

Abstract
Herein, we report the synthesis of novel 2′,2′,3′,3′-tetrafluorinated nucleoside analogs along with their phosphoramidate prodrugs. A tetrafluoro ribose moiety was coupled with different Boc/benzoyl-protected nucleobases under Mitsunobu conditions. After deprotection, tetrafluorinated nucleosides 13b, 14b, 20b–22b were reacted with phenyl-(isopropoxy-L-alaninyl)-phosphorochloridate to afford corresponding monophosphate prodrugs 24b–28b. All synthesized compounds were evaluated against several DNA and RNA viruses including HIV, HBV, HCV, Ebola and Zika viruses.
Keywords: Nucleoside, Prodrug, Antiviral, Fluorine, Mitsunobu
Graphical abstract
Incorporation of fluorine(s) into potential drug candidates continues to be investigated by medicinal chemists and has shown the potential to confer favorable biological properties.1 Due to the similarity in size of fluorine (147 pm) and hydrogen (120 pm), the substitution of a single hydrogen atom by a fluorine atom induces only a minor change in steric factors. However, being the most electronegative atom in the periodic table, its incorporation results in considerable electronic changes in the molecule through inductive electron withdrawing effects. Furthermore, introduction of one or more atoms of fluorine (e.g., CF2) into a molecule is a viable bioisosteric replacement for a diverse set of functional groups such as carbonyl, ether and hydroxyl groups. Finally, a C–F bond is much stronger than a CH bond, which can increase the chemical and biological stability of the fluorine-containing compounds, compared to their hydrogen counterparts.
In the field of nucleoside analogs,2 introduction of fluorine has proven to be successful and has led to several very widely prescribed drugs such as (−)-FTC (HIV), gemcitabine (cancer), or more recently sofosbuvir (hepatitis C virus, HCV) (Figure 1). In addition to the approved fluorinated nucleosides discussed above, several fluorine-containing analogs have been reported in the literature to have attractive antiviral profiles, although some proved too toxic for human use. Thus, 2′-β-fluoro nucleosides analogs such as FIAC, FEAU, FMAU exhibit potent activities against herpes simplex virus (HSV), hepatitis B virus (HBV), varicella zoster virus (VZV), cytomegalovirus (CMV) and Epstein-Barr virus (EBV).3
Figure 1.
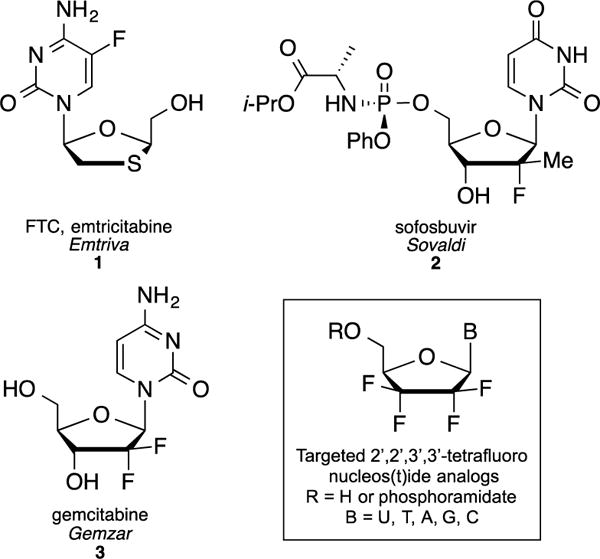
Structures of FDA approved fluorine containing nucleosides 1–3 and targeted 2′,2′,3′,3′-tetrafluoro derivatives.
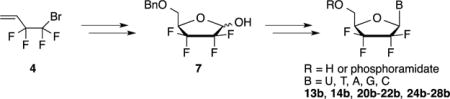
Based on these precedents, we turned our attention toward a unique series of 2′,2′,3′,3′-tetrafluorinated nucleoside analogs. Even though this type of highly fluorinated compounds is claimed in a patent published in 2000,4 their preparation is only described in a generic scheme and with no mention of any biological evaluation. Therefore, we report herein the detailed synthesis and antiviral evaluation of, not only these compounds, but also their phosphoramidate prodrugs.
2,2,3,3-Tetrafluorodideoxy ribose derivative 7 was prepared in four steps from commercially available 4-bromo-3,3,4,4-tetrafluorobut-1-ene 4 following a procedure reported by Linclau et al.5 (Scheme 1). It is noteworthy that the Sharpless dihydroxylation of the perfluorinated substrate 4 is not entirely enantioselective (reported ee 78%, observed 60%). Consequently, the 4′-position of the lactol 7, obtained after cyclization of the formylated intermediate 6, is isolated as a 8:2 mixture at the 4′-position while the α/β anomers ratio is 1:1.5. The separation of the diastereomers was not attempted at this stage. Hence, with 7 in hand, the glycosylation reaction was studied and different approaches were evaluated in order to identify the most efficient coupling conditions. Vorbrüggen type coupling of 1′-benzoyl, 1′-mesyl or 1′-triflate tetrafluorinated ribose derivative 7 with either silylated uracil or cytosine in presence of SnCl4 (or TMSOTf) only led to the complete recovery of starting material 8. Attempts to directly react the 1′-triflate derivative 8c with the sodium salt of N3-benzoyluracil was unsuccessful as well.
Scheme 1.
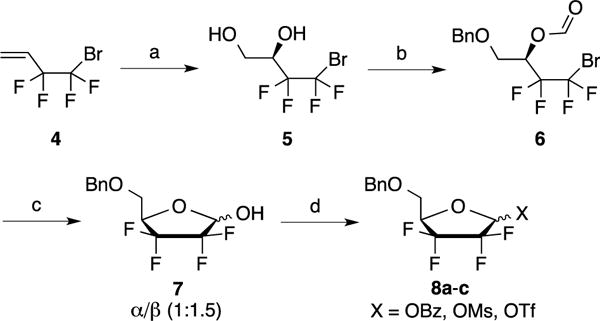
Reagents and conditions: a) K2OSO4.2H2O, (DHQ)2Pyr, K3Fe(CN)6, t-BuOH/H2O, 4 °C, 9 days, 81%; b) i) Bu2SnO, TBAI, BnBr, toluene, reflux, 82%; ii) Formic acid, DCC, DMAP, CH2Cl2, rt, 16 h, 95%; c) MeLi, THF, −78 °C, 3 h, 62%; d) BzCl, Et3N, CH2Cl2, rt, 1.5 h, 92% or MsCl, Et3N, CH2Cl2, 0 °C to rt, 16 h, 74% or Tf2O, pyr, 0 °C to rt, 16 h, quant.
In contrast, treatment of compound 7 with N3-benzoyluracil or N3–benzoylthymine under classical Mitsunobu conditions (DIAD and PPh3 in THF) afforded the desired coupling products 9a–b (60%) and 10a–b (68%) respectively as a silica gel separable mixture of α/β anomers (ratio: 1.5:1) (Scheme 2). Remarkably, the desired coupling products 9 and 10 were only observed when DIAD was added slowly at −20 °C. Performing this reaction at higher temperatures led to the formation of several unidentified side products as determined by LC-MS. The isolated β anomers 9b and 10b were debenzoylated using methanolic ammonia to afford intermediates 11b (91%) and 12b (96%), which were subsequently treated with BCl3 to give uracil and thymine analogs 13b (94%) and 14b (85%), respectively. Preparation of the adenine analog was initially attempted by coupling of protected lactol 7 with 6-chloropurine using the Mitsunobu conditions described above (Scheme 3). However, even though the desired coupling product 15 was obtained in 34% yield, separation of the α/β isomers, at this stage, or after 6-amination of the purine ring (compound 16), was found to be challenging.
Scheme 2.
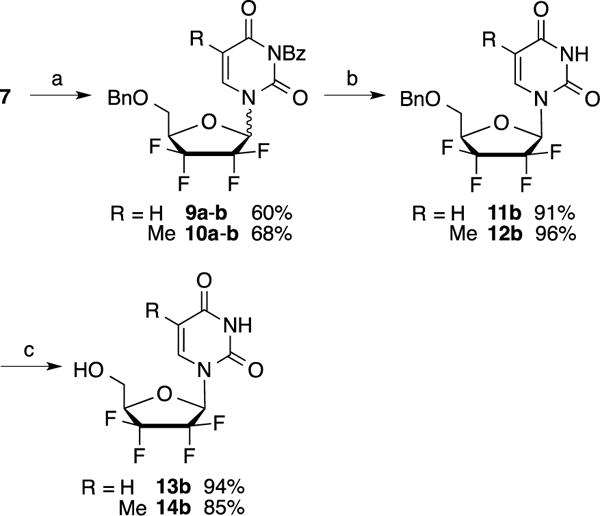
Reagents and conditions: a) N3-BzU or N3-BzT, DIAD, PPh3, THF, −20 °C to rt, 2 h (overall yields for both anomers); b) NH3, MeOH, 4 °C, 2 h; c) BCl3, DCM, −20 °C to rt, 3.5 h.
Scheme 3.
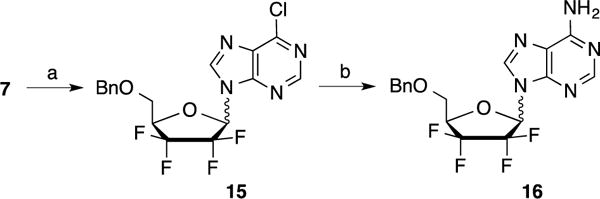
Reagents and conditions: a) 6-chloropurine, DIAD, PPh3, THF, 2 h, rt, 34%; b) NH3/MeOH, 80 °C, 16 h, 86%.
Therefore, we then examined the coupling of lactol 7 with bisBoc adenine.6 Under classical Mitsunobu conditions, the reaction afforded products 17a–b in 58% yield (Scheme 4) as a silica gel separable mixture of α/β anomers (1.5:1). Further treatment of the β-anomer 17b with BCl3 afforded adenine nucleoside analog 20b in 96% yield. Likewise, the coupling with bis-Boc-2-amino-6-benzolyloxypurine7 afforded a mixture of anomers 18 (48%) that were separable by silica gel chromatography. The β-isomer 18b was then fully deprotected in one step to give 21b in 40% yield using BCl3. Finally, the preparation of cytosine analogs 19a–b was performed in a similar manner using bisBoc-cytosine8. The separated β-anomer 19b was subsequently deprotected to afford nucleoside analog 22b in 82% yield.
Scheme 4.
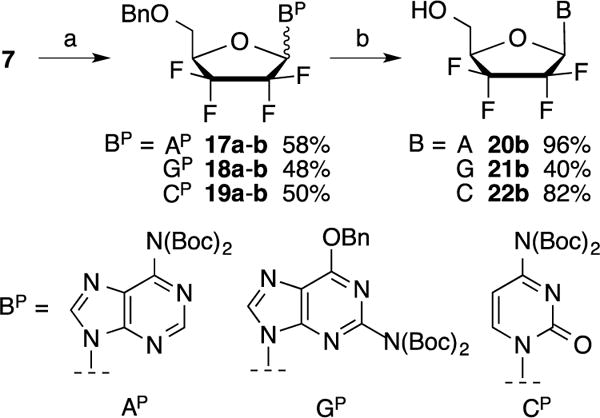
Reagents and conditions: a) Protected base (Bp), DIAD, PPh3, THF, −20 °C to rt, 2 h (overall yields for both anomers); b) BCl3, DCM, −20 °C to rt, 3.5 h.
Identification of both α and β anomers was determined by 1H-NMR and 2D-NOE experiments after Mitsunobu coupling. In all cases, clear correlations were observed between 1′-protons and 4′-protons of β anomers while α anomers exhibited correlations between 1′-protons and 5′-protons. Additionally, in the case of purine analogs 17 and 18, correlations between 8-protons and 5′-protons were also noted (Figure 2).
Figure 2.
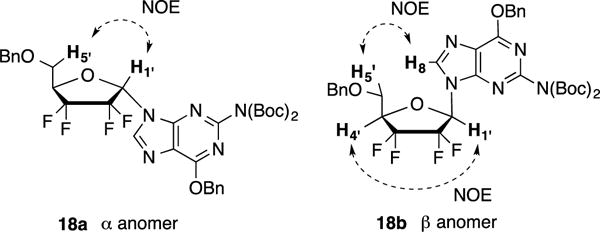
Anomer assignment for compound 18a and 18b via NOE experiments
In order to express their therapeutic effect, nucleoside analogs usually rely on cellular kinases to be phosphorylated onto their active 5′-triphosphate forms. Among the three consecutive phosphorylations, the first phosphate addition has often been identified as the limiting step which led to the development of “protected” monophosphate nucleosides, or nucleoside prodrugs, that can by-pass this first activation step.9 Unlike, 5′-mono, di or triphosphate nucleosides, theses prodrugs are capable of crossing the cell membrane to deliver a monophosphate nucleoside after enzymatic degradation of their biolabile group. The concept of monophosphate or phosphonate prodrugs has been clinically validated with the FDA-approval of drugs such as sofosbuvir 3 (HCV) or tenofovir disoproxyl fumarate (TDF) (HIV and HBV).
Thus, 5′-monophosphate prodrugs of nucleosides 13b, 14b and 20b–22b were prepared by reaction with the chloro phosphoramidate 23 via two different strategies, depending on the nature of the nucleobase (Scheme 5).10 For free-NH2 containing analogs, such as adenine, guanine and cytosine derivatives 20b, 21b and 22b, t-BuMgCl was used to favor alkoxide formation and minimize side reactions with the amino group while the pyrimidine prodrugs 24b and 25b were prepared by treatment of 13b and 14b with N-methylimidazole (NMI). Full conversions were usually observed after 3 h at room temperature affording prodrugs 24b–28b, as a RP and SP mixture, in yields ranging from 49% to 64%. Due to the fact that the synthesis of the tetrafluorinated sugar 7 is not completely diastereoselective at the 4′-position, 31P NMR spectra for these compounds indicated the presence of four non separable diastereoisomers in a 4:1:1:4 ratio.
Scheme 5.
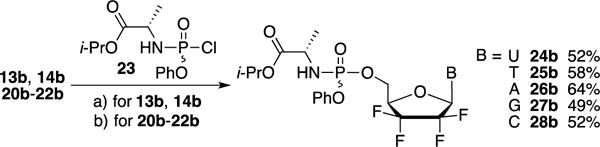
Reagents and conditions: a) NMI, THF, rt, 3 h; b) t-BuMgCl, THF, rt, 3 h.
To determine the effects of the tetrafluoro substitution, the energetics of ring conformations were investigated using quantum mechanical calculations (Figure 3).
Figure 3.
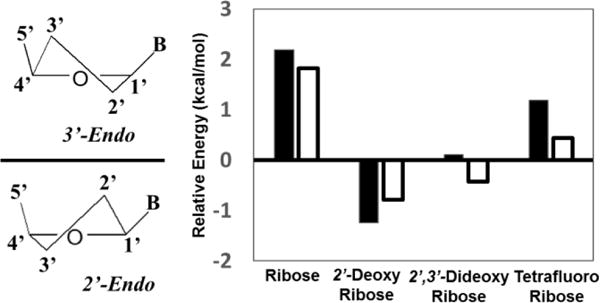
Energetics of ring conformations for differently substituted nucleoside analogs. The y-axis is the relative energy of E[2′-endo] – E[3′-endo] for each analog. The dark bars represent analogs with an uracil base, and white bars are analogs with an adenine base. Nucleoside analogs that display positive values favor the 3′-endo conformation and negative values indicate that the 2′-endo conformer is more stable.
Consistent with previous data, the ribose and 2′-deoxyribose rings exhibited lower energies in the 3′-endo and 2′-endo conformers, respectively.11 The 3′-endo and 2′-endo conformers were equal in energy for the 2′,3′-dideoxyribose U and A analogs. The tetrafluoro-2′,3′-dideoxyribose U and A analogs preferred the 3′-endo conformation by 0.4 – 1.2 kcal/mol. This result suggests that the tetrafluorinated ribose moiety might be a closer mimic of a ribose rather than a 2′,3′-dideoxyribose.
Consequently, nucleosides 13b, 14b, 20b–22b and their corresponding phosphoramidate prodrugs 24b–28b were evaluated against a panel of DNA and RNA viruses. Hence, the screening was performed against HIV-112 up to 100 μM and HBV12 (HepAD38), HCV13 (clone B replicon), Ebola14 (Zaire ebolavirus replicon) and Zika15 (cytopathic effect reduction assay) viruses up to 20 μM. In addition, cytotoxicity was determined in primary human peripheral blood mononuclear (PBM) cells, human lymphoblastoid CEM, African Green monkey Vero cells and HepG2 cells.16 Except for compound 26b that exhibited slight toxicities in human PBM (CC50 = 27 μM) and CEM (CC50 = 14 μM) cells, none of these derivatives exhibited significant activity against these viruses nor toxicities in PBM, CEM and Vero cells up to 100 μM.
In conclusion, five new 2′,2′,3′,3′-tetrafluorodideoxy nucleoside analogs along with their corresponding phosphoramidate prodrugs were synthesized from tetrafluorinated lactol 7 using a key Mitsunobu reaction. We demonstrated that the use of Boc or Bz-protected bases as glycosylation partners in this reaction facilitated the separation of both α and β anomers. None of the synthesized compounds showed marked activity when tested against HIV-1, HBV, HCV, Ebola or Zika viruses.
Supplementary Material
Highlights.
The synthesis of novel 2′,2′,3′,3′-tetrafluoro nucleoside analogs is described
Glycosylation reactions are achieved via Mitsunobu coupling
Energetics of ring conformation are compared to differently substituted ribose
Evaluation of Tetrafluoro nucleosides in vitro against a panel of DNA and RNA viruses
Acknowledgments
This work was supported in part by NIH Grant 5P30-AI-50409 (CFAR). Dr. Schinazi is the Chairman and a major shareholder of Cocrystal Pharma, Inc. Emory received no funding from Cocrystal Pharma, Inc. to perform this work and vice versa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data (experimental details, NMR data and HRMS) associated with this article can be found, in the online version.
References
- 1.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 2.(a) Liu P, Sharon A, Chu CK. J Fluorine Chem. 2008;129:743–766. doi: 10.1016/j.jfluchem.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wójtowicz-Rajchel H. J Fluorine Chem. 2012;143:11–48. [Google Scholar]
- 3.(a) Wright JA, Taylor NF, Fox JJ. J Org Chem. 1969;34:2632–2636. doi: 10.1021/jo01261a031. [DOI] [PubMed] [Google Scholar]; (b) Reichman U, Watanabe KA, Fox JJ. Carbohydr Res. 1975;42:233–240. doi: 10.1016/s0008-6215(00)84265-2. [DOI] [PubMed] [Google Scholar]; (c) Watanabe KA, Su TL, Klein RS, Chu CK, Matsuda A, Chun MW, Lopez C, Fox JJ. J Med Chem. 1983;26:152–156. doi: 10.1021/jm00356a007. [DOI] [PubMed] [Google Scholar]; (d) Watanabe KA, Su TL, Reichman U, Greenberg N, Lopez C, Fox JJ. J Med Chem. 1984;27:91–94. doi: 10.1021/jm00367a020. [DOI] [PubMed] [Google Scholar]; (e) Perlman ME, Watanabe KA, Schinazi RF, Fox JJ. J Med Chem. 1985;28:741–748. doi: 10.1021/jm00383a009. [DOI] [PubMed] [Google Scholar]; (f) Su TL, Watanabe KA, Schinazi RF, Fox JJ. J Med Chem. 1986;29:151–154. doi: 10.1021/jm00151a025. [DOI] [PubMed] [Google Scholar]
- 4.Dimagno SG, Nebr L. Heavily fluorinated sugar analogs. 6,013,790. US Patent. 2000 Jan 11;
- 5.(a) Boydell AJ, Vinader V, Linclau B. Angew Chem Int Ed. 2004;43:5677–5679. doi: 10.1002/anie.200460746. [DOI] [PubMed] [Google Scholar]; (b) Linclau B, Boydell AJ, Timofte RS, Brown KJ, Vinader V, Weymouth-Wilson AC. Org Biomol Chem. 2009;7:803–814. doi: 10.1039/b817260a. [DOI] [PubMed] [Google Scholar]
- 6.Dey S, Garner P. J Org Chem. 2000;65:7697–7699. doi: 10.1021/jo000983i. [DOI] [PubMed] [Google Scholar]
- 7.Kramer RA, Bleicher KH, Wennemers H. Helv Chim Acta. 2012;95:2621–2634. [Google Scholar]
- 8.Porcheddu A, Giacomelli G, Piredda I, Carta M, Nieddu G. Eur J Org Chem. 2008;34:5786–5797. [Google Scholar]
- 9.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Chem Rev. 2014;114:9154–9218. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGuigan C, Harris SA, Daluge SM, Gudmundsson KS, McLean EW, Burnette TC, Marr H, Hazen R, Condreay LD, Johnson L, De Clercq E, Balzarini J. J Med Chem. 2005;48:3504–3515. doi: 10.1021/jm0491400. [DOI] [PubMed] [Google Scholar]
- 11.Huang M, Giese TJ, Lee TS, York DM. J Chem Theory Comput. 2014;10:1538–1545. doi: 10.1021/ct401013s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stuyver LJ, Lostia S, Adams M, Mathew JS, Pai BS, Grier J, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854–3860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuyver LJ, Whitaker T, McBrayer TR, Hernandez-Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J, Rachakonda S, Watanabe KA, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2003;47:244–254. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uebelhoer LS, Albariño CG, McMullan LK, Chakrabarti AK, Vincent JP, Nichol ST, Towner JS. Antiviral Res. 2014;106:86–94. doi: 10.1016/j.antiviral.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Zmurko J, Marques RE, Schols D, Verbeken E, Kaptein SJF, Neyts J. PLoS Negl Trop Dis. 2016;10:1–15. doi: 10.1371/journal.pntd.0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MW, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061–1067. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854–3860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.