Subjects, NIAID, NIH. All subjects were adult and gave informed written consent.
Supporting Information
A clinical strain collection was analyzed for PSM-mec production by RP-HPLC/MS of stationary-phase culture filtrates. The horizontal line shows the mean. Colored dots show the production in the strains used in this study.
(TIF)
Data are not significantly different between the two groups at any time point.
(TIF)
(TIF)
(A,B) Mortality in the mouse bacteremia model at different doses of strains SE620 and RP62A, respectively. (C) Concentration of the inflammatory cytokine CXCL-1 (TNF-α) in mouse blood at 12 h. Note only one mouse could be used for the group infected with strain SE620, as the others died very early. (A-C) n = 5 in every group.
(TIF)
Data Availability
All relevant data, except for the complete set of microarray data (see below) are within the paper and its Supporting Information files. The complete set of microarray data was deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE85265.
Funding Statement
This study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), U.S. National Institutes of Health (NIH) (https://www.niaid.nih.gov/about/dir), grant AI001080-15 (to MO). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Becker K, Heilmann C, Peters G. Coagulase-negative staphylococci. Clin Microbiol Rev. 2014;27(4):870–926. PubMed Central PMCID: PMCPMC4187637. 10.1128/CMR.00109-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung GY, Otto M. Understanding the significance of Staphylococcus epidermidis bacteremia in babies and children. Curr Opin Infect Dis. 2010;23(3):208–16. PubMed Central PMCID: PMCPMC2874874. 10.1097/QCO.0b013e328337fecb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchant EA, Boyce GK, Sadarangani M, Lavoie PM. Neonatal sepsis due to coagulase-negative staphylococci. Clin Dev Immunol. 2013;2013:586076 PubMed Central PMCID: PMCPMC3674645. 10.1155/2013/586076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vassallo M, Dunais B, Roger PM. Antimicrobial lock therapy in central-line associated bloodstream infections: a systematic review. Infection. 2015;43(4):389–98. 10.1007/s15010-015-0738-1 [DOI] [PubMed] [Google Scholar]
- 5.Otto M. Staphylococcus epidermidis—the 'accidental' pathogen. Nat Rev Microbiol. 2009;7(8):555–67. PubMed Central PMCID: PMCPMC2807625. 10.1038/nrmicro2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otto M. Virulence factors of the coagulase-negative staphylococci. Front Biosci. 2004;9:841–63. [DOI] [PubMed] [Google Scholar]
- 7.Bochud PY, Calandra T. Pathogenesis of sepsis: new concepts and implications for future treatment. BMJ. 2003;326(7383):262–6. PubMed Central PMCID: PMCPMC1125122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung GY, Joo HS, Chatterjee SS, Otto M. Phenol-soluble modulins—critical determinants of staphylococcal virulence. FEMS Microbiol Rev. 2014;38(4):698–719. PubMed Central PMCID: PMCPMC4072763. 10.1111/1574-6976.12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raad I, Alrahwan A, Rolston K. Staphylococcus epidermidis: emerging resistance and need for alternative agents. Clin Infect Dis. 1998;26(5):1182–7. [DOI] [PubMed] [Google Scholar]
- 10.Otto M. Coagulase-negative staphylococci as reservoirs of genes facilitating MRSA infection: Staphylococcal commensal species such as Staphylococcus epidermidis are being recognized as important sources of genes promoting MRSA colonization and virulence. Bioessays. 2013;35(1):4–11. PubMed Central PMCID: PMCPMC3755491. 10.1002/bies.201200112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee SS, Chen L, Joo HS, Cheung GY, Kreiswirth BN, Otto M. Distribution and regulation of the mobile genetic element-encoded phenol-soluble modulin PSM-mec in methicillin-resistant Staphylococcus aureus. PLoS One. 2011;6(12):e28781 PubMed Central PMCID: PMCPMC3236207. 10.1371/journal.pone.0028781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Queck SY, Khan BA, Wang R, Bach TH, Kretschmer D, Chen L, et al. Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 2009;5(7):e1000533 PubMed Central PMCID: PMCPMC2712073. 10.1371/journal.ppat.1000533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaito C, Saito Y, Ikuo M, Omae Y, Mao H, Nagano G, et al. Mobile genetic element SCCmec-encoded psm-mec RNA suppresses translation of agrA and attenuates MRSA virulence. PLoS Pathog. 2013;9(4):e1003269 PubMed Central PMCID: PMCPMC3617227. 10.1371/journal.ppat.1003269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung GY, Villaruz AE, Joo HS, Duong AC, Yeh AJ, Nguyen TH, et al. Genome-wide analysis of the regulatory function mediated by the small regulatory psm-mec RNA of methicillin-resistant Staphylococcus aureus. Int J Med Microbiol. 2014;304(5–6):637–44. PubMed Central PMCID: PMCPMC4087065. 10.1016/j.ijmm.2014.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikuo M, Nagano G, Saito Y, Mao H, Sekimizu K, Kaito C. Inhibition of exotoxin production by mobile genetic element SCCmec-encoded psm-mec RNA is conserved in staphylococcal species. PLoS One. 2014;9(6):e100260 PubMed Central PMCID: PMCPMC4057442. 10.1371/journal.pone.0100260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, et al. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol. 2013;15(8):1427–37. PubMed Central PMCID: PMCPMC4784422. 10.1111/cmi.12130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grosz M, Kolter J, Paprotka K, Winkler AC, Schafer D, Chatterjee SS, et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol. 2014;16(4):451–65. PubMed Central PMCID: PMCPMC3969633. 10.1111/cmi.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaito C, Saito Y, Nagano G, Ikuo M, Omae Y, Hanada Y, et al. Transcription and translation products of the cytolysin gene psm-mec on the mobile genetic element SCCmec regulate Staphylococcus aureus virulence. PLoS Pathog. 2011;7(2):e1001267 PubMed Central PMCID: PMCPMC3033363. 10.1371/journal.ppat.1001267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novick RP, Ross HF, Projan SJ, Kornblum J, Kreiswirth B, Moghazeh S. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 1993;12(10):3967–75. PubMed Central PMCID: PMCPMC413679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan BA, Yeh AJ, Cheung GY, Otto M. Investigational therapies targeting quorum-sensing for the treatment of Staphylococcus aureus infections. Expert Opin Investig Drugs. 2015;24(5):689–704. 10.1517/13543784.2015.1019062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, et al. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell. 2008;32(1):150–8. PubMed Central PMCID: PMCPMC2575650. 10.1016/j.molcel.2008.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187(7):2426–38. PubMed Central PMCID: PMCPMC1065214. 10.1128/JB.187.7.2426-2438.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klingenberg C, Ronnestad A, Anderson AS, Abrahamsen TG, Zorman J, Villaruz A, et al. Persistent strains of coagulase-negative staphylococci in a neonatal intensive care unit: virulence factors and invasiveness. Clin Microbiol Infect. 2007;13(11):1100–11. 10.1111/j.1469-0691.2007.01818.x [DOI] [PubMed] [Google Scholar]
- 24.Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55(1):58–63. 10.1016/j.plasmid.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 25.Qin L, McCausland JW, Cheung GY, Otto M. PSM-Mec-A Virulence Determinant that Connects Transcriptional Regulation, Virulence, and Antibiotic Resistance in Staphylococci. Front Microbiol. 2016;7:1293 PubMed Central PMCID: PMCPMC4992726. 10.3389/fmicb.2016.01293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, et al. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis. 2006;194(12):1761–70. 10.1086/509506 [DOI] [PubMed] [Google Scholar]
- 27.Vuong C, Gerke C, Somerville GA, Fischer ER, Otto M. Quorum-sensing control of biofilm factors in Staphylococcus epidermidis. J Infect Dis. 2003;188(5):706–18. 10.1086/377239 [DOI] [PubMed] [Google Scholar]
- 28.Joo HS, Otto M. The isolation and analysis of phenol-soluble modulins of Staphylococcus epidermidis. Methods Mol Biol. 2014;1106:93–100. 10.1007/978-1-62703-736-5_7 [DOI] [PubMed] [Google Scholar]
- 29.Yao Y, Vuong C, Kocianova S, Villaruz AE, Lai Y, Sturdevant DE, et al. Characterization of the Staphylococcus epidermidis accessory-gene regulator response: quorum-sensing regulation of resistance to human innate host defense. J Infect Dis. 2006;193(6):841–8. 10.1086/500246 [DOI] [PubMed] [Google Scholar]
- 30.Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M. Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci U S A. 2007;104(22):9469–74. PubMed Central PMCID: PMCPMC1890518. 10.1073/pnas.0702159104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A clinical strain collection was analyzed for PSM-mec production by RP-HPLC/MS of stationary-phase culture filtrates. The horizontal line shows the mean. Colored dots show the production in the strains used in this study.
(TIF)
Data are not significantly different between the two groups at any time point.
(TIF)
(TIF)
(A,B) Mortality in the mouse bacteremia model at different doses of strains SE620 and RP62A, respectively. (C) Concentration of the inflammatory cytokine CXCL-1 (TNF-α) in mouse blood at 12 h. Note only one mouse could be used for the group infected with strain SE620, as the others died very early. (A-C) n = 5 in every group.
(TIF)
Data Availability Statement
All relevant data, except for the complete set of microarray data (see below) are within the paper and its Supporting Information files. The complete set of microarray data was deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE85265.