

Abstract
The carcinogen 2-acetylaminofluorene forms two major DNA adducts: N-(2′-deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) and its deacetylated derivative, N-(2′-deoxyguanosin-8-yl)-2-aminofluorene (dG-AF). Although the dG-AAF and dG-AF adducts are distinguished only by the presence or absence of an acetyl group, they have profoundly different effects on DNA replication. dG-AAF poses a strong block to DNA synthesis and primarily induces frameshift mutations in bacteria, resulting in the loss of one or two nucleotides during replication past the lesion. dG-AF is less toxic and more easily bypassed by DNA polymerases, albeit with an increased frequency of misincorporation opposite the lesion, primarily resulting in G → T transversions. We present three crystal structures of bacteriophage T7 DNA polymerase replication complexes, one with dG-AAF in the templating position and two others with dG-AF in the templating position. Our crystallographic data suggest why a dG-AAF adduct blocks replication more strongly than does a dG-AF adduct and provide a possible explanation for frameshift mutagenesis during replication bypass of a dG-AAF adduct. The dG-AAF nucleoside adopts a syn conformation that facilitates the intercalation of its fluorene ring into a hydrophobic pocket on the surface of the fingers subdomain and locks the fingers in an open, inactive conformation. In contrast, the dG-AF base at the templating position is not well defined by the electron density, consistent with weak binding to the polymerase and a possible interchange of this adduct between the syn and anti conformations.
Numerous carcinogenic aromatic amines, including a variety of environmental and dietary carcinogens and heterocyclic aromatic amines present in tobacco smoke condensate, are known to react with DNA to form adducts at the C8 position of guanine (1). 2-Acetylaminofluorene (AAF) is the best-studied example of this class of carcinogen (2). Originally developed as a pesticide, toxicity tests showed that this compound and related derivatives are potent liver carcinogens (3). Thus, the compound was never introduced as a pesticide. Instead, AAF has become a model compound for the study of the mutagenic and carcinogenic effects of aromatic amines (4).
Metabolic activation of AAF in vivo generates intermediates that form two related adducts bound to the C8 position of guanine DNA: the N-(2′-deoxyguanosin-8-yl)-AAF (dG-AAF) adduct and the corresponding deacetylated N-(2′-deoxyguanosin-8-yl)-2-aminofluorene (dG-AF) derivative (Fig. 1) (3). The mutagenic consequences of these adducts are quite distinct in Escherichia coli. The dG-AF adduct predominately produces randomly distributed base-substitution mutations (5, 6), whereas the dG-AAF adduct results in frameshift mutations that frequently target specific repetitive sequences (4, 7-9). In vitro studies using templates modified with either a dG-AF or dGAAF adduct have shown that the 2-aminofluorene (AF) adduct is bypassed much more readily than the corresponding AAF adduct by a variety of DNA polymerases (10, 11). It has been suggested that the differences in mutagenic effects may be related to the diverse replication properties of these adducts. Moreover, most spectral, enzymatic, and theoretical studies suggest that the AF structure produces much less distortion in the DNA helix than does the closely related AAF adduct (12-16) and that these structural differences are responsible for the different biological consequences.
Fig. 1.

Schematic representation of dG-AAF and dG-AF. The anti conformation (a) is energetically favored for unmodified dG. dG-AF (b) can be in anti conformation or syn conformation. However, for dG-AAF (c), the anti conformation is strongly unfavorable because of the steric hindrance between the acetyl group (circled in green) and the sugar moiety (circled in pink).
NMR and molecular mechanics studies of a dG-AAF adduct in double-stranded DNA have confirmed that a major structural distortion is caused by this adduct. These spectral studies have shown that the glycosidic torsion angle of the modified nucleotide adopts a syn conformation with the modified guanine unpaired and lying outside the double helix (17). This distortion of the DNA structure is consistent with an earlier base-displacement model proposed on the basis of circular dichroism studies of the DNA lesion (18) in which the fluorene ring of dG-AAF partially inserts between adjacent base pairs, causing a local disruption of base-pairing. This model is termed the insertion-denaturation model (19, 20).
Studies of the deacylated dG-AF adduct in double-stranded DNA by similar methods have revealed two interchangeable conformers of the modified nucleoside (21-24). In one conformer, the fluorene ring of dG-AF is oriented outside the DNA helix, where it does not perturb base-pairing. In this outside-binding conformation, the modified guanosine adopts an anti conformation and base-pairs with the complementary DNA strand. In the alternative conformer of dG-AF, the fluorene ring stacks inside the double helix, and the guanosine adopts a syn conformation in which it is exposed and unpaired in a manner similar to the dG-AAF adduct.
The highly distorting syn conformation of the dG-AAF adduct has been used to explain why this modification poses a strong block to replication and is not efficiently copied by a polymerase (11). It is possible that the ability of the dG-AAF adduct to halt replication may also promote frameshift mutagenesis by providing sufficient time for the realignment of primer and template DNA strands to form a slipped mispair with the lesioned nucleotide looped out of the template strand (25). Similarly, the syn conformation of dG-AF would presumably interfere with base-pairing in the active site of a DNA polymerase, whereas the relatively undistorted anti conformation of the dG-AF adduct structure could template the insertion of dCMP or dAMP opposite the lesion, possibly explaining error-prone bypass of the lesion (see below). Although it has been assumed that the structural differences between the AAF and AF adducts are responsible for the observed replicative differences, the molecular mechanism that produces these differences is not known in any detail. To date, the structures of dG-AAF and dG-AF adducts in DNA have provided the only detailed physical information supporting the proposed mechanisms for their mutagenic bypass in DNA (26). No structural studies are available showing either a dG-AF or dG-AAF adduct in the context of a replicative polymerase.
A number of high-fidelity DNA polymerases have been examined for their ability to bypass a dG-AAF or dG-AF adduct in vitro (3). Nucleotide insertion is strongly blocked across from a dG-AAF adduct during primer extension reactions catalyzed by the Klenow fragment of E. coli DNA polymerase I, T7 DNA polymerase, and T4 DNA polymerase (10, 11, 27), and misinsertion of dAMP is favored over insertion of the correct nucleotide (dCMP) (28, 29). The dG-AF lesion dramatically slows DNA synthesis, but this lesion is eventually bypassed (10, 11). Although the correct nucleotide (dCTP) is preferentially incorporated across from a dG-AF adduct, dATP also is incorporated, to a lesser extent, at this site (29). Compared with the insertion of dCMP opposite dG, the incorporation across from either a dG-AAF or dG-AF adduct by a proofreading-deficient T7 DNA polymerase is ≈106- or 104-fold slower, respectively (30).
Interestingly, the Klenow fragment polymerase binds to DNA containing a dG-AAF adduct with 5- to 10-fold higher affinity than it does to unmodified DNA or DNA containing a dG-AF adduct (10). The addition of nucleotide substrates that can base-pair with the templating base normally enhances DNA binding affinity by inducing the closure of the polymerase active site around the substrates (10, 31). However, the affinity of the Klenow fragment for DNA containing a dG-AAF adduct is unaffected by the addition of dCTP or any other nucleotide, suggesting that the lesion does not support base-pairing interactions within the polymerase active site (10, 32). Furthermore, the substrate-induced conformational change of the polymerase is not observed when nucleotides are added to the complex of the Klenow fragment bound to DNA containing a dG-AAF adduct (30). Additional support for a distorted conformation of dGAAF-modified DNA in complex with a DNA polymerase comes from footprinting studies of T7 DNA polymerase in complex with native and AAF-lesioned DNAs (33). In contrast to a dG-AAF lesion, DNA containing a dG-AF adduct binds the Klenow fragment less strongly, and the polymerase can undergo a conformational change upon the addition of dCTP (30).
The different binding modes of DNA templates containing a dG-AAF or dG-AF adduct to a DNA polymerase are likely to account for the strikingly different mutagenic spectra caused by these lesions. Here we present crystal structures of the replicative T7 DNA polymerase (consisting of the gp5 catalytic subunit and its processivity factor, E. coli thioredoxin) in complex with dG-AAF- and dG-AF-modified DNA substrates that reveal the basis for the strong blockage of DNA synthesis by a dG-AAF adduct and suggest why bypass of a dG-AF adduct can occur with a high probability of misinsertion opposite the lesion.
Methods
Preparation of Modified DNAs and Replication Complexes. A 26-mer template strand containing a single dG was designed (5′-CCCGATCACACTACCAATCACTCTCC-3′), along with a complementary primer strand (5′-GGAGAGTGATTGGTAGTGTGA-3′), and both oligonucleotides were chemically synthesized. N-acetoxy-AAF was obtained from the National Cancer Institute repository operated by the Midwest Research Institute (Midland, TX). The 26-mer template strand and 21-mer primer strand were purified by electrophoresis on denaturing polyacrylamide gels. The template strand was reacted with N-acetoxy-AAF to form a single dG-AAF adduct and then purified by reverse-phase HPLC (34). A portion of the dG-AAF oligonucleotide was deacetylated by incubating in 1 M sodium hydroxide and 0.25 M 2-mercaptoethanol for 2 h at 37°C. The resulting DNA containing dG-AF was purified again by HPLC.
An exonuclease-deficient T7 DNA polymerase (Δ118-123 T7) was purified as described in ref. 35. Replication complexes were assembled with either the dG-AAF- or dG-AF-containing DNA template by incubating T7 DNA polymerase (0.1 mM protein) with a stoichiometric amount of the DNA template and primer strands in buffer containing 0.5 mM 2′,3′-dideoxythymidine triphosphate (ddTTP) and 10 mM incoming nucleotide, either 2′,3′-dideoxycytidine triphosphate (ddCTP) or 2′,3′-dideoxyadenosine triphosphate (ddATP). Complexes with either ddCTP or ddATP as the incoming nucleotide were separately assembled and crystallized for the dG-AF complex. For the dG-AAF DNA, a binary complex with T7 DNA polymerase lacking an incoming nucleotide also was assembled for crystallization.
Crystallization and X-Ray Structure Determination. Complexes of T7 DNA polymerase with DNA were crystallized under conditions described in ref. 35, and the crystals were harvested in the presence or absence of added ddCTP or ddATP and then frozen. X-ray diffraction data were collected at station F1 of the Cornell High Energy Synchrotron Source (CHESS) (Ithaca, NY). The x-ray data were indexed, integrated, and scaled by using the hkl 2000 package (36), and the structures were determined by the molecular replacement method by using the program epmr (37). A model of a native ternary complex of T7 DNA polymerase (Protein Data Bank ID code 1T7P) was positioned in the unit cell after nucleotides within the polymerase active site and residues from the surrounding fingers subdomain were removed. These regions around the active site were rebuilt into the omit electron density calculated from the partial model of the complex, and the resulting crystallographic models were refined by using the program cns (38) (Table 1). The coordinates for the dG-AAF complex (PDB ID code 1X9M), the dG-AF complex grown in the presence of ddCTP (PDB ID code 1X9S), and the dG-AF complex grown in ddATP (PDB ID code 1X9W) have been deposited in the Protein Data Bank.
Table 1. X-ray data collection and refinement statistics.
| Complex
|
|||
|---|---|---|---|
| dG-AAF | dG-AF/ddCTP | dG-AF/ddATP | |
| Data collection | |||
| Resolution, Å | 50-2.1 | 50-2.7 | 50-2.3 |
| Space group | P21212 | P21212 | P21212 |
| Cell dimensions, Å | |||
| a | 105.109 | 104.579 | 105.944 |
| b | 212.308 | 211.103 | 213.559 |
| c | 52.070 | 51.991 | 52.168 |
| Unique reflections | 69,183 | 33,030 | 51,286 |
| Rsym,* % | 5.6 (21.8) | 4.1 (7.6) | 6.2 (22.3) |
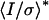 * *
|
21.3 (3.5) | 35.4 (16.0) | 17.7 (3.0) |
| Completeness,* % | 94.6 (84.1) | 95.1 (85.6) | 94.5 (88.4) |
| Refinement | |||
| Resolution, Å | 50-2.1 | 20-2.7 | 50-2.3 |
| R/Rfree,† % | 21.3/23.6 | 22.9/28.2 | 23.1/25.8 |
| Bond, Å | 0.007 | 0.007 | 0.006 |
| Angle, ° | 1.3 | 1.2 | 1.2 |
| Average B factors, Å2 | 33.4 | 31.4 | 36.2 |
| rms for main-chain atoms | 0.94 | 0.73 | 0.77 |
| rms for side-chain atoms | 2.89 | 2.92 | 1.39 |
Values in parentheses are for the last resolution shell.
R = (Σ|Fo| −|Fc|)/ΣFo, where|Fo| and|Fc| are observed and calculated structure factor amplitudes. Rfree is calculated with 5% of randomly selected reflections that were not used in refinement.
Results
Structure of the dG-AAF-Containing Replication Complex. When dG-AAF is in the templating position of a replication complex with T7 DNA polymerase, the modified nucleoside is flipped out of the polymerase active site and bound to the surface of the fingers subdomain (Fig. 2). The syn conformation of the dGAAF nucleoside (Fig. 2c) facilitates the insertion of the fluorene ring into a hydrophobic pocket behind the O-helix of the fingers subdomain (Fig. 2d). The fluorene adduct stacks against Phe-528 and other aliphatic side chains that are normally buried in the core of the fingers subdomain (Fig. 2 b and d and Fig. 4, which is published as supporting information on the PNAS web site). The acetyl group of dG-AAF is not contacted by the polymerase and lies outside the hydrophobic pocket with the oxygen atom facing solvent. The N2 amino group of the guanine base donates a hydrogen bond to Asp-534, and N7 of guanine accepts a hydrogen bond from Arg-566 (Fig. 2d). These electrostatic contacts with guanine further stabilize the interaction of the DNA adduct with the fingers.
Fig. 2.

dG-AAF adopts the syn conformation with the fluorene ring intercalated into the polymerase fingers domain. (a) dG-AAF lies outside the polymerase active site, and the fingers domain of the polymerase is in an open conformation. The T7 gene 5 protein (gray) and the thioredoxin (green) processivity factor are shown as ribbons, with the O-helix within the fingers highlighted in red. The primer strand of the DNA is in light red, the template is in yellow, and dG-AAF is in cyan and green. The region around the dG-AAF binding site is circled. (b) Enlarged view of the circled region in a. The fluorene ring (green) of dG-AAF (cyan and green) is inserted into the fingers domain between helices L, O, O1, O2, and P. (c) The simulated annealing omit electron density around the region of the syn dG-AAF of the dG-AAF-containing complex is shown in stereo, contoured at 2.5 σ above the mean value. (d) Interaction between dG-AAF and the protein. Hydrophobic side chains (gold) of the fingers form a pocket around the fluorene ring. Two charged residues (pink), Arg-566 and Asp-534, form hydrogen bonding interactions with the G base.
In comparison with a native ternary replication complex (35), T7 DNA polymerase in complex with dG-AAF-modified DNA adopts an open, distorted conformation of the fingers. This change in conformation involves a hinge-like rotation of the fingers subdomain away from the polymerase active site and repacking of helices within the fingers in response to the intercalation of the dG-AAF nucleoside. The fingers are rotated ≈35° away from the fully closed position that is observed in complexes with native DNA and a bound nucleotide (39). There is no evidence for a bound ddCTP molecule in the crystal structure of the dG-AAF complex, despite our having grown the crystals in the presence of 10 mM ddCTP. In fact, repacking of the fingers subdomain caused by the insertion of the AAF adduct pushes the O-helix into the active site, where the side chain of Tyr-530 partially occludes the nucleotide-binding site (Fig. 3). This finding is consistent with the high affinity of DNA containing a dG-AAF adduct for the related Klenow fragment polymerase and the failure of added nucleotides to influence the stability of the complex (10). We subsequently determined another structure of the dG-AAF complex by using crystals grown in the absence of ddCTP (data not shown) and found that the addition of ddCTP during crystallization has no effect on the structure of the complex.
Fig. 3.

Distortion of the O-helix and stacking of the side chain of Tyr-530 with the primer/template DNA. In the dG-AAF/ddCTP (a) and dG-AF/ddCTP (b) complexes (Table 1), this residue (dark gray) stacks with the 3′ base of the primer and occupies the position where the base of the incoming nucleotide would bind in a catalytically active, closed complex (c). The incoming nucleotide in the closed complex is shown in green in c. An incoming nucleotide is modeled in a according to its position in the active closed ternary complex and is outlined in black.
The C terminus of the O-helix is distorted in the dG-AAF-DNA complex, with three C-terminal residues displaced from an α-helical geometry. The adjoining helices O1 and O2 also pack in slightly different orientations in comparison with the structure of the fingers bound to native DNA. All other regions of the structure, including the DNA template and primer strands, can be superimposed well on structures of T7 DNA polymerase in complex with an unmodified DNA and a nucleotide.
Structure of a dG-AF-Containing Replication Complex. The overall structure of T7 DNA polymerase in complex with DNA containing a dG-AF adduct is very similar to that of the dG-AAF complex described above (Table 1 and Fig. 3), but there are two notable differences. First, electron density corresponding to the dG-AF nucleotide is absent from several different structures determined by using crystals grown in the presence of ddCTP (2.7-Å resolution) or ddATP (2.4-Å resolution) (Table 1; see Methods for crystallization conditions). Although the structure of the protein and the bound DNA is well defined by the x-ray experiment, the electron density around the dG-AF adduct is of limited quality. However, the crystal structure clearly shows that the dG-AF nucleoside does not bind in the polymerase active site and that the side chain of Tyr-530 occupies the binding site for the template base in a closed ternary complex. Sketchy electron density on the surface of the polymerase in the dG-AF complex is suggestive of the 5′ end of the template strand extending away from the normal binding site for the template base. However, the fluorene ring of the dG-AF adduct is not intercalated into the hydrophobic pocket on the surface of the fingers where the syn dG-AAF nucleotide binds. In the dG-AF structure, the surrounding nonpolar side chains rotate inward to fill the pocket.
The fingers of the polymerase in the dG-AF complex are in an open conformation, closely resembling their overall position in complexes with dG-AAF DNA, and, likewise, there is no evidence of a bound nucleotide for crystals grown in the presence of 10 mM ddCTP or ddATP (Fig. 3). Harvesting crystals in the continued presence of ddCTP (5 mM) also had no effect on the structure. It is surprising, therefore, that crystals of the dG-AF complex grew only in the presence of ddCTP or ddATP. This observation suggests that the incoming nucleotide stabilizes the polymerase-DNA complex during some stage of the crystallization experiment but that the open conformation of the polymerase lacking bound nucleotide is energetically preferred.
We have modeled the anti conformation of the dG-AF nucleoside in the active site of the closed conformation of T7 DNA polymerase by superimposing the modified guanine on the crystal structure of a native dG-ddCTP complex (40). By rotating about the C8-N2 bond, the fluorene ring of the bound dG-AF adduct can be positioned so that it does not clash with the closed fingers of the polymerase. However, after the nucleotide binds to the templating position, rotation about the C8-N2 bond would be restricted by clashes between the N2 proton and the sugar phosphate backbone of the dG-AF nucleoside, as well as by interference between the fluorene ring and neighboring protein groups as the nucleotide transits through unfavorable conformations. In the syn conformation, the aromatic fluorene ring of the dG-AF adduct might favorably stack with the base pair adjacent to the polymerase active site, effectively competing with binding of the guanine template in the anti conformation seen in native complexes (35, 40). These restrictions on positioning the nucleotide adduct for base-pairing interactions with the incoming nucleotide may explain the observed pause in DNA synthesis opposite dG-AF (29).
Discussion
dG-AAF and dG-AF Interfere with Nucleotide Binding and Induced Fit. T7 DNA polymerase and other polymerases undergo a conformational change during nucleotide incorporation in which the fingers subdomain opens and closes by a hinge-like motion (31). The open conformation allows the active site to sample nucleotides until the fingers close around a nucleotide correctly paired with a template base (35, 41). Bulky lesions such as a cis-syn cyclobutane thymine dimer in a DNA template shift the conformational equilibrium toward the open conformation of the fingers, slowing DNA synthesis (42). The fingers of T7 DNA polymerase are in an open conformation in complexes with dG-AAF and dG-AF templating DNA synthesis, and there is an additional distortion of the O-helix (Fig. 3 and Fig. 5, which is published as supporting information on the PNAS web site).
NMR studies of double-stranded DNA containing a dG-AAF adduct show that it exists predominantly in a syn conformation with its fluorene ring intercalated between base pairs (17). In complex with T7 DNA polymerase, the dG-AAF nucleoside maintains the syn conformation, but the adduct binds in a surprising location, inserting in a hydrophobic pocket behind the O-helix of the fingers subdomain. This mode of interaction with the polymerase halts DNA synthesis by locking the fingers in an open conformation (Fig. 2) and causing the Tyr-530 side chain to encroach on the nucleotide binding site (Fig. 3). Intercalation of the fluorenyl moiety of dG-AAF causes a repacking of hydrophobic residues within the core of the fingers subdomain (Fig. 2c), distorting the C terminus of the O-helix. This conformational change brings Tyr-530 into the active site of the polymerase and prevents nucleotide substrates from binding (Fig. 3). This shift of Tyr-530 might explain the strong block of DNA synthesis by a dG-AAF adduct (29) and the failure of nucleotide substrates to interact with the related Klenow fragment polymerase in complex with dG-AAF-modified DNA (30).
Computational analyses of dG, dG-AAF, and dG-AF have shown that these nucleosides can adopt syn and anti conformations, with different preferences that are thought to influence their coding potential (4, 43). dG-AAF strongly favors the syn conformation because of steric clashes between the acetyl group and the ribose sugar in the anti conformation (Fig. 1b). The deacylated dG-AF nucleoside has only a slight preference for the syn conformation, and it interchanges between syn and anti conformations in double-stranded DNA (Fig. 1) (21-24). This dynamic disorder might underlie the weaker binding affinity of dG-AF for T7 DNA polymerase and the failure to observe the dG-AF nucleotide in the crystal structure. Although unmodified purine nucleosides also transition between syn and anti conformations (44), the added fluorenyl moiety of dG-AF might interfere with movement of the DNA and positioning of the modified template base in the polymerase active site. We constructed a model of dG-AF paired with dCTP in the active site of T7 DNA polymerase on the basis of a crystal structure of a native dG·dCTP complex (40). The model shows that anti dG-AF can be positioned in the closed polymerase with no significant steric or electrostatic clashes. However, in the syn conformation, the fluorene ring of dG-AF could stack into the polymerase active site, binding nonproductively and effectively competing with the binding of the guanine templating base of dG-AF (anti).
The deacetylated dG-AF nucleoside is not visible in the electron density calculated for several different crystal structures, suggesting weak binding to the polymerase. It is clear that the fluorene ring of dG-AF has not intercalated into the hydrophobic pocket on the surface of the finger where dG-AAF binds (Figs. 2 and 3), presumably because the N-acetyl group of dG-AAF constrains it to adopt a syn conformation compatible with binding to the pocket. Our results do not exclude the possibility that the dG-AF adduct binds to the pocket with low affinity and potentially slows lesion bypass. The crystallographic results for T7 DNA polymerase are in agreement with the weaker binding of the Klenow fragment to DNA modified with dG-AF in comparison with dG-AAF (10). Furthermore, by using an exonuclease protection assay that measures the rate of DNA dissociation (35) (data not shown), we find that complexes of T7 DNA polymerase with dG-AF-modified DNA are less stable than complexes with dG-AAF. The addition of dCTP was found to induce a conformational change in the Klenow fragment bound to dG-AF in the active site that is similar to that caused by dCTP binding to a native dG template (30). However, DNA containing the dG-AF lesion failed to bind the incoming nucleotide in the polymerase active site when crystals were grown in the presence of 10 mM ddCTP or ddATP, the nucleotides that are preferentially inserted opposite dG-AF (29).
Structural Insights into the Mutagenic Potentials of dG-AAF and dG-AF. In bacteria, there is a preference for a dG-AAF adduct to cause frameshift mutations and for a dG-AF adduct to cause base substitution mutations, mostly G → T transversions (4). The different mutagenic outcomes of replication bypass of dG-AAF and dG-AF lesions in DNA have been ascribed to differences in the internal conformation and flexibility of these modified nucleosides (Fig. 1), with dG-AAF causing greater distortion of DNA structure than dG-AF (18, 25, 26, 45). Our structural studies of these adducts in the context of a DNA polymerase reported here verify some aspects of this model and offer additional insights about a surprising mechanism for inducing replication blockage.
Single-turnover kinetic studies show that T7 DNA polymerase inserts dCMP opposite dG-AF and dG-AAF ≈104- and 106-fold more slowly, respectively, than it does opposite an unmodified dG (29). dAMP is misincorporated across from dG-AF adducts, albeit at a slower rate than that at which dCMP is incorporated, whereas dAMP is the preferred nucleotide opposite a dG-AAF (29). In the syn conformation of dG-AF, the guanine base would lie outside the templating position, where it is unable to pair with an incoming nucleotide. This situation would resemble synthesis past an abasic site, which causes DNA synthesis to pause and favors the eventual insertion of dAMP (46, 47). A similar situation could also hold for incorporation opposite a dG-AAF adduct in the syn conformation, allowing it to slowly misincorporate dAMP across from the modified nucleoside. However, insertion by the A-rule would likely require the fluorenyl moiety of dG-AAF to dissociate from the polymerase so that the fingers could close and assist with positioning, activating the (unpaired) nucleotide for incorporation. In contrast to dG-AF, a dG-AAF nucleoside adopts the anti conformation infrequently, and it supports templated DNA synthesis at a vanishingly small rate. Thus, it may appear that there is a preference for dAMP incorporation across from dG-AAF.
Residues participating in the hydrophobic interactions that stabilize dG-AAF binding in the fingers subdomain are located far away from the polymerase active site, yet they play a major role in blocking the enzyme function. However, the role of Tyr-530 described here is more direct. In the native ternary complex of T7 DNA polymerase, Tyr-530 stacks against the templating base that pairs with the incoming nucleotide (35). The analogous residue (Tyr-671) of the Thermus aquaticus DNA polymerase plays the same role, acting as a chaperone or “positioning device” for the templating base in the polymerase active site (41). In crystal structures of T7 DNA polymerase complexed to a thymine photodimer, the lesioned base is swung out of the helix (42). Here, this residue fills in the “pseudoabasic site” created when the bulky thymine dimer adduct cannot be accommodated in the polymerase active site. The analogous residue (Tyr-714) of Bacillus stearothermophilus DNA polymerase is reported to act as a flexible chaperone that guides the transport of primer-template base pairs through the polymerase during DNA synthesis (48). The chaperoning function of Tyr-530 takes an interesting turn in the dG-AAF- and dG-AF-containing complexes reported here, where the O-helix is pushed toward the active site by the adduct and the side chain of Tyr-530 blocks the binding of nucleotides. Removal of this bulky side chain could relieve the blockage and perhaps facilitate untemplated bypass of dG-AAF. In fact, replacement of the analogous residue in the Klenow fragment polymerase (Tyr-766) with a smaller serine residue allows the polymerase to undergo a nucleotide-induced conformational change when bound to dG-AAF-modified DNA, which presumably leads to the enhanced ability of this mutant to bypass this lesion (32).
The binding of dG-AAF on the surface of the fingers subdomain provides several key elements for the formation of a slipped mispair that is compatible with deletion frameshift mutagenesis during lesion bypass (25). First, the stable binding of the DNA template outside the polymerase active site dramatically slows DNA synthesis, providing time for the primer strand to realign with the lesioned nucleotide looped out of the template. Second, the fingers are held in an open conformation that imposes fewer restraints on the 3′ end of the primer, allowing it to dissociate and establish alternative base-pairing interactions with the template. Finally, the interactions of the polymerase with the dG-AAF nucleotide interfere with or prevent base-pairing to the lesion while keeping the DNA anchored to the polymerase. These factors increase the likelihood of forming a slipped mispair that excludes the lesioned nucleotide from the duplex while preventing dissociation of the DNA substrate from the polymerase during lesion bypass.
In summary, it has long been suspected that the structural difference between the dG-AAF and dG-AF adducts in duplex DNA somehow were correlated to the differences in replicative and mutagenic properties of these adducts in cellular systems. However, it was not clear how the structures that formed in double-stranded DNA are related to the structures at the primer-template junction bound in the active site of DNA polymerase before and during nucleotide insertion opposite the lesion. The crystal structures reported here provide a molecular basis for the observed effects of these DNA adducts on replication and reveal a correspondence between the structures of the adducts in the polymerase active site and those in duplex DNA.
Supplementary Material
Acknowledgments
X-ray diffraction experiments were conducted at the Cornell High Energy Synchrotron Source, which is supported by National Science Foundation Grant DMR 97-13424 and National Institutes of Health Grant RR-01646, and at the Harvard-Armenise Structural Biology Center (Boston). This work was supported National Institutes of Health Grants GM55390 (to T.E.), GM54397 (to C.C.R.), and CA40605 (to L.J.R.) and Postdoctoral Fellowship GM065746 (to Y.L.). T.E. is the Hsien Wu and Daisy Yen Wu Professor of Biological Chemistry and Molecular Pharmacology.
Author contributions: S.D., Y.L., D.J., C.C.R., L.J.R., and T.E. designed research; S.D., Y.L., D.J., and L.D. performed research; S.D., Y.L., D.J., C.C.R., and L.J.R. analyzed data; S.D., Y.L., D.J., L.J.R., and T.E. wrote the paper.
Abbreviations: AAF, 2-acetylaminofluorene; dG-AAF, N-(2′-deoxyguanosin-8-yl)-AAF; dGAF, N-(2′-deoxyguanosin-8-yl)-2-aminofluorene; ddATP, 2′,3′-dideoxyadenosine triphosphate; ddCTP, 2′,3′-dideoxycytidine triphosphate.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes IX9M (dG-AAF complex), IX9S (dG-AF complex grown in the presence of ddCTP), and 1X9W (dG-AF complex grown in ddATP)].
References
- 1.Sugimura, T. (2000) Carcinogenesis 21, 387-395. [DOI] [PubMed] [Google Scholar]
- 2.King, C. M. (1985) in Prostaglandins, Leukotrienes, and Cancer, ed. Marnett, L. J. (Martinus-Nyhoff, New York), Vol. 2, pp. 1-37. [Google Scholar]
- 3.Heflich, R. H. & Neft, R. E. (1994) Mutat. Res. 318, 73-114. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann, G. R. & Fuchs, R. P. (1997) Chem. Res. Toxicol. 10, 347-359. [DOI] [PubMed] [Google Scholar]
- 5.Bichara, M. & Fuchs, R. P. (1985) J. Mol. Biol. 183, 341-351. [DOI] [PubMed] [Google Scholar]
- 6.Gupta, P. K., Lee, M.-S. & King, C. M. (1988) Carcinogenesis 9, 1337-1345. [DOI] [PubMed] [Google Scholar]
- 7.Koffel-Schwartz, N., Verdier, J. M., Bichara, M., Freund, A. M., Daune, M. P. & Fuchs, R. P. (1984) J. Mol. Biol. 177, 33-51. [DOI] [PubMed] [Google Scholar]
- 8.Tebbs, R. S. & Romano, L. J. (1994) Biochemistry 33, 8998-9006. [DOI] [PubMed] [Google Scholar]
- 9.Tan, X., Suzuki, N., Grollman, A. P. & Shibutani, S. (2002) Biochemistry 41, 14255-14262. [DOI] [PubMed] [Google Scholar]
- 10.Dzantiev, L. & Romano, L. J. (1999) J. Biol. Chem. 274, 3279-3284. [DOI] [PubMed] [Google Scholar]
- 11.Belguise-Valladier, P. & Fuchs, R. P. (1995) J. Mol. Biol. 249, 903-913. [DOI] [PubMed] [Google Scholar]
- 12.van Houte, L. P. A., Bokma, J. T., Lutgerink, J. T., Westra, J. G., Retel, J. & van Grondelle, R. (1987) Carcinogenesis 8, 759-766. [DOI] [PubMed] [Google Scholar]
- 13.van Houte, L. P. A., Westra, J. G., Retel, J., van Grondelle, R. & Blok, J. (1988) Carcinogenesis 9, 1222-1226. [DOI] [PubMed] [Google Scholar]
- 14.Hingerty, B. E. & Broyde, S. (1986) J. Biomol. Struct. Dyn. 4, 365-372. [DOI] [PubMed] [Google Scholar]
- 15.Belguise-Valladier, P. & Fuchs, R. P. P. (1991) Biochemistry 30, 10091-10100. [DOI] [PubMed] [Google Scholar]
- 16.Singer, B. & Grunberger, D. (1984) Molecular Biology of Mutagens and Carcinogens (Plenum, New York).
- 17.O'Handley, S. F., Sanford, D. G., Xu, R., Lester, C. C., Hingerty, B. E., Broyde, S. & Krugh, T. R. (1993) Biochemistry 32, 2481-2497. [DOI] [PubMed] [Google Scholar]
- 18.Grunberger, D., Nelson, J. H., Cantor, C. R. & Weinstein, I. B. (1970) Proc. Natl. Acad. Sci. USA 66, 488-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs, R. P. & Daune, M. (1971) FEBS Lett. 14, 206-208. [DOI] [PubMed] [Google Scholar]
- 20.Fuchs, R. P., Schwartz, N. & Daune, M. P. (1981) Nature 294, 657-659. [DOI] [PubMed] [Google Scholar]
- 21.Eckel, L. M. & Krugh, T. R. (1994) Biochemistry 33, 13611-13624. [DOI] [PubMed] [Google Scholar]
- 22.Cho, B. P., Beland, F. A. & Marques, M. M. (1994) Biochemistry 33, 1373-1384. [DOI] [PubMed] [Google Scholar]
- 23.Mao, B., Hingerty, B. E., Broyde, S. & Patel, D. J. (1998) Biochemistry 37, 95-106. [DOI] [PubMed] [Google Scholar]
- 24.Mao, B., Hingerty, B. E., Broyde, S. & Patel, D. J. (1998) Biochemistry 37, 81-94. [DOI] [PubMed] [Google Scholar]
- 25.Shibutani, S. & Grollman, A. P. (1993) J. Biol. Chem. 268, 11703-11710. [PubMed] [Google Scholar]
- 26.Patel, D. J., Mao, B., Gu, Z., Hingerty, B. E., Gorin, A., Basu, A. K. & Broyde, S. (1998) Chem. Res. Toxicol. 11, 391-407. [DOI] [PubMed] [Google Scholar]
- 27.Strauss, B. S. & Wang, J. (1990) Carcinogenesis 11, 2103-2109. [DOI] [PubMed] [Google Scholar]
- 28.Shibutani, S. & Grollman, A. P. (1997) Mutat. Res. 376, 71-78. [DOI] [PubMed] [Google Scholar]
- 29.Lindsley, J. E. & Fuchs, R. P. (1994) Biochemistry 33, 764-772. [DOI] [PubMed] [Google Scholar]
- 30.Dzantiev, L. & Romano, L. J. (2000) Biochemistry 39, 5139-5145. [DOI] [PubMed] [Google Scholar]
- 31.Doublie, S., Sawaya, M. R. & Ellenberger, T. (1999) Structure Fold. Des. 7, R31-R35. [DOI] [PubMed] [Google Scholar]
- 32.Lone, S. & Romano, L. J. (2003) Biochemistry 42, 3826-3834. [DOI] [PubMed] [Google Scholar]
- 33.Burnouf, D. Y. & Fuchs, R. P. (1998) Mutat. Res. 407, 35-45. [DOI] [PubMed] [Google Scholar]
- 34.Shibutani, S., Gentles, R., Johnson, F. & Grollman, A. P. (1991) Carcinogenesis 12, 813-818. [DOI] [PubMed] [Google Scholar]
- 35.Doublie, S., Tabor, S., Long, A. M., Richardson, C. C. & Ellenberger, T. (1998) Nature 391, 251-258. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 37.Kissinger, C. R., Gehlharr, D. K. & Fogel, D. B. (1999) Acta Crystallogr. D 55, 484-491. [DOI] [PubMed] [Google Scholar]
- 38.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 39.Doublie, S. & Ellenberger, T. (1998) Curr. Opin. Struct. Biol. 8, 704-712. [DOI] [PubMed] [Google Scholar]
- 40.Brieba, L. J., Eichman, B. F., Kokoska, R. J., Doublie, S., Kunkel, T. A. & Ellenberger, T. (2004) EMBO J. 23, 3452-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li, Y., Korolev, S. & Waksman, G. (1998) EMBO J. 17, 7514-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, Y., Dutta, S., Doublie, S., Bdour, H. M., Taylor, J. S. & Ellenberger, T. (2004) Nat. Struct. Mol. Biol. 11, 784-790. [DOI] [PubMed] [Google Scholar]
- 43.Lipkowitz, K. B., Chevalier, T., Widdifield, M. & Beland, F. A. (1982) Chem. Biol. Interact. 40, 57-76. [DOI] [PubMed] [Google Scholar]
- 44.Foloppe, N., Hartmann, B., Nilsson, L. & MacKerell, A. D., Jr. (2002) Biophys. J. 82, 1554-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kriek, E. (1992) J. Cancer Res. Clin. Oncol. 118, 481-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strauss, B. S. (1991) BioEssays 13, 79-84. [DOI] [PubMed] [Google Scholar]
- 47.Hogg, M., Wallace, S. S. & Doublie, S. (2004) EMBO J. 23, 1483-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson, S. J., Taylor, J. S. & Beese, L. S. (2003) Proc. Natl. Acad. Sci. USA 100, 3895-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}